
Custom Array Comparative Genomic Hybridization: the Importance of DNA Quality, an Expert Eye, and Variant Validation


Abstract
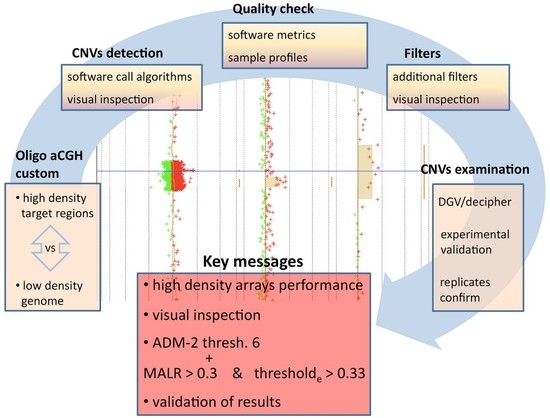
:
1. Introduction
2. Results
2.1. Sample Quality and Design Reliability
2.2. High Density Design Performance
2.3. Comparison between Algorithms and Filters
2.4. Software Algorithms Calls and Visual Inspection
3. Discussion
4. Materials and Methods
4.1. Microarray Design
4.2. Data Analysis and Structural Variant Detection
4.3. Statistical Analysis
4.4. Detected Variants Classification and Validation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shaffer, L.G.; Bejjani, B.A.; Torchia, B.; Kirkpatrick, S.; Coppinger, J.; Ballif, B.C. The identification of microdeletion syndromes and other chromosome abnormalities: Cytogenetic methods of the past, new technologies for the future. Am. J. Med. Genet. C Semin. Med. Genet. 2007, 145C, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Ballif, B.C.; Hornor, S.A.; Jenkins, E.; Madan-Khetarpal, S.; Surti, U.; Jackson, K.E.; Asamoah, A.; Brock, P.L.; Gowans, G.C.; Conway, R.L.; et al. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2-p12.2. Nat. Genet. 2007, 39, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Hansen, S.; Selzer, R.R.; Cheng, Z.; Regan, R.; Hurst, J.A.; Stewart, H.; Price, S.M.; Blair, E.; Hennekam, R.C.; et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006, 38, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Coppinger, J.; McDonald-McGinn, D.; Zackai, E.; Shane, K.; Atkin, J.F.; Asamoah, A.; Leland, R.; Weaver, D.D.; Lansky-Shafer, S.; Schmidt, K.; Feldman, H.; et al. Identification of familial and de novo microduplications of 22q11.21-q11.23 distal to the 22q11.21 microdeletion syndrome region. Hum. Mol. Genet. 2009, 18, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Iafrate, A.J.; Feuk, L.; Rivera, M.N.; Listewnik, M.L.; Donahoe, P.K.; Qi, Y.; Scherer, S.W.; Lee, C. Detection of large-scale variation in the human genome. Nat. Genet. 2004, 36, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.H.; Ben-Dor, A.; Tsalenko, A.; Sampas, N.; Rodriguez-Revenga, L.; Tran, C.W.; Scheffer, A.; Steinfeld, I.; Tsang, P.; Yamada, N.A.; et al. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 2008, 82, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Shearer, B.M.; Thorland, E.C.; Gonzales, P.R.; Ketterling, R.P. Evaluation of a commercially available focused aCGH platform for the detection of constitutional chromosome anomalies. Am. J. Med. Genet. A 2007, 143A, 2357–2370. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; de Leeuw, R.J.; Dosanjh, N.S.; Kimm, L.R.; Cheng, Z.; Horsman, D.E.; MacAulay, C.; Ng, R.T.; Brown, C.J.; Eichler, E.E.; et al. A comprehensive analysis of common copy-number variations in the human genome. Am. J. Hum. Genet. 2007, 80, 91–104. [Google Scholar] [CrossRef] [PubMed]
- De Smith, A.J.; Tsalenko, A.; Sampas, N.; Scheffer, A.; Yamada, N.A.; Tsang, P.; Ben-Dor, A.; Yakhini, Z.; Ellis, R.J.; Bruhn, L.; et al. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: Implications for association studies of complex diseases. Hum. Mol. Genet. 2007, 16, 2783–2794. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.W.; Shaw, C.A.; Yu, W.; Li, J.; Ou, Z.; Patel, A.; Yatsenko, S.A.; Cooper, M.L.; Furman, P.; Stankiewicz, P.; et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet. Med. 2005, 7, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Ballif, B.C.; Kashork, C.D.; Heilstedt, H.A.; Howard, L.A.; Cai, W.W.; White, L.D.; Liu, W.; Beaudet, A.L.; Bejjani, B.A.; et al. Development of a comparative genomic hybridization microarray and demonstration of its utility with 25 well-characterized 1p36 deletions. Hum. Mol. Genet. 2003, 12, 2145–2152. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Irons, M.; Miller, D.T.; Cheung, S.W.; Lip, V.; Sheng, X.; Tomaszewicz, K.; Shao, H.; Fang, H.; Tang, H.S.; et al. Development of a focused oligonucleotide-array comparative genomic hybridization chip for clinical diagnosis of genomic imbalance. Clin. Chem. 2007, 53, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Bittel, D.C.; Kibiryeva, N.; Zwick, D.L.; Cooley, L.D. Validation of the Agilent 244 K oligonucleotide array-based comparative genomic hybridization platform for clinical cytogenetic diagnosis. Am. J. Clin. Pathol. 2009, 132, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.; Li, A.; Valentin, D.; Nowak, N.J.; Zhao, H.; Li, P. Analytical and clinical validity of whole-genome oligonucleotide array comparative genomic hybridization for pediatric patients with mental retardation and developmental delay. Am. J. Med. Genet. A 2008, 146, 1942–1954. [Google Scholar] [CrossRef] [PubMed]
- Neill, N.J.; Torchia, B.S.; Bejjani, B.A.; Shaffer, L.G.; Ballif, B.C. Comparative analysis of copy number detection by whole-genome BAC and oligonucleotide array CGH. Mol. Cytogenet. 2010, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, S.K.; Riska, S.M.; Klee, E.W.; Thorland, E.C.; Kay, N.E.; Thibodeau, S.N.; Parker, A.S.; Eckel-Passow, J.E. Experimental designs for array comparative genomic hybridization technology. Cytogenet. Genome Res. 2013, 139, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Przybytkowski, E.; Aguilar-Mahecha, A.; Nabavi, S.; Tonellato, P.J.; Basik, M. Ultradense array CGH and discovery of micro-copy number alterations and gene fusions in the cancer genome. Methods Mol. Biol. 2013, 973, 15–38. [Google Scholar] [PubMed]
- Carter, N.P. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat. Genet. 2007, 39, S16–S21. [Google Scholar] [CrossRef] [PubMed]
- Leo, A.; Walker, A.M.; Lebo, M.S.; Hendrickson, B.; Scholl, T.; Akmaev, V.R. A GC-wave correction algorithm that improves the analytical performance of aCGH. J. Mol. Diagn. 2012, 14, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Commo, F.; Ferté, C.; Soria, J.C.; Friend, S.H.; André, F.; Guinney, J. Impact of centralization on aCGH-based genomic profiles for precision medicine in oncology. Ann. Oncol. 2015, 26, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Boone, P.M.; Bacino, C.A.; Shaw, C.A.; Eng, P.A.; Hixson, P.M.; Pursley, A.N.; Kang, S.H.; Yang, Y.; Wiszniewska, J.; Nowakowska, B.A.; et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum. Mutat. 2010, 31, 1326–1342. [Google Scholar] [CrossRef] [PubMed]
- Hackmann, K.; Kuhlee, F.; Betcheva-Krajcir, E.; Kahlert, A.K.; Mackenroth, L.; Klink, B.; di Donato, N.; Tzschach, A.; Kast, K.; Wimberger, P.; et al. Ready to clone: CNV detection and breakpoint fine-mapping in breast and ovarian cancer susceptibility genes by high-resolution array CGH. Breast Cancer Res. Treat. 2016, 159, 585–590. [Google Scholar] [CrossRef] [PubMed]
- La Cognata, V.; Morello, G.; Gentile, G.; D’Agata, V.; Criscuolo, C.; Cavalcanti, F.; Cavallaro, S. A customized high-resolution array-comparative genomic hybridization to explore copy number variations in Parkinson‘s disease. Neurogenetics 2016, 17, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Tayeh, M.K.; Chin, E.L.; Miller, V.R.; Bean, L.J.; Coffee, B.; Hegde, M. Targeted comparative genomic hybridization array for the detection of single- and multiexon gene deletions and duplications. Genet. Med. 2009, 11, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, Y.; Emi, M.; Hashimoto-Tamaoki, T.; Ohmuraya, M.; Sato, A.; Tsujimura, T.; Hasegawa, S.; Nakano, T.; Nasu, M.; Pastorino, S.; et al. High-density array-CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc. Natl. Acad. Sci. USA 2016, 113, 13432–13437. [Google Scholar] [CrossRef] [PubMed]
- Wiszniewska, J.; Bi, W.; Shaw, C.; Stankiewicz, P.; Kang, S.H.; Pursley, A.N.; Lalani, S.; Hixson, P.; Gambin, T.; Tsai, C.H.; et al. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur. J. Hum. Genet. 2014, 22, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Du, R.; Li, S.; Zhang, F.; Jin, L.; Wang, H. Evaluation of copy number variation detection for a SNP array platform. BMC Bioinform. 2014, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Siggberg, L.; Ala-Mello, S.; Linnankivi, T.; Avela, K.; Scheinin, I.; Kristiansson, K.; Lahermo, P.; Hietala, M.; Metsähonkala, L.; Kuusinen, E.; et al. High-resolution SNP array analysis of patients with developmental disorder and normal array CGH results. BMC Med. Genet. 2012, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Wajnberg, G.; Carvalho, B.S.; Ferreira, C.G.; Passetti, F. Combined analysis of SNP array data identifies novel CNV candidates and pathways in ependymoma and mesothelioma. BioMed Res. Int. 2015, 2015, 902419. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, A.; Treff, N.R.; Cimadomo, D.; Tao, X.; Upham, K.; Ubaldi, F.M.; Rienzi, L.; Scott, R.T. Comparison of array comparative genomic hybridization and quantitative real-time PCR-based aneuploidy screening of blastocyst biopsies. Eur. J. Hum. Genet. 2015, 23, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, A.W.; Hodes-Wertz, B.; McCulloh, D.H.; Munné, S.; Grifo, J.A. Discrepant diagnosis rate of array comparative genomic hybridization in thawed euploid blastocysts. J. Assist. Reprod. Genet. 2016, 33, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Zanda, M.; Onengut-Gumuscu, S.; Walker, N.; Shtir, C.; Gallo, D.; Wallace, C.; Smyth, D.; Todd, J.A.; Hurles, M.E.; Plagnol, V.; et al. A genome-wide assessment of the role of untagged copy number variants in type 1 diabetes. PLoS Genet. 2014, 10, e1004367. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wu, S.Y.; Amato, K.; di Adamo, A.; Li, P. Spectrum of cytogenomic abnormalities revealed by array comparative genomic hybridization on products of conception culture failure and normal karyotype samples. J. Genet. Genom. 2016, 43, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Lantieri, F.; Gimelli, S.; Viaggi, C.; Stathaki, E.; Malacarne, M.; Santamaria, G.; Grossi, A.; Coviello, D.; Ceccherini, I. Copy Number Variations in Candidate Regions Confirm Genetic Heterogeneity in Hirschsprung Disease. Unpublished work. 2017. [Google Scholar]
- Jiang, Q.; Ho, Y.Y.; Hao, L.; Nichols Berrios, C.; Chakravarti, A. Copy number variants in candidate genes are genetic modifiers of hirschsprung disease. PLoS ONE 2011, 6, e21219. [Google Scholar] [CrossRef] [PubMed]
- Byerly, S.; Sundin, K.; Raja, R.; Stanchfield, J.; Bejjani, B.A.; Shaffer, L.G. Effects of ozoneexposure during microarray posthybridization washes and scanning. J. Mol. Diagn. 2009, 11, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.; Feuk, L.; Scherer, S.W. The database of genomic variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef] [PubMed]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selected Pairs | Groups Comparisons | N | All Log2ratios | Log2ratios > |0.3| | p-Value |
|---|---|---|---|---|---|
| Mean r | Mean r | ||||
| All samples | replicated | 37 | 0.18 | 0.42 | 1.8 × 10−9 |
| random | 37 | 0.07 | 0.14 | 0.0036 | |
| p-value | 0.004 | 4.8 × 10−5 | |||
| Only pairs with at least one excellent quality sample (DLRS ≤ 0.2) | replicated | 24 | 0.23 * | 0.53 § | 2.01 × 10−8 |
| random | 24 | 0.09 ** | −0.17 §§ | 0.0057 | |
| p-value | 0.0018 | 6.8 × 10−6 | |||
| Pairs with no excellent quality sample (DLRS > 0.2) | replicated | 13 | 0.09 * | 0.21 § | 0.003 |
| random | 13 | 0.05 ** | 0.09 §§ | 0.1594 | |
| p-value | 0.2635 | 0.1492 |
| Sample ID | DLRS | Chromosomal Region (chr:start–end) | CNV Type | # Probes | Detection Algorithm | Fuzzy Zero | Visual Inspection Classification | Reported on DGV | Reported on Decipher | Validated | Replicate | True Variants † | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ADM-2, Threshold 6 | ADM-2, Threshold 8 | ||||||||||||
| HSCR000 | 0.148 | 9:110381888–110401999 | gain | 9 | Y | Y | Y | likely | N | N | Y | confirmed | yes |
| HSCR000 | 0.148 | 10:43435867–60812533 | loss | 849 | Y | Y | Y | known | N | N | known | confirmed | known |
| HSCR000 | 0.148 | 10:43572551–43573368 | gain | 3 | N | N | N | unlikely | N | N | not confirmed | no | |
| HSCR037 | 0.120 | 10:43589687–62786887 | loss | 544 | Y | Y | Y | known | N | N | known | known | |
| HSCR005 | 0.226 | 7:84217007–84225649 | loss | 4 | Y | - | - | likely | Y (freq < 1%) | N | Y | yes | |
| HSCR005 | 0.226 | 10:43679892–43680816 | loss | 5 | Y | - | Y | likely | N | N | N | no | |
| HSCR005 * | 0.226 | 21:9833187–11096086 | loss | 4 | N | - | N | possible | N | N | unknown | ||
| HSCR006 | 0.276 | 10:43679612–43680816 | loss | 6 | N | - | - | likely | N | N | N | no | |
| HSCR006 | 0.276 | 10:43685614–43715348 | gain | 78 | N | N | - | unlikely | N | N | unknown | ||
| HSCR006 | 0.276 | 19:5822193–5832504 | gain | 13 | Y | - | - | unlikely | N | N | unknown | ||
| HSCR009 | 0.176 | 10:43691613–43713132 | gain | 50 | N | N | N | unlikely | N | N | unknown | ||
| HSCR009 | 0.176 | 19:5825458–5831976 | gain | 9 | Y | Y | Y | unlikely | N | N | unknown | ||
| HSCR010 * | 0.211 | 15:20848460–22432687 | gain | 5 | Y | - | - | likely | Y (freq ≥ 5%) | N | not excluded | yes | |
| HSCR014 | 0.221 | 8:32532001–32532545 | gain | 2 | Y | - | Y | unlikely | N | N | unknown | ||
| HSCR014 * | 0.221 | 10:29939955–30822470 | gain | 3 | Y | - | Y | possible | N | N | unknown | ||
| HSCR014 * | 0.221 | 12:80226392–80589429 | gain | 2 | Y | - | Y | possible | N | N | unknown | ||
| HSCR014 | 0.221 | 22:22417683–23228483 | loss | 15 | Y | Y | Y | likely | Y (freq ≥ 5%) | N | yes | ||
| HSCR016 | 0.117 | 5:69288477–70309855 | gain | 3 | Y | - | - | likely | Y (freq ≥ 5%) | N | not excluded | yes | |
| HSCR016 | 0.117 | 22:25672585–25892401 | gain | 5 | Y | - | Y | likely | Y (freq ≥ 5%) | Y (3 inds.) | not excluded | yes | |
| HSCR018 § | 0.172 | 9:109336464–109348467 | gain | 6 | - | - | - | likely | N | N | Y | yes | |
| HSCR019 * | 0.122 | 1:146638075–147824207 | loss | 4 | Y | Y | Y | likely | N | Y (1q21.1 recurrent microdel) | Y | confirmed with a different size | yes |
| HSCR033* | 0.229 | 15:21162691–22173977 | loss | 3 | Y | Y | Y | likely | Y (freq ≥ 5%) | N | yes | ||
| HSCR036 | 0.177 | 22:22781091–23228483 | loss | 8 | Y | Y | Y | likely | Y (freq ≥ 5%) | N | yes | ||
| HSCR039 | 0.217 | 3:51458492–51665134 | loss | 62 | N | N | - | unlikely | N | N | not confirmed | no | |
| HSCR039 | 0.217 | 6:148651353–150170473 | loss | 52 | N | N | - | unlikely | N | N | not confirmed | no | |
| HSCR039 | 0.217 | 9:110130442–110370427 | loss | 99 | N | N | - | unlikely | N | N | not confirmed | no | |
| HSCR043 § | 0.175 | 9:109273643–109275694 | loss | 2 | - | - | - | likely | N | N | Y | yes | |
| HSCR045 § | 0.271 | 7:84594683–84607065 | loss | 6 | - | - | - | unlikely | N | N | N | no | |
| HSCR045 § | 0.271 | 8:32597644–32598929 | loss | 3 | - | - | - | likely | N | N | Y | yes | |
| HSCR045 | 0.271 | 10:43679612–43680816 | loss | 6 | Y | - | Y | likely | N | N | N | no | |
| HSCR045 | 0.271 | 19:5819037–18310693 | gain | 25 | Y | - | - | unlikely | N | N | unknown | ||
| HSCR058 | 0.243 | 22:18661724–18920001 | gain | 7 | Y | - | Y | unlikely | Y (freq ≥ 5%) | N | not evaluable | yes | |
| HSCR064 * | 0.192 | 15:20848460–22173977 | loss | 4 | Y | Y | Y | likely | Y (freq ≥ 5%) | N | yes | ||
| HSCR126 | 0.176 | 19:4205366–18310693 | gain | 26 | N | - | - | unlikely | N | N | unknown | ||
| HSCR146 * | 0.122 | 15:58257674–59009890 | gain | 2 | Y | Y | Y | likely | N | N | Y | yes | |
| HSCR146 | 0.122 | 19:30888070–30891329 | gain | 2 | Y | - | Y | likely | N | N | N | no | |
| HSCR160 * | 0.200 | 15:20848460–22173977 | gain | 4 | Y | - | Y | likely | Y (freq ≥ 5%) | N | yes | ||
| HSCR162 *,§ | 0.184 | 9:43659247–43659512 | loss | 2 | - | - | - | likely | Y (freq ≥ 5%) | N | confirmed with a different size | yes | |
| HSCR181 * | 0.150 | 15:20848460–22432687 | loss | 5 | N | - | - | possible | Y (freq ≥ 5%) | N | not excluded | yes | |
| HSCR181 | 0.150 | 21:14629063–48080926 | gain | 245 | Y | Y | Y | known | N | N | known | confirmed | known |
| HSCR183 | 0.138 | 22:22781091–23228483 | loss | 8 | Y | Y | Y | likely | Y (freq ≥ 5%) | N | yes | ||
| HSCR195 | 0.158 | 9:112078131–112089193 | loss | 5 | Y | - | - | likely | N | N | inconclusive | confirmed with a different size | yes |
| HSCR217 | 0.168 | 16:82200334–82202467 | gain | 2 | Y | - | Y | likely | N | N | Y | yes | |
| HSCR228 § | 0.158 | 22:25672585–25892401 | gain | 5 | - | - | - | likely | Y (freq ≥ 5%) | Y (3 inds.) | not excluded | yes | |
| HSCR231* | 0.164 | 15:21162691–22432687 | gain | 4 | Y | - | Y | unlikely | Y (freq ≥ 5%) | N | yes | ||
| HSCR312 | 0.215 | 3:50161771–50618134 | gain | 143 | N | - | - | unlikely | N | N | unknown | ||
| HSCR312 | 0.215 | 4:41748211–41753993 | gain | 16 | N | - | - | unlikely | N | N | unknown | ||
| HSCR312 | 0.215 | 10:43550696–43621994 | gain | 196 | N | - | - | unlikely | N | N | unknown | ||
| HSCR312 | 0.215 | 10:43684681–43718450 | gain | 86 | N | N | N | unlikely | N | N | unknown | ||
| HSCR312 | 0.215 | 14:36983123–36994136 | gain | 14 | Y | - | - | unlikely | N | N | unknown | ||
| HSCR312 | 0.215 | 19:5821171–5832504 | gain | 15 | N | N | N | unlikely | N | N | unknown | ||
| HSCR323 | 0.253 | 13:78465278–78484576 | gain | 30 | N | - | - | unlikely | N | N | unknown | ||
| HSCR331 | 0.172 | 19:5822193–5832928 | gain | 14 | N | - | - | unlikely | N | N | not excluded | unknown | |
| HSCR335 * | 0.183 | 15:20848460–22173977 | gain | 4 | Y | - | - | possible | Y (freq ≥ 5%) | N | not excluded | yes | |
| HSCR335 | 0.183 | 22:18628019–18807881 | gain | 6 | Y | - | Y | unlikely | N | N | not excluded | unknown | |
| HSCR335 | 0.183 | 22:20345868–20499789 | gain | 4 | Y | - | Y | unlikely | Y (freq ≥ 5%) | N | not excluded | yes | |
| HSCR335 | 0.183 | 22:21494163–21704972 | gain | 5 | Y | - | Y | unlikely | N | N | not excluded | unknown | |
| HSCR349 | 0.220 | 3:51452049–51647312 | loss | 59 | N | N | - | unlikely | N | N | unknown | ||
| HSCR349 * | 0.220 | 7:63449575–75986814 | loss | 25 | N | - | - | unlikely | N | N | unknown | ||
| HSCR349 | 0.220 | 10:43573685–43574005 | gain | 2 | Y | - | Y | unlikely | N | N | N | no | |
| HSCR374 | 0.266 | 10:43473690–43474033 | gain | 4 | Y | - | Y | unlikely | N | N | N | no | |
| HSCR380 | 0.123 | 22:16054691–18807881 | gain | 23 | Y | Y | Y | known | N | N | known | known | |
| HSCR380 | 0.123 | 22:20345868–20659606 | gain | 5 | Y | Y | Y | unlikely | N | N | unknown | ||
| HSCR380 | 0.123 | 22:21494163–21704972 | gain | 5 | Y | Y | Y | unlikely | N | N | unknown | ||
| HSCR382 | 0.235 | 10:43474436–43483543 | loss | 29 | N | - | - | unlikely | N | N | unknown | ||
| HSCR382 | 0.235 | 10:43630181–43636329 | gain | 31 | N | - | - | unlikely | N | N | unknown | ||
| HSCR382 * | 0.235 | 15:20190548–22173977 | gain | 5 | Y | - | - | possible | Y (freq ≥ 5%) | N | yes | ||
| HSCR391 | 0.173 | 21:14629063–48080926 | gain | 245 | Y | Y | Y | known | N | N | known | confirmed with a different size | known |
| HSCR403 §§ | 0.111 | 4:41746863–41751291 | loss | 11 | N | - | - | likely | N | N | Y | yes | |
| HSCR403 *,§§ | 0.111 | 9:43659247–43659512 | gain | 2 | Y | - | Y | likely | Y (freq ≥ 5%) | N | yes | ||
| HSCR403 | 0.111 | 22:18661724–18807881 | gain | 5 | Y | - | - | possible | N | N | not excluded | unknown | |
| HSCR403 | 0.111 | 22:21494163–21704972 | gain | 5 | Y | - | Y | unlikely | N | N | confirmed and not excluded | yes | |
| HSCR403 | 0.111 | 22:23056562–23228483 | loss | 3 | Y | - | Y | likely | Y (freq ≥ 5%) | N | confirmed with a different size | yes | |
| HSCR409 * | 0.139 | 15:20848460–22173977 | gain | 4 | Y | Y | Y | likely | Y (freq ≥ 5%) | N | yes | ||
| HSCR412 | 0.204 | 22:20345868–21778882 | loss | 26 | N | - | - | unlikely | N | N | not confirmed | no | |
| HSCR414 * | 0.156 | 15:20848460–22432687 | loss | 5 | N | - | - | possible | Y (freq ≥ 5%) | N | yes | ||
| HSCR415 | 0.195 | 9:113025039–113029430 | loss | 2 | Y | Y | Y | likely | Y (freq ≥ 5%) | Y (1 ind.) ‡ | yes | ||
| HSCR421 * | 0.166 | 9:43659247–43659512 | loss | 2 | Y | Y | Y | likely | Y (freq ≥ 5%) | N | confirmed | yes | |
| HSCR421 | 0.166 | 22:25672585–25892401 | loss | 5 | Y | Y | Y | likely | Y (freq ≥ 5%) | Y (3 inds.) | not excluded | yes | |
| HSCR426 * | 0.111 | 9:43659247–43659512 | loss | 2 | Y | - | Y | likely | Y (freq ≥ 5%) | N | not confirmed and confirmed | yes | |
| HSCR481 * | 0.248 | 5:7656467–8124532 | loss | 2 | Y | - | Y | possible | N | N | not confirmed | no | |
| HSCR481 | 0.248 | 19:31954093–31966036 | loss | 5 | Y | - | - | likely | N | N | Y | not evaluable | yes |
| HSCR481 | 0.248 | 21:14629063–48080926 | gain | 245 | Y | Y | Y | known | N | N | known | confirmed | known |
| Comparison Groups | True Calls | Not Confirmed | Unknown | Total | p-Value Likely/Possible vs. Unlikely or Called vs. Not Called by the Software * | p-Values Thresholde ≥ 0.33 vs. below * |
|---|---|---|---|---|---|---|
| Likely/possible | 39 | 5 | 4 | 48 | 0.0003 † | |
| Unlikely | 4 | 8 | 23 | 35 | ||
| ADM-2_th6 ≥ 0.333 | 35 | 6 | 12 | 53 | 1.0000 | 0.0033 †† |
| ADM-2_th6 < 0.333 | 3 | 6 | 15 | 24 | ||
| NO ADM-2_th6 (visual only) * | 5 | 1 | 0 | 6 | ||
| ADM-2_th8 ≥ 0.333 | 18 | 0 | 3 | 21 | 0.5346 | 0.0001 †† |
| ADM-2_th8 < 0.333 | 0 | 4 | 5 | 9 | ||
| NO ADM-2_th8 | 25 | 9 | 19 | 53 | ||
| Fuzzy ≥ 0.333 | 28 | 6 | 8 | 42 | 0.5230 | 0.2000 |
| Fuzzy < 0.333 | 0 | 1 | 4 | 5 | ||
| NO Fuzzy | 15 | 6 | 15 | 36 | ||
| Total | 43 | 13 | 27 | 83 |
| Kind of Probes | Candidate Region | Locus | # of Features * | # of Unique Probes * | Average Space (nt) * |
|---|---|---|---|---|---|
| Selected | RET | 10q11.2 | 813 | 8333 | 312 |
| 9q31 | 9q31 | 1824 | 2501 | ||
| 9p24.1 | 9p24.1 | 142 | 3521 | ||
| PHOX2B | 4p13 | 49 | 508 | ||
| NRG1 | 8p12 | 473 | 501 | ||
| SEMA3A/SEMA3D | 7q21.11 | 468 | 2506 | ||
| rs12707682 | 40 | 500 | |||
| 6q25.1 | 6q25.1 | 714 | 3501 | ||
| 21q22 | 21q22 | 202 | 48,297 | ||
| 3p21 | 3p21 | 1141 | 3503 | ||
| 19q12 | 19q12 | 1085 | 3502 | ||
| NRTN | 19p13.3 | 18 | 806 | ||
| 16q23.3 | 16q23.3 | 714 | 3501 | ||
| NKX2-1 | 14q13 | 17 | 812 | ||
| SOX10 | 22q13 | 27 | 823 | ||
| 22q11.2 | 22q11.2 | 162 | 49,383 | ||
| ECE1 | 1p36.1 | 103 | 806 | ||
| ZEB2 | 2q22.3 | 165 | 923 | ||
| EDNRB | 13q22 | 112 | 804 | ||
| GDNF | 5p13.1-p12 | 42 | 810 | ||
| EDN3 | 20q13.2-q13.3 | 44 | 808 | ||
| Genome | 3149 | 3130 | 971,074 | ||
| Replicates | 301 × 5 = 1505 | 301 | |||
| Normalization | 1262 | 1262 | |||
| Agilent controls | 1482 | ||||
| Total | 15,748 | 13,026 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lantieri, F.; Malacarne, M.; Gimelli, S.; Santamaria, G.; Coviello, D.; Ceccherini, I. Custom Array Comparative Genomic Hybridization: the Importance of DNA Quality, an Expert Eye, and Variant Validation. Int. J. Mol. Sci. 2017, 18, 609. https://doi.org/10.3390/ijms18030609
Lantieri F, Malacarne M, Gimelli S, Santamaria G, Coviello D, Ceccherini I. Custom Array Comparative Genomic Hybridization: the Importance of DNA Quality, an Expert Eye, and Variant Validation. International Journal of Molecular Sciences. 2017; 18(3):609. https://doi.org/10.3390/ijms18030609
Chicago/Turabian StyleLantieri, Francesca, Michela Malacarne, Stefania Gimelli, Giuseppe Santamaria, Domenico Coviello, and Isabella Ceccherini. 2017. "Custom Array Comparative Genomic Hybridization: the Importance of DNA Quality, an Expert Eye, and Variant Validation" International Journal of Molecular Sciences 18, no. 3: 609. https://doi.org/10.3390/ijms18030609
APA StyleLantieri, F., Malacarne, M., Gimelli, S., Santamaria, G., Coviello, D., & Ceccherini, I. (2017). Custom Array Comparative Genomic Hybridization: the Importance of DNA Quality, an Expert Eye, and Variant Validation. International Journal of Molecular Sciences, 18(3), 609. https://doi.org/10.3390/ijms18030609