
Molecular Imaging of Neuroinflammation in Neurodegenerative Dementias: The Role of In Vivo PET Imaging

Abstract
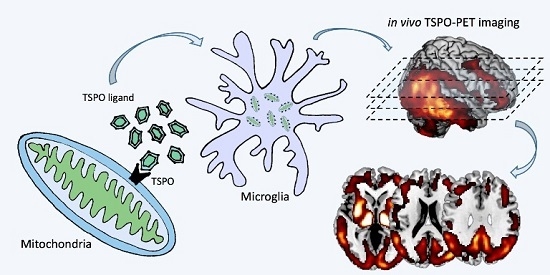
:
{kind=link}
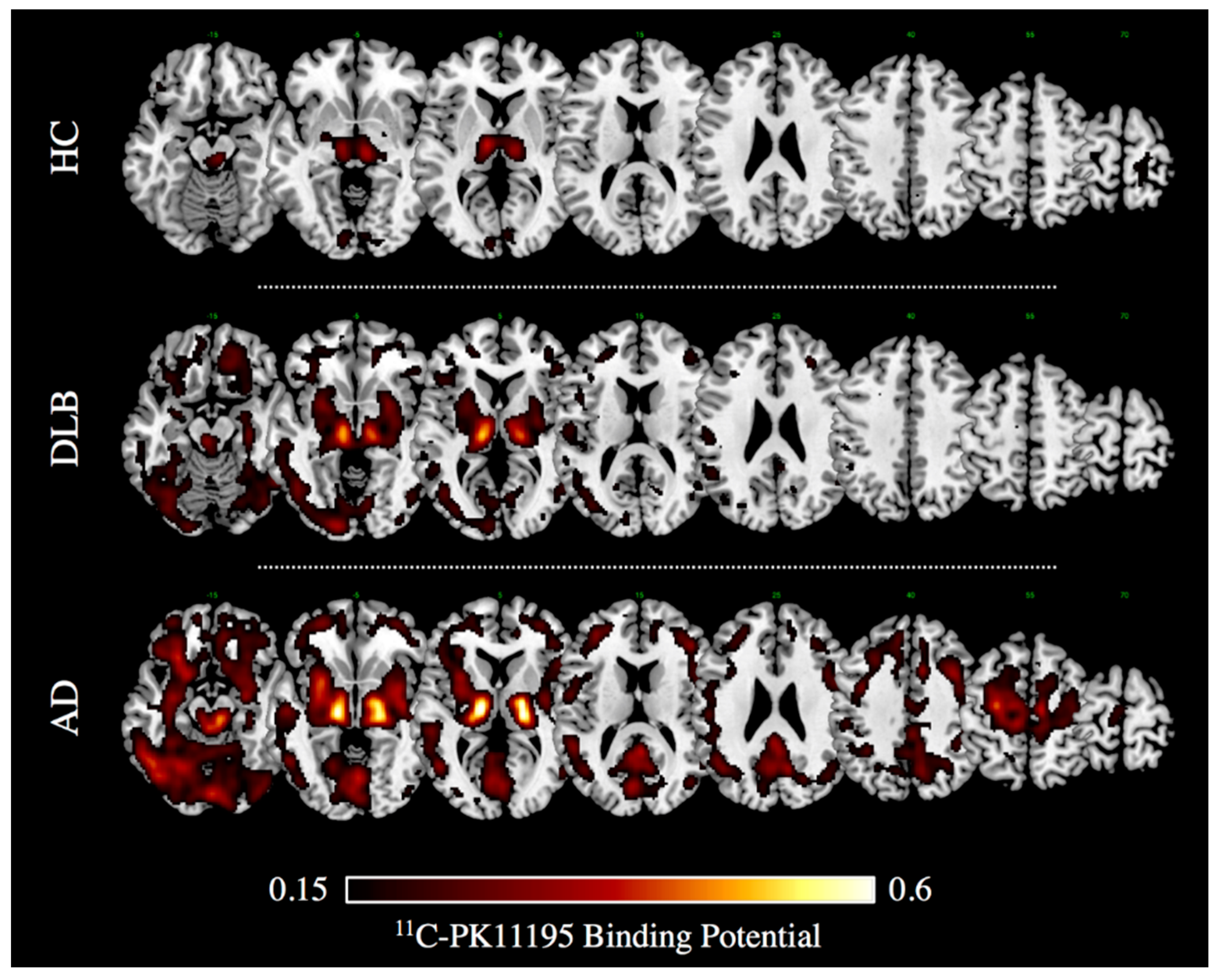{kind=link}
1. Introduction
2. Molecular Basis of Neuroinflammation in Neurodegeneration
3. PET Molecular Imaging of Neuroinflammation
4. In Vivo Pet Evidence in Neurodegenerative Dementias
4.1. Alzheimer’s Dementia and Prodromal Alzheimer’s Disease
4.2. Frontotemporal Lobar Degeneration and Parkinsonisms
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pievani, M.; Filippini, N.; van den Heuvel, M.P.; Cappa, S.F.; Frisoni, G.B. Brain connectivity in neurodegenerative diseases—From phenotype to proteinopathy. Nat. Rev. Neurol. 2014, 10, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; del Tredici, K.; Lee, V.M.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Pasqualetti, G.; Brooks, D.J.; Edison, P. The role of neuroinflammation in dementias. Curr. Neurol. Neurosci. Rep. 2015, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhang, Y.; Chen, Y.; Zhu, J.; Yang, Y.; Zhang, H.L. Role of microglia in neurological disorders and their potentials as a therapeutic target. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 5, 419–432. [Google Scholar] [CrossRef]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Czeh, M.; Gressens, P.; Kaindl, A.M. The yin and yang of microglia. Dev. Neurosci. 2011, 33, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Stefaniak, J.; O’Brien, J. Imaging of neuroinflammation in dementia: A review. J. Neurol. Neurosurg. Psychiatry 2016, 87, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, M.; Shinotoh, H.; Wszolek, Z.K.; Strongosky, A.J.; Shimada, H.; Arakawa, R.; Higuchi, M.; Ikoma, Y.; Yasuno, F.; Fukushi, K.; et al. In vivo detection of neuropathologic changes in presymptomatic MAPT mutation carriers: A PET and MRI study. Park. Relat. Disord. 2010, 16, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Lant, S.B.; Robinson, A.C.; Thompson, J.C.; Rollinson, S.; Pickering-Brown, S.; Snowden, J.S.; Davidson, Y.S.; Gerhard, A.; Mann, D.M. Patterns of microglial cell activation in frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 2014, 40, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Santillo, A.F.; Gambini, J.P.; Lannfelt, L.; Långström, B.; Ulla-Marja, L.; Kilander, L.; Engler, H. In vivo imaging of astrocytosis in Alzheimer’s disease: An 11C-l-deuteriodeprenyl and PIB PET study. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 2202–2208. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.F.; Schöll, M.; Almkvist, O.; Wall, A.; Engler, H.; Långström, B.; Nordberg, A. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-l-deprenyl: A multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 2012, 53, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Choo, I.L.; Carter, S.F.; Schöll, M.L.; Nordberg, A. Astrocytosis measured by 11C-deprenyl PET correlates with decrease in gray matter density in the parahippocampus of prodromal Alzheimer’s patients. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2120–2126. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vieitez, E.; Saint-Aubert, L.; Carter, S.F.; Almkvist, O.; Farid, K.; Schöll, M.; Chiotis, K.; Thordardottir, S.; Graff, C.; Wall, A.; et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain 2016, 139, 922–936. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Rawji, K.S.; Mishra, M.K.; Michaels, N.J.; Rivest, S.; Stys, P.K.; Yong, V.W. Immunosenescence of microglia and macrophages: Impact on the ageing central nervous system. Brain 2016, 139 Pt 3, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.È.; Lecours, C.; Samson, L.; Sánchez-Zafra, V.; Sierra, A. From the Cajal alumni Achúcarro and Río-Hortega to the rediscovery of never-resting microglia. Front. Neuroanat. 2015, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 54, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D. M1 and M2 macrophages: Oracles of health and disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.N.; Lambracht-Washington, D.; Yu, G.; Xia, W. Genomics of Alzheimer disease: A review. JAMA Neurol. 2016, 73, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; El Khoury, J. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H. TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 2013, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.H.; Tavitian, B. Noninvasive molecular imaging of neuroinflammation. J. Cereb. Blood Flow Metab. 2012, 32, 1393–1415. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Lopresti, B.J.; Wiley, C.A. Molecular imaging of microglia/macrophages in the brain. Glia 2013, 61, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.J.; Middleton, R.J.; Hatty, C.R.; Kam, W.W.; Chan, R.; Pham, T.; Harrison-Brown, M.; Dodson, E.; Veale, K.; Banati, R.B. The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol. 2014, 24, 631–653. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Lopresti, B.J.; Wiley, C.A. The peripheral benzodiazepine receptor (Translocator protein 18 kDa) in microglia: From pathology to imaging. Prog. Neurobiol. 2006, 80, 308–322. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, R.; Papadopoulos, V.; Rammes, G.; Baghai, T.C.; Fan, J.; Akula, N.; Groyer, G.; Adams, D.; Schumacher, M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2010, 9, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Lavisse, S.; Guillermier, M.; Hérard, A.S.; Petit, F.; Delahaye, M.; van Camp, N.; Ben Haim, L.; Lebon, V.; Remy, P.; Dollé, F.; et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J. Neurosci. 2012, 8, 10809–10818. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.S.; Logan, J.; Volkow, N.D.; Wang, G.J. Translational neuroimaging: Positron emission tomography studies of monoamine oxidase. Mol. Imaging Biol. 2005, 7, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Cosenza-Nashat, M.; Zhao, M.L.; Suh, H.S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, F.; Boutin, H.; van Camp, N.; Dolle, F.; Tavitian, B. Nuclear imaging of neuroinflammation: A comprehensive review of 11C-PK11195 challengers. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 2304–2319. [Google Scholar] [CrossRef] [PubMed]
- Varley, J.; Brooks, D.J.; Edison, P. Imaging neuroinflammation in Alzheimer’s disease and other dementias: Recent advances and future directions. Alzheimers Dement. 2015, 11, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R.A.; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Roncaroli, F.; Su, Z.; Herholz, K.; Gerhard, A.; Turkheimer, F.E. TSPO expression in brain tumours: Is TSPO a target for brain tumour imaging? Clin. Transl. Imaging 2016, 4, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Cerami, C.; Perani, D. Imaging neuroinflammation in ischemic stroke and in the atherosclerotic vascular disease. Curr. Vasc. Pharmacol. 2015, 13, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Airas, L.; Rissanen, E.; Rinne, J. Imaging of microglial activation in MS using PET: Research use and potential future clinical application. Mult. Scler. 2017, 23, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Yokokura, M.; Terada, T.; Bunai, T.; Nakaizumi, K.; Takebayashi, K.; Iwata, Y.; Yoshikawa, E.; Futatsubashi, M.; Suzuki, K.; Mori, N.; et al. Depiction of microglial activation in aging and dementia: Positron emission tomography with 11C-DPA713 versus 11C-(R)PK11195. J. Cereb. Blood Flow Metab. 2016, 37, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, F.; Ota, M.; Kosaka, J.; Ito, H.; Higuchi, M.; Doronbekov, T.K.; Nozaki, S.; Fujimura, Y.; Koeda, M.; Asada, T.; et al. Increased binding of peripheral benzodiazepine receptor in Alzheimer’s disease measured by positron emission tomography with 11C-DAA1106. Biol. Psychiatry 2008, 64, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, W.C.; Lyoo, C.H.; Liow, J.S.; Snow, J.; Page, E.; Jenko, K.J.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; Turner, R.S.; et al. Distinct patterns of increased translocator protein in posterior cortical atrophy and amnestic Alzheimer’s disease. Neurobiol. Aging 2017, 51, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, W.C.; Lyoo, C.H.; McGwier, M.; Snow, J.; Jenko, K.J.; Kimura, N.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 2013, 136, 2228–2238. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, W.C.; Lyoo, C.H.; Liow, J.S.; Wei, M.; Snow, J.; Page, E.; Jenko, K.J.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. 11C-PBR28 binding to translocator protein increases with progression of Alzheimer’s disease. Neurobiol. Aging 2016, 44, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Gulyás, B.; Vas, A.; Tóth, M.; Takano, A.; Varrone, A.; Cselényi, Z.; Schain, M.; Mattsson, P.; Halldin, C. Age and disease related changes in the translocator protein (TSPO) system in the human brain: Positron emission tomography measurements with 11C-vinpocetine. Neuroimage 2011, 56, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 2016, 139 Pt 4, 1252–1264. [Google Scholar] [CrossRef] [PubMed]
- Suridjan, I.; Pollock, B.G.; Verhoeff, N.P.; Voineskos, A.N.; Chow, T.; Rusjan, P.M.; Lobaugh, N.J.; Houle, S.; Mulsant, B.H.; Mizrahi, R. In vivo imaging of grey and white matter neuroinflammation in Alzheimer’s disease: A positron emission tomography study with a novel radioligand, 18F-FEPPA. Mol. Psychiatry 2015, 20, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Varrone, A.; Oikonen, V.; Forsberg, A.; Joutsa, J.; Takano, A.; Solin, O.; Haaparanta-Solin, M.; Nag, S.; Nakao, R.; Al-Tawil, N.; et al. Positron emission tomography imaging of the 18 kDa translocator protein (TSPO) with 18F-FEMPA in Alzheimer’s disease patients and control subjects. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, Y.; Ikoma, Y.; Yasuno, F.; Suhara, T.; Ota, M.; Matsumoto, R.; Nozaki, S.; Takano, A.; Kosaka, J.; Zhang, M.R.; et al. Quantitative analyses of 18F-FEDAA1106 binding to peripheral benzodiazepine receptors in living human brain. J. Nucl. Med. 2006, 47, 43–50. [Google Scholar] [PubMed]
- Takano, A.; Gulyás, B.; Varrone, A.; Karlsson, P.; Sjoholm, N.; Larsson, S.; Jonsson, C.; Odh, R.; Sparks, R.; Al Tawil, N.; et al. Biodistribution and radiation dosimetry of the 18 kDa translocator protein (TSPO) radioligand 18F-FEDAA1106: A human whole-body PET study. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Varrone, A.; Mattsson, P.; Forsberg, A.; Takano, A.; Nag, S.; Gulyás, B.; Borg, J.; Boellaard, R.; Al-Tawil, N.; Eriksdotter, M.; et al. In vivo imaging of the 18 kDa translocator protein (TSPO) with 18F-FEDAA1106 and PET does not show increased binding in Alzheimer’s disease patients. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.J.; Gunn, R.N.; Rabiner, E.A.; Bennacef, I.; Fujita, M.; Kreisl, W.C.; Innis, R.B.; Pike, V.W.; Reynolds, R.; Matthews, P.M.; et al. Mixed-affinity binding in humans with 18 kDa translocator protein ligands. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2011, 52, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Dodel, R.; Spottke, A.; Gerhard, A.; Reuss, A.; Reinecker, S.; Schimke, N.; Trenkwalder, C.; Sixel-Döring, F.; Herting, B.; Kamm, C.; et al. Minocycline 1-year therapy in multiple-system-atrophy: Effect on clinical symptoms and 11C-(R)-PK11195 PET (MEMSA-trial). Mov. Disord. 2010, 25, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Yeo, A.J.; Gunn, R.N.; Song, K.; Wadsworth, G.; Lewis, A.; Rhodes, C.; Pulford, D.J.; Bennacef, I.; Parker, C.A.; et al. An 18 kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 2012, 32, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Turkheimer, F.E.; Rizzo, G.; Bloomfield, P.S.; Howes, O.; Zanotti-Fregonara, P.; Bertoldo, A.; Veronese, M. The methodology of TSPO imaging with positron emission tomography. Biochem. Soc. Trans. 2015, 43, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Turkheimer, F.E.; Edison, P.; Pavese, N.; Roncaroli, F.; Anderson, A.N.; Hammers, A.; Gerhard, A.; Hinz, R.; Tai, Y.F.; Brooks, D.J. Reference and target region modeling of 11C-(R)-PK11195 brain studies. J. Nucl. Med. 2007, 48, 158–167. [Google Scholar] [PubMed]
- Yaqub, M.; van Berckel, B.N.; Schuitemaker, A.; Hinz, R.; Turkheimer, F.E.; Tomasi, G.; Lammertsma, A.A.; Boellaard, R. Optimization of supervised cluster analysis for extracting reference tissue input curves in (R)-11C-PK11195 brain PET studies. J. Cereb. Blood Flow Metab. 2012, 32, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.N.; Pavese, N.; Edison, P.; Tai, Y.F.; Hammers, A.; Gerhard, A.; Brooks, D.J.; Turkheimer, F.E. A systematic comparison of kinetic modelling methods generating parametric maps for 11C-(R)-PK11195. Neuroimage 2007, 36, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, G.; Edison, P.; Bertoldo, A.; Roncaroli, F.; Singh, P.; Gerhard, A.; Cobelli, C.; Brooks, D.J.; Turkheimer, F.E. Novel reference region model reveals increased microglial and reduced vascular binding of 11C-(R)-PK11195 in patients with Alzheimer’s disease. J. Nucl. Med. 2008, 49, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Engler, H.; Lundberg, P.O.; Ekbom, K.; Nennesmo, I.; Nilsson, A.; Bergström, M.; Tsukada, H.; Hartvig, P.; Långström, B. Multitracer study with positron emission tomography in Creutzfeldt-Jakob disease. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Engler, H.; Blomquist, G.; Scott, B.; Wall, A.; Aquilonius, S.M.; Långström, B.; Askmark, H. Evidence for astrocytosis in ALS demonstrated by 11C-l-deprenyl-D2 PET. J. Neurol. Sci. 2007, 255, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Kumlien, E.; Nilsson, A.; Hagberg, G.; Långström, B.; Bergström, M. PET with 11C-deuterium-deprenyl and 18F-FDG in focal epilepsy. Acta Neurol. Scand. 2001, 103, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Postnov, A.; Bormans, G.; Versijpt, J.; Vandenbulcke, M.; Van Laere, K. Decreased in vivo availability of the cannabinoid type 2 receptor in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Giovacchini, G.; Liow, J.S.; Bhattacharjee, A.K.; Greenstein, D.; Schapiro, M.; Hallett, M.; Herscovitch, P.; Eckelman, W.C.; Carson, R.E.; et al. Imaging neuroinflammation in Alzheimer’s disease with radiolabeled arachidonic acid and PET. J. Nucl. Med. 2008, 49, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Groom, G.N.; Junck, L.; Foster, N.L.; Frey, K.A.; Kuhl, D.E. PET of peripheral benzodiazepine binding sites in the microgliosis of Alzheimer’s disease. J. Nucl. Med. 1995, 36, 2207–2210. [Google Scholar] [PubMed]
- Kropholler, M.A.; Boellaard, R.; van Berckel, B.N.; Schuitemaker, A.; Kloet, R.W.; Lubberink, M.J.; Jonker, C.; Scheltens, P.; Lammertsma, A.A. Evaluation of reference regions for (R)-11C-PK11195 studies in Alzheimer’s disease and mild cognitive impairment. J. Cereb. Blood Flow Metab. 2007, 27, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.A.; Lopresti, B.J.; Venneti, S.; Price, J.; Klunk, W.E.; DeKosky, S.T.; Mathis, C.A. Carbon 11-labeled Pittsburgh compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch. Neurol. 2009, 66, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Versijpt, J.J.; Dumont, F.; van Laere, K.J.; Decoo, D.; Santens, P.; Audenaert, K.; Achten, E.; Slegers, G.; Dierckx, R.A.; Korf, J. Assessment of neuroinflammation and microglial activation in Alzheimer’s disease with radiolabelled PK11195 and single photon emission computed tomography. Eur. Neurol. 2003, 50, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Edison, P.; Archer, H.A.; Gerhard, A.; Hinz, R.; Pavese, N.; Turkheimer, F.E.; Hammers, A.; Tai, Y.F.; Fox, N.; Kennedy, A.; et al. Microglia, amyloid, and cognition in Alzheimer’s disease: An 11C-(R)PK11195-PET and 11C-PIB-PET study. Neurobiol. Dis. 2008, 32, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Aman, Y.; Ahmed, I.; Chetelat, G.; Landeau, B.; Ray Chaudhuri, K.; Brooks, D.J.; Edison, P. Influence of microglial activation on neuronal function in Alzheimer’s and Parkinson’s disease dementia. Alzheimers Dement. 2015, 11, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Ninan, S.; Atkinson, R.; Fan, Z.; Brooks, D.J.; Edison, P. Does microglial activation influence hippocampal volume and neuronal function in Alzheimer’s disease and Parkinson’s disease dementia? J. Alzheimers Dis. 2016, 51, 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Okello, A.A.; Brooks, D.J.; Edison, P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain 2015, 138, 3685–3698. [Google Scholar] [CrossRef] [PubMed]
- Okello, A.; Edison, P.; Archer, H.A.; Turkheimer, F.E.; Kennedy, J.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.; Rossor, M.; et al. Microglial activation and amyloid deposition in mild cognitive impairment: A PET study. Neurology 2009, 72, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Schuitemaker, A.; Kropholler, M.A.; Boellaard, R.; van der Flier, W.M.; Kloet, R.W.; van der Doef, T.F.; Knol, D.L.; Windhorst, A.D.; Luurtsema, G.; Barkhof, F.; et al. Microglial activation in Alzheimer’s disease: An (R)-11C-PK11195 positron emission tomography study. Neurobiol. Aging 2013, 34, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Yokokura, M.; Mori, N.; Yagi, S.; Yoshikawa, E.; Kikuchi, M.; Yoshihara, Y.; Wakuda, T.; Sugihara, G.; Takebayashi, K.; Suda, S.; et al. In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, F.; Kosaka, J.; Ota, M.; Higuchi, M.; Ito, H.; Fujimura, Y.; Nozaki, S.; Takahashi, S.; Mizukami, K.; Asada, T.; et al. Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with 11C-DAA1106. Psychiatry Res. 2012, 203, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Cerami, C.; Crespi, C.; Della Rosa, P.A.; Dodich, A.; Marcone, A.; Magnani, G.; Coppi, E.; Falini, A.; Cappa, S.F.; Perani, D. Brain changes within the visuo-spatial attentional network in posterior cortical atrophy. J. Alzheimers Dis. 2015, 43, 385–395. [Google Scholar] [PubMed]
- Cagnin, A.; Rossor, M.; Sampson, E.L.; Mackinnon, T.; Banati, R.B. In vivo detection of microglial activation in frontotemporal dementia. Ann. Neurol. 2004, 56, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Watts, J.; Trender-Gerhard, I.; Turkheimer, F.; Banati, R.B.; Bhatia, K.; Brooks, D.J. In vivo imaging of microglial activation with 11C-(R)-PK11195 PET in corticobasal degeneration. Mov. Disord. 2004, 19, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Trender-Gerhard, I.; Turkheimer, F.; Quinn, N.P.; Bhatia, K.P.; Brooks, D.J. In vivo imaging of microglial activation with 11C-(R)-PK11195 PET in progressive supranuclear palsy. Mov. Disord. 2006, 21, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Iannaccone, S.; Cerami, C.; Alessio, M.; Garibotto, V.; Panzacchi, A.; Olivieri, S.; Gelsomino, G.; Moresco, R.M.; Perani, D. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with 11C-(R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.L.; Willemsen, A.T.; Doorduin, J.; de Vries, E.F.; Dierckx, R.A.; Leenders, K.L. 11C-PK11195 PET: Quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat. Disord. 2010, 16, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Y.; Shi, X.; Ma, C. An overview on therapeutics attenuating amyloid β level in Alzheimer’s disease: Targeting neurotransmission, inflammation, oxidative stress and enhanced cholesterol levels. Am. J. Transl. Res. 2016, 8, 246–269. [Google Scholar] [PubMed]
- Budni, J.; Garcez, M.L.; de Medeiros, J.; Cassaro, E.; Bellettini-Santos, T.; Mina, F.; Quevedo, J. The anti-inflammatory role of minocycline in Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Venigalla, M.; Sonego, S.; Gyengesi, E.; Sharman, M.J.; Münch, G. Novel promising therapeutics against chronic neuroinflammation and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2016, 95, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Ratchford, J.N.; Endres, C.J.; Hammoud, D.A.; Pomper, M.G.; Shiee, N.; McGready, J.; Pham, D.L.; Calabresi, P.A. Decreased microglial activation in MS patients treated with glatiramer acetate. J. Neurol. 2012, 259, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Sucksdorff, M.; Rissanen, E.; Tuisku, J.; Nuutinen, S.; Paavilainen, T.; Rokka, J.; Rinne, J.; Airas, L. Evaluation of the effect of fingolimod treatment on microglial activation using serial PET imaging in multiple sclerosis. J. Nucl. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Vivash, L.; O’Brien, T.J. Imaging microglial activation with TSPO PET: Lighting up neurologic diseases? J. Nucl. Med. 2016, 57, 165–168. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerami, C.; Iaccarino, L.; Perani, D. Molecular Imaging of Neuroinflammation in Neurodegenerative Dementias: The Role of In Vivo PET Imaging. Int. J. Mol. Sci. 2017, 18, 993. https://doi.org/10.3390/ijms18050993
Cerami C, Iaccarino L, Perani D. Molecular Imaging of Neuroinflammation in Neurodegenerative Dementias: The Role of In Vivo PET Imaging. International Journal of Molecular Sciences. 2017; 18(5):993. https://doi.org/10.3390/ijms18050993
Chicago/Turabian StyleCerami, Chiara, Leonardo Iaccarino, and Daniela Perani. 2017. "Molecular Imaging of Neuroinflammation in Neurodegenerative Dementias: The Role of In Vivo PET Imaging" International Journal of Molecular Sciences 18, no. 5: 993. https://doi.org/10.3390/ijms18050993
APA StyleCerami, C., Iaccarino, L., & Perani, D. (2017). Molecular Imaging of Neuroinflammation in Neurodegenerative Dementias: The Role of In Vivo PET Imaging. International Journal of Molecular Sciences, 18(5), 993. https://doi.org/10.3390/ijms18050993