
Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging

Abstract
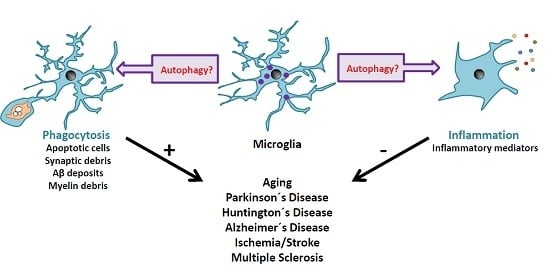
:
1. Introduction
2. Autophagy: The Facts
3. Technical Pitfalls in Autophagy Research
4. Autophagy in the Central Nervous System (CNS)
4.1. Aging
4.2. Acute Neurodegeneration—Ischemia/Stroke
4.3. Chronic Neurodegeneration—Alzheimer’s, Parkinson’s, and Huntington’s Diseases
4.4. Autoimmunity-Mediated Secondary Neurodegeneration—Multiple Sclerosis
5. Autophagy in Microglia
6. Autophagy and Microglial Phagocytosis
6.1. LC3-Associated Phagocytosis
6.2. Autophagy Modulation of Phagocytosis Efficiency
7. Autophagy and Microglial Phagocytosis in Aging and Neurodegeneration
7.1. Apoptotic Cells
7.2. Amyloid-β
7.3. Synaptic Pruning
7.4. Myelin Debris
8. Autophagy and Microglial Inflammation
Autophagy and Inflammasomes
9. Autophagy and Microglial Inflammation during Aging and Neurodegeneration
9.1. Aging
9.2. Acute Neurodegeneration—Ischemia/Stroke
9.3. Chronic Neurodegeneration—Alzheimer’s, Parkinson’s, and Huntington’s Diseases
9.4. Autoimmunity-Mediated Secondary Neurodegeneration—Multiple Sclerosis
10. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| CNS | Central nervous system |
| MTORC1 | Mechanistic target of rapamycin 1 |
| ULK-1 | Unc-51 like autophagy activating kinase 1 |
| BECN-1 | Beclin-1 |
| Vps34 | Vacuolar protein sorting 34 |
| PI3P | Phosphatidyl-inositol-3-phosphate |
| ATG | Autophagy-related gene |
| LC3 | Microtubule-associated light chain 3 |
| SQSTM-1 | Sequestosome 1 |
| NBR1 | Neighbor of BRCA1 |
| NIX | NIP-3 like protein X |
| NCCD | Nomenclature committee on cell death |
| 3-MA | 3-methyladenine |
| PI3K | Phophatidyl-inositol-3-kinase |
| CR | Caloric restriction |
| PD | Parkinson’s Disease |
| SNpc | Substantia nigra pars compacta |
| DA | Dopamine |
| PINK-1 | PTEN-induced putative kinase-1 |
| TFEB | Transcription factor EB |
| HD | Huntington’s Disease |
| mHTT | Mutant huntingtin |
| HTT | Huntingtin |
| AD | Alzheimer’s Disease |
| Aβ | amyloid-β |
| PS-1 | Presenilin-1 |
| TSC-1 | Tuberous sclerosis complex-1 |
| mdivi-1 | Mitochondrial division inhibitor-1 |
| tMCAO | Transient middle cerebral artery occlusion |
| MS | Multiple sclerosis |
| EAE | Experimental autoimmune encephalomyelitis |
| DC | Dendritic cell |
| LAP | LC3-associated phagocytosis |
| TLR | Toll-like receptor |
| TIM4 | T cell immunoglobulin mucin protein 4 |
| SLE | Systemic lupus erythemathosus |
| TREM-2 | Triggering receptor expressed on myeloid cells 2 |
| FTD | Frontotemporal dementia |
| ASD | Autism spectrum disorders |
| PNS | Peripheral nervous system |
| GFP | Green fluorescent protein |
| ASC | Apoptosis-associated speck-like protein containing CARD |
| ROS | Radical oxygen species |
| IL-1β | Interleukin-1β |
| IL-18 | Interleukin-18 |
| IL-6 | Interleukin-6 |
| GM-CSF | Granulocyte-macrophage colony stimulating factor |
| TNF-α | Tumor necrosis factor-α |
| IL-10 | Interleukin-10 |
| TGF-β | Transforming growth factor-β |
| LPS | Lipopolysaccharide |
| NLRP3 | nucleotide-binding domain, leucine-rich-repeat-containing, pyrin-domain-containing 3 |
| MPP+ | 1-Methyl-4-phenylpyridinium |
| MPTPp | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine/probenecid |
| CB2R | Cannabinoid receptor 2 |
| pMCAO | Permanent middle cerebral artery occlusion |
| GSK-3β | Glycogen synthase kinase-3 β |
| iNOS | Inducible nitric oxide synthase |
References
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Stefanis, L. Chaperone mediated autophagy in aging: Starve to prosper. Ageing Res. Rev. 2016, 32, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2014, 7, 673–682. [Google Scholar] [CrossRef]
- Tasset, I.; Cuervo, A.M. Role of chaperone-mediated autophagy in metabolism. FEBS J. 2016, 283, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S. Regulation of autophagy by MTOR-dependent and MTOR-independent pathways: Autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Inagaki, F. Mechanisms of autophagy. Annu. Rev. Biophys. 2015, 44, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Osawa, T.; Fujioka, Y.; Noda, N.N. Structural biology of the core autophagy machinery. Curr. Opin. Struct. Biol. 2016, 43, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: A key role for RAB7. Biochim. Biophys. Acta. 2013, 1833, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E. Maturation of autophagic vacuoles in mammalian cells. Autophagy 2005, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [PubMed]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell. Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy in protein and organelle turnover. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 397–402. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Munz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta. 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.-U. Calpain-mediated cleavage of ATG5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Florey, O.; Overholtzer, M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol. 2012, 22, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; Yuan, J. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; König, J.; Höhn, A.; Jung, T.; Grune, T. Macroautophagy is impaired in old murine brain tissue as well as in senescent human fibroblasts. Redox Biol. 2016, 10, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chu, X.; Yin, M.; Liu, X.; Yuan, H.; Niu, Y.; Fu, L. MTOR and autophagy in normal brain aging and caloric restriction ameliorating age-related cognition deficits. Behav. Brain Res. 2014, 264, 82–90. [Google Scholar] [CrossRef]
- De Cabo, R.; Carmona-Gutierrez, D.; Bernier, M.; Hall Michael, N.; Madeo, F. The search for antiaging interventions: From elixirs to fasting regimens. Cell 2014, 157, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.M.; Schafer, M.J.; LeBrasseur, N.K. Energetic interventions for healthspan and resiliency with aging. Exp. Gerontol. 2016, 86, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.M.; Herbert, R.L.; Longo, D.L.; Allison, D.B.; Young, J.E.; Bryant, M.; et al. Impact of caloric restriction on health and survival in rhesus monkeys from the nia study. Nature 2012, 489, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Mejías-Peña, Y.; Estebanez, B.; Rodriguez-Miguelez, P.; Fernandez-Gonzalo, R.; Almar, M.; de Paz, J.; González-Gallego, J.; Cuevas, M. Impact of resistance training on the autophagy-inflammation-apoptosis crosstalk in elderly subjects. Aging 2017, 9, 408–418. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Sumpter, J.R.; Levine, B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012, 8, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Blomgren, K.; Kroemer, G. Autophagy in acute brain injury. Nat. Rev. Neurosci. 2016, 17, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-D.; Sheng, R.; Zhang, L.-S.; Han, R.; Zhang, X.; Zhang, X.-D.; Han, F.; Fukunaga, K.; Qin, Z.-H. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 2008, 4, 762–769. [Google Scholar] [CrossRef]
- Jeong, J.H.; Yu, K.S.; Bak, D.H.; Lee, J.H.; Lee, N.S.; Jeong, Y.G.; Kim, D.K.; Kim, J.J.; Han, S.Y. Intermittent fasting is neuroprotective in focal cerebral ischemia by minimizing autophagic flux disturbance and inhibiting apoptosis. Exp. Ther. Med. 2016, 12, 3021–3028. [Google Scholar] [PubMed]
- Buckley, K.M.; Hess, D.L.; Sazonova, I.Y.; Periyasamy-Thandavan, S.; Barrett, J.R.; Kirks, R.; Grace, H.; Kondrikova, G.; Johnson, M.H.; Hess, D.C.; et al. Rapamycin up-regulation of autophagy reduces infarct size and improves outcomes in both permanent MCAL, and embolic MCAO, murine models of stroke. Exp. Transl. Stroke Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, M.; Hadley, G.; Xilouri, M.; Hoyte, L.C.; Nagel, S.; McMenamin, M.M.; Tsaknakis, G.; Watt, S.M.; Drakesmith, C.W.; Chen, R.; et al. TSC1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat. Med. 2013, 19, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Zhang, X.Y.; Han, R.; Zhang, T.T.; Chen, C.; Qin, Z.H.; Sheng, R. The endoplasmic reticulum stress inhibitor salubrinal inhibits the activation of autophagy and neuroprotection induced by brain ischemic preconditioning. Acta. Pharmacol. Sin. 2013, 34, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.-R.; Wang, X.; Hu, W.-W.; et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar] [CrossRef]
- Zheng, Y.Q.; Liu, J.X.; Li, X.Z.; Xu, L.; Xu, Y.G. RNA interference-mediated downregulation of beclin1 attenuates cerebral ischemic injury in rats. Acta. Pharmacol. Sin. 2009, 30, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- DuBoff, B.; Feany, M.; Götz, J. Why size matters—Balancing mitochondrial dynamics in Alzheimer's disease. Trends Neurosci. 2013, 36, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Whyte, L.S.; Lau, A.A.; Hemsley, K.M.; Hopwood, J.J.; Sargeant, T.J. Endo-lysosomal and autophagic dysfunction: A driving factor in Alzheimer’s disease? J. Neurochem. 2017, 140, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.; Bandyopadhyay, U.; Jiang, Y.; et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011, 134, 258–277. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012, 135, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. The Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, A.J.; Pallanck, L.J. PINK1/Parkin mitophagy and neurodegeneration—What do we really know in vivo? Curr. Opin. Genet. Dev. 2017, 44, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to α-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Björklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Harjes, P.; Wanker, E.E. The hunt for huntingtin function: Interaction partners tell many different stories. Trends Biochem. Sci. 2003, 28, 425–433. [Google Scholar] [CrossRef]
- Caviston, J.P.; Holzbaur, E.L.F. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009, 19, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.D.O.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Humbert, S. The biology of huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. The regulation of autophagosome dynamics by Huntingtin and HAP1 is disrupted by expression of mutant Huntingtin, leading to defective cargo degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of MTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.; Menzies, F.M.; Renna, M.; Acevedo-Arozena, A.; Corrochano, S.; Sadiq, O.; Brown, S.D.; Rubinsztein, D.C. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2010, 19, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Ashe, T.D.; Morrison, B.E.; Soriano, K.R.; Au, J.; Roque, R.A.V.; Lazarowski, E.R.; Damian, V.A.; Masliah, E.; la Spada, A.R. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Magliozzi, R.; Ciccarelli, O.; Geurts, J.J.G.; Reynolds, R.; Martin, R. Exploring the origins of grey matter damage in multiple sclerosis. Nat. Rev. Neurosci. 2015, 16, 147–158. [Google Scholar] [CrossRef] [PubMed]
- González, H.; Pacheco, R. T cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J. Neuroinflammation. 2014, 11, 201. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.R.; Li, C.; Yang, Q.; Li, G.; Garcia, I.G.; Ju, S.; Roodman, D.G.; Windle, J.J.; Zhang, X.; Lu, B. Autophagy promotes T cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012, 19, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Fox, H.S.; Flynn, C.T.; Moore, C.S.; Hebb, A.L.O.; Frausto, R.F.; Bhan, V.; Kiosses, W.B.; Whitton, J.L.; Robertson, G.S.; et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 2009, 5, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Igci, M.; Baysan, M.; Yigiter, R.; Ulasli, M.; Geyik, S.; Bayraktar, R.; Bozgeyik, İ.; Bozgeyik, E.; Bayram, A.; Cakmak, E.A. Gene expression profiles of autophagy-related genes in multiple sclerosis. Gene 2016, 588, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Parillon, X.; Zeng, S.; Han, S.; Eissa, N.T. Deficiency of autophagy in dendritic cells protects against experimental autoimmune encephalomyelitis. J. Biol. Chem. 2014, 289, 26525–26532. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; White, E. Autophagy, metabolism, and cancer. Cold Spring Harb. Symp. Quant. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Dello Russo, C.; Lisi, L.; Feinstein, D.L.; Navarra, P. MTOR kinase, a key player in the regulation of glial functions: Relevance for the therapy of multiple sclerosis. Glia 2013, 61, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Yue, Z. Autophagy and its normal and pathogenic states in the brain. Annu. Rev. Neurosci. 2014, 37, 55–78. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Oguin, T.H.; Martinez, J. The clearance of dying cells: Table for two. Cell Death Differ. 2016, 23, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol. Rev. 2016, 272, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Puleston, D.J.; Simon, A.K. Autophagy and immune senescence. Trends Mol. Med. 2016, 22, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and “garb-aging”. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Stranks, A.J.; Hansen, A.L.; Panse, I.; Mortensen, M.; Ferguson, D.J.P.; Puleston, D.J.; Shenderov, K.; Watson, A.S.; Veldhoen, M.; Phadwal, K.; et al. Autophagy controls acquisition of aging features in macrophages. J. Innate Immun. 2015, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Romao, S.; Gasser, N.; Becker, A.C.; Guhl, B.; Bajagic, M.; Vanoaica, D.; Ziegler, U.; Roesler, J.; Dengjel, J.; Reichenbach, J.; et al. Autophagy proteins stabilize pathogen-containing phagosomes for prolonged mhc ii antigen processing. J. Cell Biol. 2013, 203, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, P.; Henault, J.; Kolbeck, R.; Sanjuan, M.A. Noncanonical autophagy: One small step for LC3, one giant leap for immunity. Curr. Opin. Immunol. 2014, 26, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 α (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Cunha, L.D.; Park, S.; Yang, M.; Lu, Q.; Orchard, R.; Li, Q.-Z.; Yan, M.; Janke, L.; Guy, C.; et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 2016, 533, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.G.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Cemma, M.; Grinstein, S.; Brumell, J.H. Autophagy proteins are not universally required for phagosome maturation. Autophagy 2016, 12, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Tan, Y.-z.; Wang, H.-j.; Li, T.; He, T.; Yu, Y.; Zhang, J.; Zhang, D. Cytoprotective effect of autophagy on phagocytosis of apoptotic cells by macrophages. Exp. Cell Res. 2016, 348, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.G.B.; de Freitas Vinhas, C.; Gomes, I.N.; Azevedo, C.M.; dos Santos, R.R.; Vannier-Santos, M.A.; Veras, P.S.T. Phagocytosis is inhibited by autophagic induction in murine macrophages. Biochem. Biophys. Res. Commun. 2011, 405, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Schrijvers, D.M.; Timmermans, J.-P.; Herman, A.G.; De Meyer, G.R.Y. Phagocytosis of bacteria is enhanced in macrophages undergoing nutrient deprivation. FEBS J. 2009, 276, 2227–2240. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, D.L.; Bhattacharya, A.; Sha, Y.; Xu, Y.; Xiang, Q.; Kan, A.; Jagannath, C.; Komatsu, M.; Eissa, N.T. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity 2013, 39, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Eugenin-von Bernhardi, L.; Eugenin, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Abiega, O.; Beccari, S.; Diaz-Aparicio, I.; Nadjar, A.; Layé, S.; Leyrolle, Q.; Gómez-Nicola, D.; Domercq, M.; Pérez-Samartín, A.; Sánchez-Zafra, V.; et al. Neuronal hyperactivity disturbs atp microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol. 2016, 14, e1002466. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Théry, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Solé-Domènech, S.; Cruz, D.L.; Capetillo-Zarate, E.; Maxfield, F.R. The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res. Rev. 2016, 32, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.-Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.I.; Elguero, E.J.; Tran, J.A.; Sinatra, V.; Feany, M.B.; McCall, K. Defective phagocytic corpse processing results in neurodegeneration and can be rescued by torc1 activation. J. Neurosci. 2016, 36, 3170–3183. [Google Scholar] [CrossRef] [PubMed]
- Lucin, K.M.; O’Brien, C.E.; Bieri, G.; Czirr, E.; Mosher, K.I.; Abbey, R.J.; Mastroeni, D.F.; Rogers, J.; Spencer, B.; Masliah, E.; et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 2013, 79, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.-H.; Cho, K.; Kang, H.-J.; Jeon, E.-Y.; Kim, H.-S.; Kwon, H.-J.; Kim, H.-M.; Kim, D.-H.; Yoon, S.-Y. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2016, 36, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Cho, M.H.; Shim, W.H.; Kim, J.K.; Jeon, E.Y.; Kim, D.H.; Yoon, S.Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2016. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.; Werner, H. Myelination of the nervous system: Mechanisms and functions. Annu. Rev. Cell. Dev. Biol. 2014, 30, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Safaiyan, S.; Kannaiyan, N.; Snaidero, N.; Brioschi, S.; Biber, K.; Yona, S.; Edinger, A.L.; Jung, S.; Rossner, M.J.; Simons, M. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 2016, 19, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, J.A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M.; Griffith, M.; Hantke, J.; Macias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.Y.; Shin, Y.K.; Park, S.Y.; Park, J.Y.; Lee, H.J.; Yoo, Y.H.; Kim, J.K.; Park, H.T. Autophagic myelin destruction by schwann cells during wallerian degeneration and segmental demyelination. Glia 2016, 64, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, E.; Novikoff, A.B. Lysosomes in the rat sciatic nerve following crush. J. Cell Biol. 1965, 27, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Cai, W.; Lee, H.K.; Pellegatta, M.; Shin, Y.K.; Jang, S.Y.; Suh, D.J.; Wrabetz, L.; Feltri, M.L.; Park, H.T. Actin polymerization is essential for myelin sheath fragmentation during wallerian degeneration. J. Neurosci. 2011, 31, 2009–2015. [Google Scholar] [CrossRef] [PubMed]
- Reichert, F.; Saada, A.; Rotshenker, S. Peripheral nerve injury induces schwann cells to express two macrophage phenotypes: Phagocytosis and the galactose-specific lectin MAC-2. J. Neurosci. 1994, 14, 3231–3245. [Google Scholar] [PubMed]
- Leonhard, C.; Müller, M.; Hickey, W.F.; Ringelstein, E.B.; Kiefer, R. Lesion response of long-term and recently immigrated resident endoneurial macrophages in peripheral nerve explant cultures from bone marrow chimeric mice. Eur. J. Neurosci. 2002, 16, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Maeda, M.; Tsuda, M.; Tozaki-Saitoh, H.; Inoue, K.; Kiyama, H. Nerve injury-activated microglia engulf myelinated axons in a P2Y12 signaling-dependent manner in the dorsal horn. Glia 2010, 58, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.U.; de Vellis, J. Microglia in health and disease. J. Neurosci. Res. 2005, 81, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Hamidzadeh, K.; Christensen, S.M.; Dalby, E.; Chandrasekaran, P.; Mosser, D.M. Macrophages and the recovery from acute and chronic inflammation. Annu. Rev. Physiol. 2016, 79, 567–592. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Krüger, E. Dysfunction in protein clearance by the proteasome: Impact on autoinflammatory diseases. Semin. Immunopathol. 2015, 37, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Zhang, J.; Wang, D.; Zhao, F.; Cao, Z.; Aschner, M.; Luo, W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience 2016, 319, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.; Li, Y.; Peng, I.; Reichelt, M.; Katakam, A.K.; Noubade, R.; Roose-Girma, M.; DeVoss, J.; Diehl, L.; Graham, R.R.; et al. A crohn/’s disease variant in atg16l1 enhances its degradation by caspase 3. Nature 2014, 506, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.-Z.; Wei, W.; Ke, P.; Xu, Z.-Q.; Zhou, J.-X.; Liu, C. Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci. Ther. 2014, 20, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, A.; Meisel, A.; Planas, A.M.; Urra, X.; van de Beek, D.; Veltkamp, R. The immunology of acute stroke. Nat. Rev. Neurol. 2012, 8, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Joers, V.; Tansey, M.G.; Mulas, G.; Carta, A.R. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.K.; Yong, V.W. Myeloid cells—Targets of medication in multiple sclerosis. Nat. Rev. Neurol. 2016, 12, 539–551. [Google Scholar] [CrossRef] [PubMed]
- González, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ellrichmann, G.; Reick, C.; Saft, C.; Linker, R.A. The role of the immune system in Huntington’s disease. Clin. Dev. Immunol. 2013, 2013, 541259. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.; Ostan, R.; Borelli, V.; Castellani, G.; Franceschi, C. Inflammaging and omics in human longevity. Mech. Ageing Dev. 2016, 6374, 30261–30265. [Google Scholar] [CrossRef] [PubMed]
- Cornejo, F.; von Bernhardi, R. Age-dependent changes in the activation and regulation of microglia. In Glial Cells in Health and Disease of the CNS; von Bernhardi, R., Ed.; Springer International Publishing: Cham (ZG), Switzerland, 2016; pp. 205–226. [Google Scholar]
- Franceschi, C.; BonafÈ, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Gottfried-Blackmore, A.C.; McEwen, B.S.; Bulloch, K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kawabori, M.; Yenari, M.A. Innate inflammatory responses in stroke: Mechanisms and potential therapeutic targets. Curr. Med. Chem. 2014, 21, 2076–2097. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Yang, Z.; Zhong, L.; Zhong, S.; Xian, R.; Yuan, B. Hypoxia induces microglia autophagy and neural inflammation injury in focal cerebral ischemia model. Exp. Mol. Pathol. 2015, 98, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor nf-κb pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhu, J.; Wu, L.; Xu, G.; Dai, J.; Liu, X. Tetracycline inhibits local inflammation induced by cerebral ischemia via modulating autophagy. PLoS ONE 2012, 7, e48672. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhou, J.; Li, X.; Guo, C.a.; Fang, T.; Chen, Z. GSK-3β inhibitors suppressed neuroinflammation in rat cortex by activating autophagy in ischemic brain injury. Biochem. Biophys. Res. Commun. 2011, 411, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.-C.; Huang, H.-J.; Wang, Y.-T.; Lin, A.M.-Y. Baicalein attenuates α-synuclein aggregation, inflammasome activation and autophagy in the MPP+-treated nigrostriatal dopaminergic system in vivo. J. Ethnopharmacol. 2016, 194, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Su, C.; Qiao, C.; Bian, Y.; Ding, J.; Hu, G. Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ros clearance. Int. J. Neuropsychopharmacol. 2016, 19. [Google Scholar] [CrossRef] [PubMed]



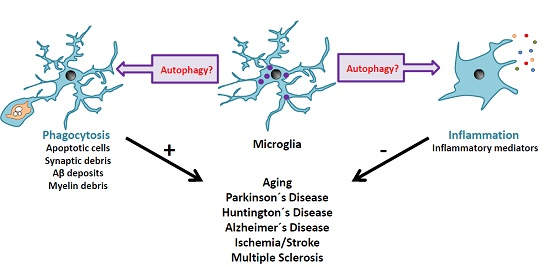{kind=link}
{kind=link}
{kind=link}
| Disease | Neurons | Microglia | ||||
|---|---|---|---|---|---|---|
| Autophagy | Phagocytosis | Inflammation | ||||
| Status | Role | Function | Role of autophagy | Function | Role of autophagy | |
| Aging | Downregulation of autophagy genes and proteins [30,31,32]. Increased MTORC1 activity [31,32]. | Autophagy inhibition (by ATG-5/7 deletion) results in spontaneous neurodegeneration [27,28]. Autophagy activation (by caloric restriction) prevents brain atrophy and enhances learning/memory [32,35]. | Clearance of apoptotic cells, Aβ, synaptic material, and myelin debris [87]. | Autophagy inhibition (by MTORC1 activation or ATG1 inhibition) prevents neurodegeneration [114]. | Inflamm-aging [92,150,151,153]. | Unknown. |
| PD | Blockade of autophagy flux [60,61]. | Autophagy activation (by TFEB activation) reduces neurodegeneration and improves motor performance [60,63]. | Clearance of apoptotic cells [87]. | Unknown. | Inflammatory mediator production [106,160,161]. | Baicalein increases LC3-II expression and attenuates inflammation [160]. Autophagy activation (MTORC1 inhibition) reduces inflammatory mediators [161]. |
| HD | Defects in autophagosome loading [72] and/or maturation [73]. | Autophagy activation (by MTORC1 inhibition and others) reduces neurodegeneration and improves motor performance [74,75,76,77]. | Clearance of apoptotic cells [87]. | Unknown. | Inflammatory mediator production [145]. | Unknown. |
| AD | Defects in autophagy flux [51,52,53]. | Autophagy activation (by cystatin b deletion) reduces Aβ load, and reduces learning/memory deficits [54]. Autophagy inhibition (by ATG-7 deletion) reduces extracellular Aβ deposition, and increases learning/memory deficits [55]. Autophagy activation (by trehalose) decreases tau inclusions and neurodegeneration [56]. | Clearance of apoptotic cells, synaptic debris, and Aβ [87,109,110,111,112]. | Autophagy inhibition (by BECN-1 downregulation) reduces Aβ phagocytosis and/or degradation [115]. Autophagy inhibition (by ATG-7 and LC3 deletion) reduces Aβ clearance [117]. | NLRP3 inflammasome activation and inflammatory mediator production [117]. | Autophagy inhibition (by LC3 and ATG-7 deletion) activates NLRP3 inflammasome and inflammatory mediator production [117]. |
| I/S | Increased autophagy markers [40,41] and mitophagy [46]. | Autophagy activation (by MTORC1 inhibition) decreases neurodegeneration [42,43]. Autophagy (by TSC1 deletion and 3-MA) and mitophagy (by mdivi-1) inhibition increases neurodegeneration [44,46]. Autophagy inhibition (by chloroquine, 3-MA, and BECN-1 deletion) decreases neurodegeneration [41,43,47]. | Clearance of apoptotic cells [87]. Phagocytosis of live cells [108]. | Unknown | Inflammatory mediator production [154,155]. | GSK-3β blockade increases LC3-II expression and reduces inflammatory mediator release [159]. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598. https://doi.org/10.3390/ijms18030598
Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. International Journal of Molecular Sciences. 2017; 18(3):598. https://doi.org/10.3390/ijms18030598
Chicago/Turabian StylePlaza-Zabala, Ainhoa, Virginia Sierra-Torre, and Amanda Sierra. 2017. "Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging" International Journal of Molecular Sciences 18, no. 3: 598. https://doi.org/10.3390/ijms18030598
APA StylePlaza-Zabala, A., Sierra-Torre, V., & Sierra, A. (2017). Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. International Journal of Molecular Sciences, 18(3), 598. https://doi.org/10.3390/ijms18030598