
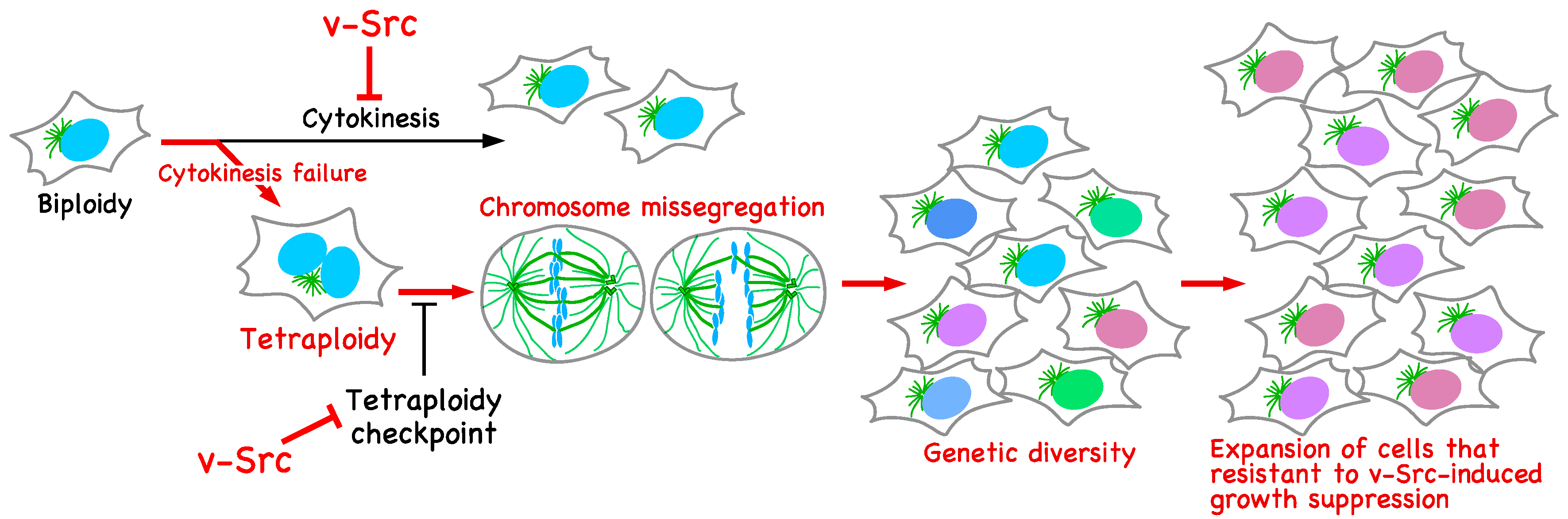
Cytokinesis Failure Leading to Chromosome Instability in v-Src-Induced Oncogenesis


Abstract
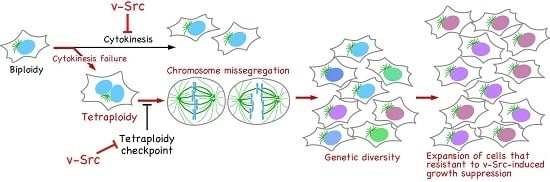
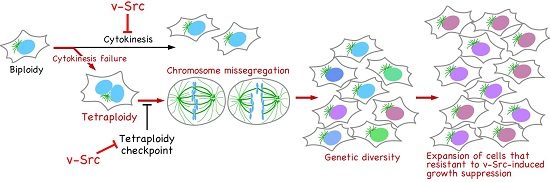
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. v-Src Suppresses Cell Proliferation and Induces Tetraploidization
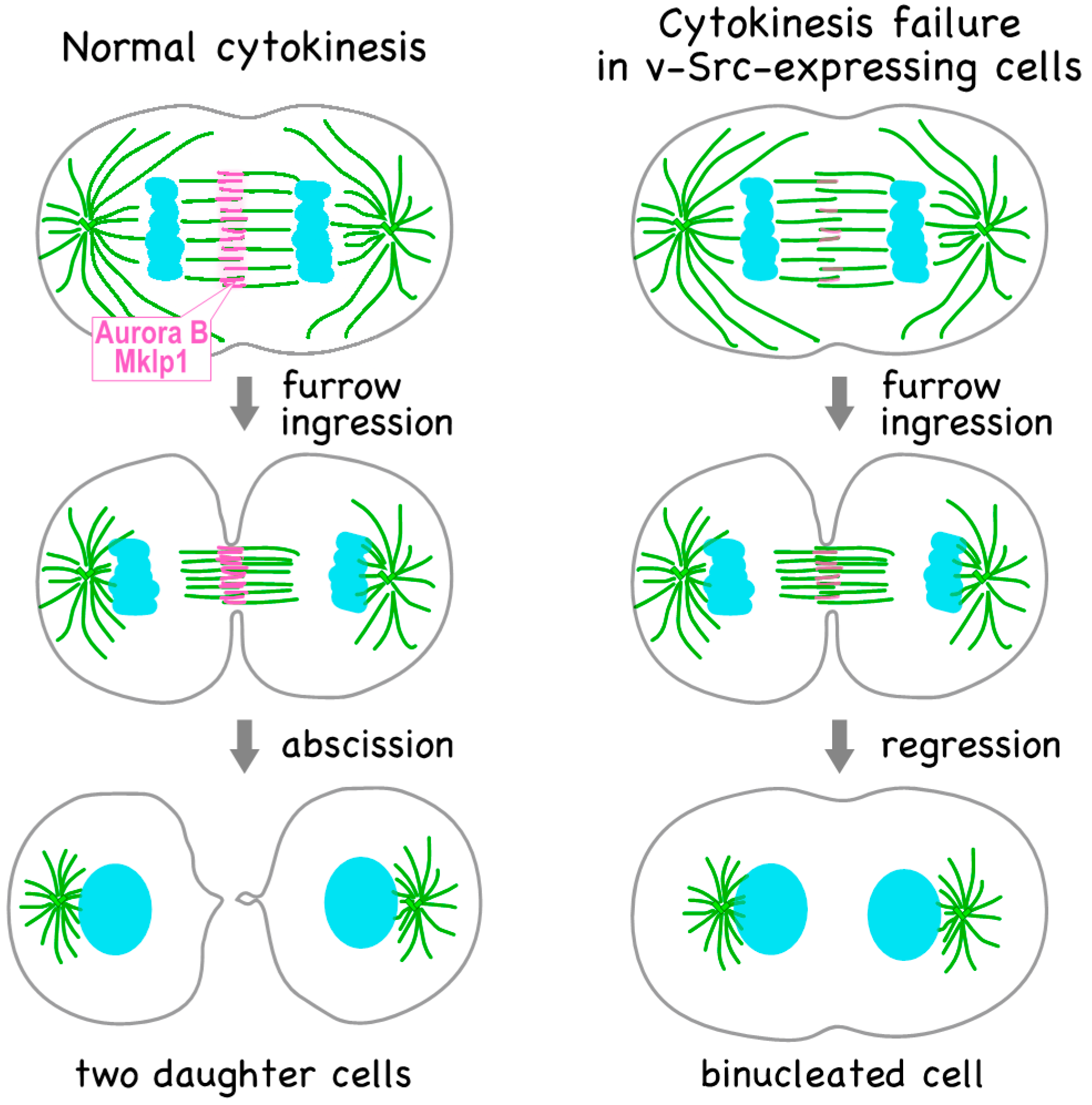
3. v-Src-Induced Cytokinesis Failure Is Caused through Delocalization of Mitotic Regulators
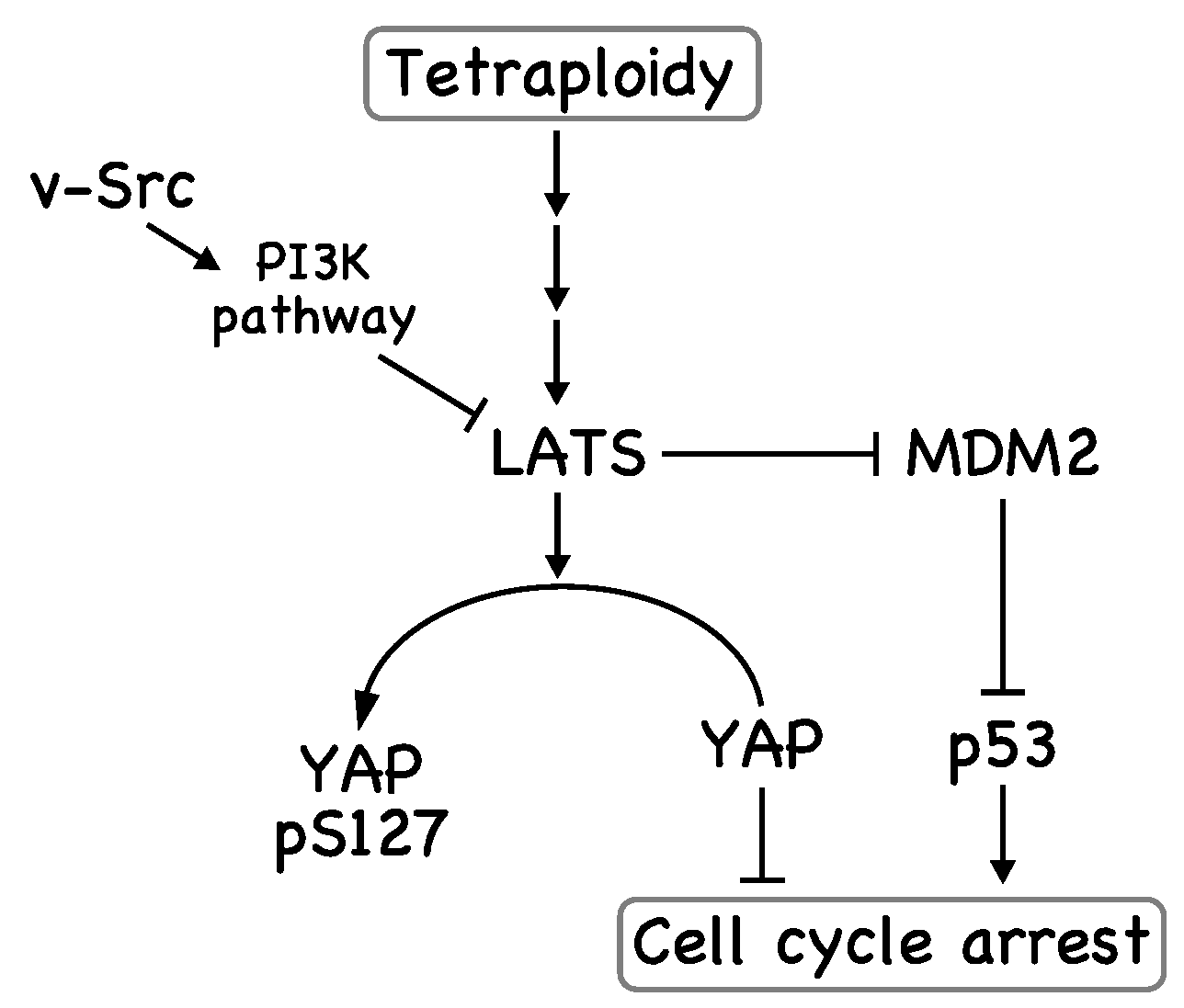
4. v-Src Attenuates the Tetraploidy Checkpoint
5. v-Src Can Induce Chromosome Instability, Generating Genetic Diversity
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Csk | C-terminal Src kinase |
| EGF | epidermal growth factor |
| CPC | chromosomal passenger complex |
| SH2 | Src homology 2 |
| SH3 | Src homology 3 |
References
- Frame, M.C. Src in cancer: Deregulation and consequences for cell behaviour. Biochim. Biophys. Acta 2002, 1602, 114–130. [Google Scholar] [CrossRef]
- Frame, M.C. Newest findings on the oldest oncogene; how activated src does it. J. Cell Sci. 2004, 117, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Soeda, S.; Nakayama, Y.; Honda, T.; Aoki, A.; Tamura, N.; Abe, K.; Fukumoto, Y.; Yamaguchi, N. v-Src causes delocalization of Mklp1, Aurora B, and INCENP from the spindle midzone during cytokinesis failure. Exp. Cell Res. 2013, 319, 1382–1397. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.W. Epidermal growth factor inhibits growth of A431 human epidermoid carcinoma in serum-free cell culture. J. Cell Biol. 1982, 93, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R. Epidermal growth factor inhibits the synthesis of the nuclear protein cyclin in A431 human carcinoma cells. Proc. Natl. Acad. Sci. USA 1984, 81, 4848–4850. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Burckhardt, J.; Curran, T.; Müller, R. Stimulation and inhibition of growth by EGF in different A431 cell clones is accompanied by the rapid induction of c-fos and c-myc proto-oncogenes. EMBO J. 1985, 4, 1193–1197. [Google Scholar] [PubMed]
- Armstrong, D.K.; Kaufmann, S.H.; Ottaviano, Y.L.; Furuya, Y.; Buckley, J.A.; Isaacs, J.T.; Davidson, N.E. Epidermal growth factor-mediated apoptosis of MDA-MB-468 human breast cancer cells. Cancer Res. 1994, 54, 5280–5283. [Google Scholar] [PubMed]
- Tikhomirov, O.; Carpenter, G. Ligand-induced, p38-dependent Apoptosis in cells expressing high levels of epidermal growth factor receptor and ErbB-2. J. Biol. Chem. 2004, 279, 12988–12996. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann-Zeh, A.; Rodriguez-Viciana, P.; Ulrich, E.; Gilbert, C.; Coffer, P.; Downward, J.; Evan, G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 1997, 385, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. The dark side of Ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef] [PubMed]
- El-Ashry, D.; Miller, D.; Kharbanda, S.; Lippman, M.E.; Kern, F. Constitutive Raf-1 kinase activity in breast cancer cells induces both estrogen-independent growth and apoptosis. Oncogene 1997, 15, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Pelengaris, S.; Rudolph, B.; Littlewood, T. Action of Myc in vivo—Proliferation and apoptosis. Curr. Opin. Genet. Dev. 2000, 10, 100–105. [Google Scholar] [CrossRef]
- Clark, W.; Gillespie, D.A. Transformation by v-Jun prevents cell cycle exit and promotes apoptosis in the absence of serum growth factors. Cell Growth Differ. 1997, 8, 371–380. [Google Scholar] [PubMed]
- Shan, B.; Lee, W.H. Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol. Cell. Biol. 1994, 14, 8166–8173. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Debbas, M.; Sabbatini, P.; Hockenbery, D.; Korsmeyer, S.; White, E. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc. Natl. Acad. Sci. USA 1992, 89, 7742–7746. [Google Scholar] [CrossRef] [PubMed]
- Welman, A.; Cawthorne, C.; Ponce-Perez, L.; Barraclough, J.; Danson, S.; Murray, S.; Cummings, J.; Allen, T.D.; Dive, C. Increases in c-Src expression level and activity do not promote the growth of human colorectal carcinoma cells in vitro and in vivo. Neoplasia 2006, 8, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Frame, M.C.; Wyke, J.A. Expression of the v-Src oncoprotein in fibroblasts disrupts normal regulation of the CDK inhibitor p27 and inhibits quiescence. Oncogene 1998, 16, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Brunton, V.G.; Ozanne, B.W.; Paraskeva, C.; Frame, M.C. A role for epidermal growth factor receptor, c-Src and focal adhesion kinase in an in vitro model for the progression of colon cancer. Oncogene 1997, 14, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Odajima, J.; Matsumura, I.; Sonoyama, J.; Daino, H.; Kawasaki, A.; Tanaka, H.; Inohara, N.; Kitamura, T.; Downward, J.; Nakajima, K.; et al. Full oncogenic activities of v-Src are mediated by multiple signaling pathways: Ras as an essential mediator for cell survival. J. Biol. Chem. 2000, 275, 24096–24105. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.L.; Jimenez, E.; Martin, G.S. v-Src generates a p53-independent apoptotic signal. Mol. Cell. Biol. 2000, 20, 9271–9280. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.; Gorgoulis, V.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.M.; Lambert, S. Replication stress-induced genome instability: The dark side of replication maintenance by homologous recombination. J. Mol. Biol. 2013, 425, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Maślikowski, B.M.; Néel, B.D.; Wu, Y.; Wang, L.; Rodrigues, N.A.; Gillet, G.; Bédard, P.-A. Cellular processes of v-Src transformation revealed by gene profiling of primary cells—Implications for human cancer. BMC Cancer 2010, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Chackalaparampil, I.; Shalloway, D. Altered phosphorylation and activation of pp60c-src during fibroblast mitosis. Cell 1988, 52, 801–810. [Google Scholar] [CrossRef]
- Morgan, D.O.; Kaplan, J.M.; Bishop, J.M.; Varmus, H.E. Mitosis-specific phosphorylation of p60c-src by p34cdc2-associated protein kinase. Cell 1989, 57, 775–786. [Google Scholar] [CrossRef]
- Zheng, X.M.; Shalloway, D. Two mechanisms activate PTPalpha during mitosis. EMBO J. 2001, 20, 6037–6049. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, K.P.; Isaacson, C.C.; Ashendel, C.L.; Geahlen, R.L.; Harrison, M.L. Characterization of the in vivo sites of serine phosphorylation on Lck identifying serine 59 as a site of mitotic phosphorylation. J. Biol. Chem. 2002, 277, 14666–14673. [Google Scholar] [CrossRef] [PubMed]
- Kuga, T.; Nakayama, Y.; Hoshino, M.; Higashiyama, Y.; Obata, Y.; Matsuda, D.; Kasahara, K.; Fukumoto, Y.; Yamaguchi, N. Differential mitotic activation of endogenous c-Src, c-Yes, and Lyn in HeLa cells. Arch. Biochem. Biophys. 2007, 466, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.; Fumagalli, S.; Courtneidgett, S.A. Requirement for Src Family Protein Tyrosine Kinases in G2 for Fibroblast Cell Division. Science 1995, 269, 1567–1569. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M.; Srethapakdi, M.; Sachar, K.S.; Kraker, A.J.; Rosen, N. Inhibition of Src kinases by a selective tyrosine kinase inhibitor causes mitotic arrest. Cancer Res. 1999, 59, 6145–6152. [Google Scholar] [PubMed]
- Ng, M.M.; Chang, F.; Burgess, D.R. Movement of membrane domains and requirement of membrane signaling molecules for cytokinesis. Dev. Cell 2005, 9, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, T.; Sahai, E.; Chardin, P.; McCormick, F.; Courtneidge, S.A.; Alberts, A.S. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell 2000, 5, 13–25. [Google Scholar] [CrossRef]
- Kasahara, K.; Nakayama, Y.; Nakazato, Y.; Ikeda, K.; Kuga, T.; Yamaguchi, N. Src signaling regulates completion of abscission in cytokinesis through ERK/MAPK activation at the midbody. J. Biol. Chem. 2007, 282, 5327–5339. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Nakayama, Y.; Kakihana, A.; Yuki, R.; Yamaguchi, N.; Yamaguchi, N. Fyn Accelerates M Phase Progression by Promoting the Assembly of Mitotic Spindle Microtubules. J. Cell. Biochem. 2016, 117, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Matsui, Y.; Takeda, Y.; Okamoto, M.; Abe, K.; Fukumoto, Y.; Yamaguchi, N. c-Src but not Fyn promotes proper spindle orientation in early prometaphase. J. Biol. Chem. 2012, 287, 24905–24915. [Google Scholar] [CrossRef] [PubMed]
- Green, R.A.; Paluch, E.; Oegema, K. Cytokinesis in Animal Cells. Annu. Rev. Cell Dev. Biol. 2012, 28, 29–58. [Google Scholar] [CrossRef] [PubMed]
- Fededa, J.P.; Gerlich, D.W. Molecular control of animal cell cytokinesis. Nat. Cell Biol. 2012, 14, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Kakae, K.; Ikeuchi, M.; Kuga, T.; Saito, Y.; Yamaguchi, N.; Nakayama, Y. v-Src-induced nuclear localization of YAP is involved in multipolar spindle formation in tetraploid cells. Cell. Signal. 2017, 30, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Kanada, M.; Nagasaki, A.; Uyeda, T.Q.P. Adhesion-dependent and Contractile Ring-independent Equatorial Furrowing during Cytokinesis in Mammalian Cells. Mol. Biol. Cell 2005, 16, 3865–3872. [Google Scholar] [CrossRef] [PubMed]
- Shafikhani, S.H.; Mostov, K.; Engel, J. Focal adhesion components are essential for mammalian cell cytokinesis. Cell Cycle 2008, 7, 2868–2876. [Google Scholar] [CrossRef] [PubMed]
- Gruneberg, U.; Neef, R.; Honda, R.; Nigg, E.A.; Barr, F.A. Relocation of Aurora B from centromeres to the central spindle at the metaphase to anaphase transition requires MKlp2. J. Cell Biol. 2004, 166, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Zhao, J.; Bibikova, M.; Leverson, J.D.; Bossy-wetzel, E.; Fan, J.; Abraham, R.T.; Jiang, W.; Jolla, L.; Diego, S.; et al. Functional Analysis of Human Microtubule-based Motor Proteins, the Kinesins and Dyneins, in Mitosis/Cytokinesis Using RNA Interference. Mol. Biol. Cell 2005, 16, 3187–3199. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.E.; Davies, T.; Joseph, N.; Mishima, M. Aurora B and 14-3-3 coordinately regulate clustering of centralspindlin during cytokinesis. Curr. Biol. 2010, 20, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Murata-Hori, M.; Tatsuka, M.; Wang, Y.L. Probing the dynamics and functions of aurora B kinase in living cells during mitosis and cytokinesis. Mol. Biol. Cell 2002, 13, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Nakayama, Y.; Okamoto, M.; Fukumoto, Y.; Yamaguchi, N. Enrichment of cell populations in metaphase, anaphase, and telophase by synchronization using nocodazole and blebbistatin: A novel method suitable for examining dynamic changes in proteins during mitotic progression. Eur. J. Cell Biol. 2012, 91, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Storchova, Z.; Pellman, D. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 2007, 17, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Margolis, R.L.; Lohez, O.D.; Andreassen, P.R. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J. Cell. Biochem. 2003, 88, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Bolgioni, A.F.; Ganem, N.J. The interplay between centrosomes and the Hippo tumor suppressor pathway. Chromosom. Res. 2016, 24, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Cornils, H.; Chiu, S.-Y.; O’Rourke, K.P.; Arnaud, J.; Yimlamai, D.; Théry, M.; Camargo, F.D.; Pellman, D. Cytokinesis Failure Triggers Hippo Tumor Suppressor Pathway Activation. Cell 2014, 158, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006, 20, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene 1994, 9, 2145–2152. [Google Scholar] [PubMed]
- Tamm, C.; Böwer, N.; Annerén, C. Regulation of mouse embryonic stem cell self-renewal by a Yes-YAP-TEAD2 signaling pathway downstream of LIF. J. Cell Sci. 2011, 124, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Wu, L.-W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.-X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 Phosphorylation by c-Abl Is a Critical Step in Selective Activation of Proapoptotic Genes in Response to DNA Damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-G.; Gumbiner, B.M. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 2015, 210, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Böttger, A.; Böttger, V.; Sparks, A.; Liu, W.L.; Howard, S.F.; Lane, D.P. Design of a synthetic Mdm2-binding mini protein that activates the p53 response in vivo. Curr. Biol. 1997, 7, 860–869. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Huang, H.; Li, J.; Zhang, J.; Gao, J.; Lu, B.; Huang, C. GADD45β mediates p53 protein degradation via Src/PP2A/MDM2 pathway upon arsenite treatment. Cell Death Dis. 2013, 4, e637. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Cosenza, M.R.; Krämer, A. Centrosome amplification, chromosomal instability and cancer: Mechanistic, clinical and therapeutic issues. Chromosom. Res. 2016, 24, 105–126. [Google Scholar] [CrossRef] [PubMed]
- Meraldi, P. Centrosomes in spindle organization and chromosome segregation: A mechanistic view. Chromosom. Res. 2016, 24, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Teitz, L.S.; Kim, D.H.; Shoshani, O.; Skaletsky, H.; Fachinetti, D.; Page, D.C.; Cleveland, D.W. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol. 2017, 19, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Soeda, S.; Tsuda, K.; Yamaguchi, C.; Aoyama, K.; Morinaga, T.; Yuki, R.; Nakayama, Y.; Yamaguchi, N.; Yamaguchi, N. Protective role for lipid modifications of Src-family kinases against chromosome missegregation. Sci. Rep. 2016, 6, 38751. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Fukumoto, Y.; Honda, T.; Kuga, T.; Saito, Y.; Yamaguchi, N.; Nakayama, Y. v-Src Causes Chromosome Bridges in a Caffeine-Sensitive Manner by Generating DNA Damage. Int. J. Mol. Sci. 2016, 17, 871. [Google Scholar] [CrossRef] [PubMed]
- Norden, C.; Mendoza, M.; Dobbelaere, J.; Kotwaliwale, C.V.; Biggins, S.; Barral, Y. The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosome breakage. Cell 2006, 125, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, S.B.; Campsteijn, C.; Vietri, M.; Schink, K.O.; Liestøl, K.; Andersen, J.S.; Raiborg, C.; Stenmark, H. ANCHR mediates Aurora-B-dependent abscission checkpoint control through retention of VPS4. Nat. Cell Biol. 2014, 16, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Steigemann, P.; Wurzenberger, C.; Schmitz, M.H.; Held, M.; Guizetti, J.; Maar, S.; Gerlich, D.W. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009, 136, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Carlton, J.G.; Caballe, A.; Agromayor, M.; Kloc, M.; Martin-Serrano, J. ESCRT-III Governs the Aurora B-Mediated Abscission Checkpoint Through CHMP4C. Science 2012, 336, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Sarangapani, K.K.; Asbury, C.L. Catch and release: How do kinetochores hook the right microtubules during mitosis? Trends Genet. 2014, 30, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Walczak, C.E.; Cai, S.; Khodjakov, A. Mechanisms of chromosome behaviour during mitosis. Nat. Rev. Mol. Cell Biol. 2010, 11, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Wan, X.; Hirel, C.B.; Salmon, E.D. Aurora Kinase Promotes Turnover of Kinetochore Microtubules to Reduce Chromosome Segregation Errors. Curr. Biol. 2006, 16, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, A.L.; Lan, W.; Stukenberg, P.T. Aurora B Is Enriched at Merotelic Attachment Sites, Where It Regulates MCAK. Curr. Biol. 2006, 16, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.M.; Sokolsky, T.; Tucker, C.M.; Chan, L.Y.; Boselli, M.; Dunham, M.J.; Amon, A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 2007, 317, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010, 188, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, R.; Hernando, E.; Díaz-Rodríguez, E.; Teruya-Feldstein, J.; Cordón-Cardo, C.; Lowe, S.W.; Benezra, R. Mad2 Overexpression Promotes Aneuploidy and Tumorigenesis in Mice. Cancer Cell 2007, 11, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Rowald, K.; Mantovan, M.; Passos, J.; Buccitelli, C.; Mardin, B.R.; Korbel, J.O.; Jechlinger, M.; Sotillo, R. Negative Selection and Chromosome Instability Induced by Mad2 Overexpression Delay Breast Cancer but Facilitate Oncogene-Independent Outgrowth. Cell Rep. 2016, 15, 2679–2691. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, R.; Schvartzman, J.M.; Socci, N.D.; Benezra, R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 2010, 464, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Mulla, W.A.; Kucharavy, A.; Tsai, H.J.; Rubinstein, B.; Conkright, J.; McCroskey, S.; Bradford, W.D.; Weems, L.; Haug, J.S.; et al. Targeting the adaptability of heterogeneous aneuploids. Cell 2015, 160, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, D.; Burrell, R.A.; Kanu, N.; McGranahan, N.; Howell, M.; Parker, P.J.; Downward, J.; Swanton, C.; Kschischo, M. Chromosomal instability selects gene copy-number variants encoding core regulators of proliferation in ER+ Breast cancer. Cancer Res. 2014, 74, 4853–4863. [Google Scholar] [CrossRef] [PubMed]
- Brdika, T.; Pavlistova, D.; Albrecht, L.; Bruyns, E.; Schraven, B. Phosphoprotein Associated with Glycosphingolipid-enriched Microdomains (PAG), a Novel Ubiquitously Expressed Transmembrane Adaptor Protein, Binds the Protein Tyrosine Kinase Csk and Is Involved in Regulation of T Cell Activation. J. Exp. Med. 2000, 191, 1591–1604. [Google Scholar] [CrossRef]
- Tarakhovsky, A.; Okada, M.; Kawabuchi, M.; Satomi, Y.; Takao, T.; Shimonishi, Y.; Nada, S.; Nagai, K. Transmembrane phosphoprotein Cbp regulates the activities of Src-familytyrosine kinases. Nature 2000, 404, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Hrdinka, M.; Horejsi, V. PAG—A multipurpose transmembrane adaptor protein. Oncogene 2014, 33, 4881–4892. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Yamamichi, N.; Saitoh, K.; Watanabe, A.; Ito, T.; Yamamichi-Nishina, M.; Mizutani, M.; Yahagi, N.; Suzuki, T.; Sasakawa, C.; et al. Kinetics of v-Src-induced epithelial-mesenchymal transition in developing glandular stomach. Oncogene 2003, 22, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Guarino, M. Src signaling in cancer invasion. J. Cell. Physiol. 2010, 223, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Sideridou, M.; Zakopoulou, R.; Evangelou, K.; Liontos, M.; Kotsinas, A.; Rampakakis, E.; Gagos, S.; Kahata, K.; Grabusic, K.; Gkouskou, K.; et al. Cdc6 expression represses E-cadherin transcription and activates adjacent replication origins. J. Cell Biol. 2011, 195, 1123–1140. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, S.A.; Rooney, C.; Liontos, M.; Connolly, Y.; Zoumpourlis, V.; Whetton, A.D.; Gorgoulis, V.G.; Malliri, A. Src-Induced Disassembly of Adherens Junctions Requires Localized Phosphorylation and Degradation of the Rac Activator Tiam1. Mol. Cell 2009, 33, 639–653. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayama, Y.; Soeda, S.; Ikeuchi, M.; Kakae, K.; Yamaguchi, N. Cytokinesis Failure Leading to Chromosome Instability in v-Src-Induced Oncogenesis. Int. J. Mol. Sci. 2017, 18, 811. https://doi.org/10.3390/ijms18040811
Nakayama Y, Soeda S, Ikeuchi M, Kakae K, Yamaguchi N. Cytokinesis Failure Leading to Chromosome Instability in v-Src-Induced Oncogenesis. International Journal of Molecular Sciences. 2017; 18(4):811. https://doi.org/10.3390/ijms18040811
Chicago/Turabian StyleNakayama, Yuji, Shuhei Soeda, Masayoshi Ikeuchi, Keiko Kakae, and Naoto Yamaguchi. 2017. "Cytokinesis Failure Leading to Chromosome Instability in v-Src-Induced Oncogenesis" International Journal of Molecular Sciences 18, no. 4: 811. https://doi.org/10.3390/ijms18040811
APA StyleNakayama, Y., Soeda, S., Ikeuchi, M., Kakae, K., & Yamaguchi, N. (2017). Cytokinesis Failure Leading to Chromosome Instability in v-Src-Induced Oncogenesis. International Journal of Molecular Sciences, 18(4), 811. https://doi.org/10.3390/ijms18040811