The Peroxisome-Mitochondria Connection: How and Why?


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
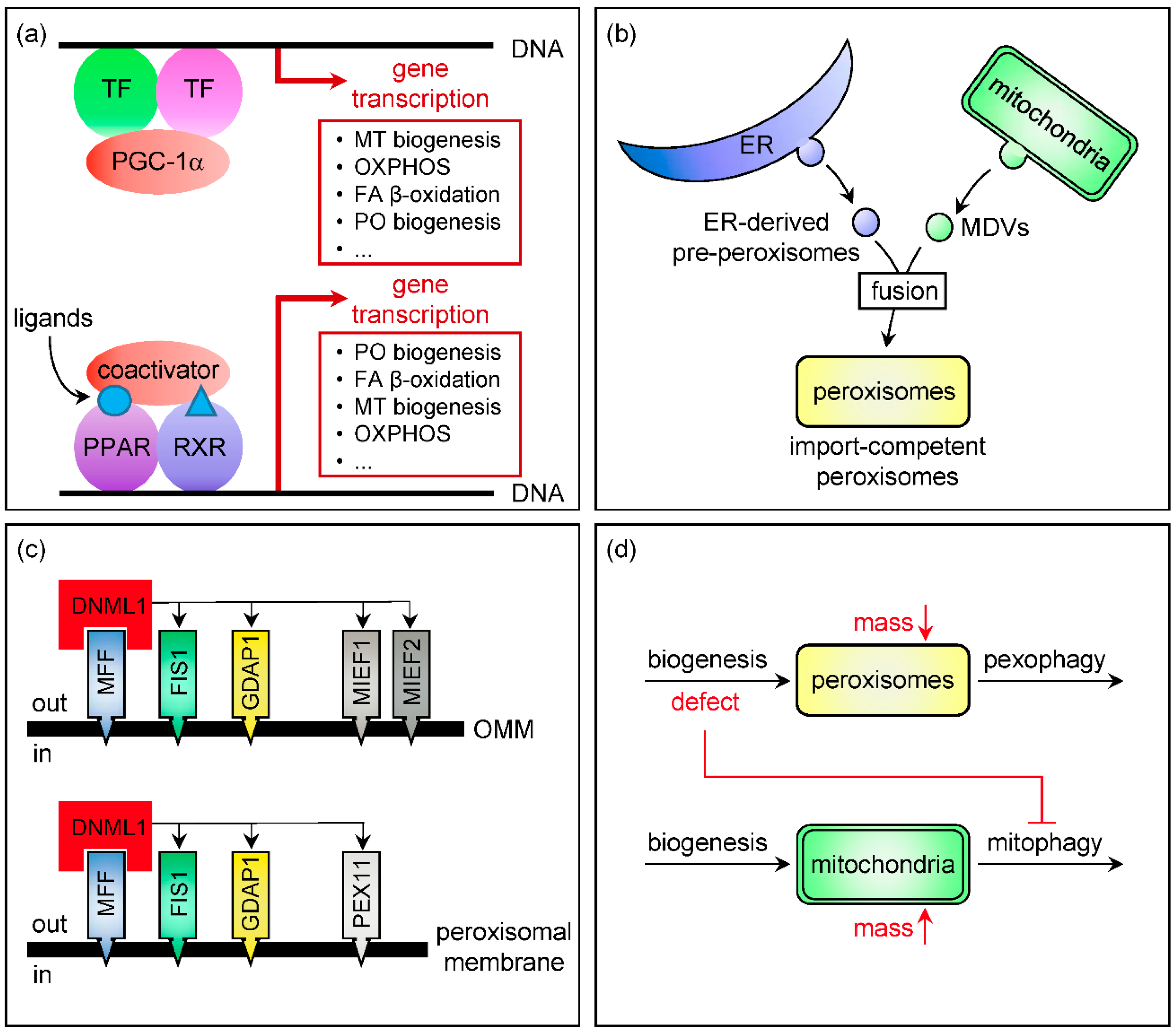
2. Control of Peroxisomal and Mitochondrial Abundance
2.1. Transcriptional Control of Organelle Biogenesis
2.2. Organelle Multiplication by Fission
2.3. Organellophagy
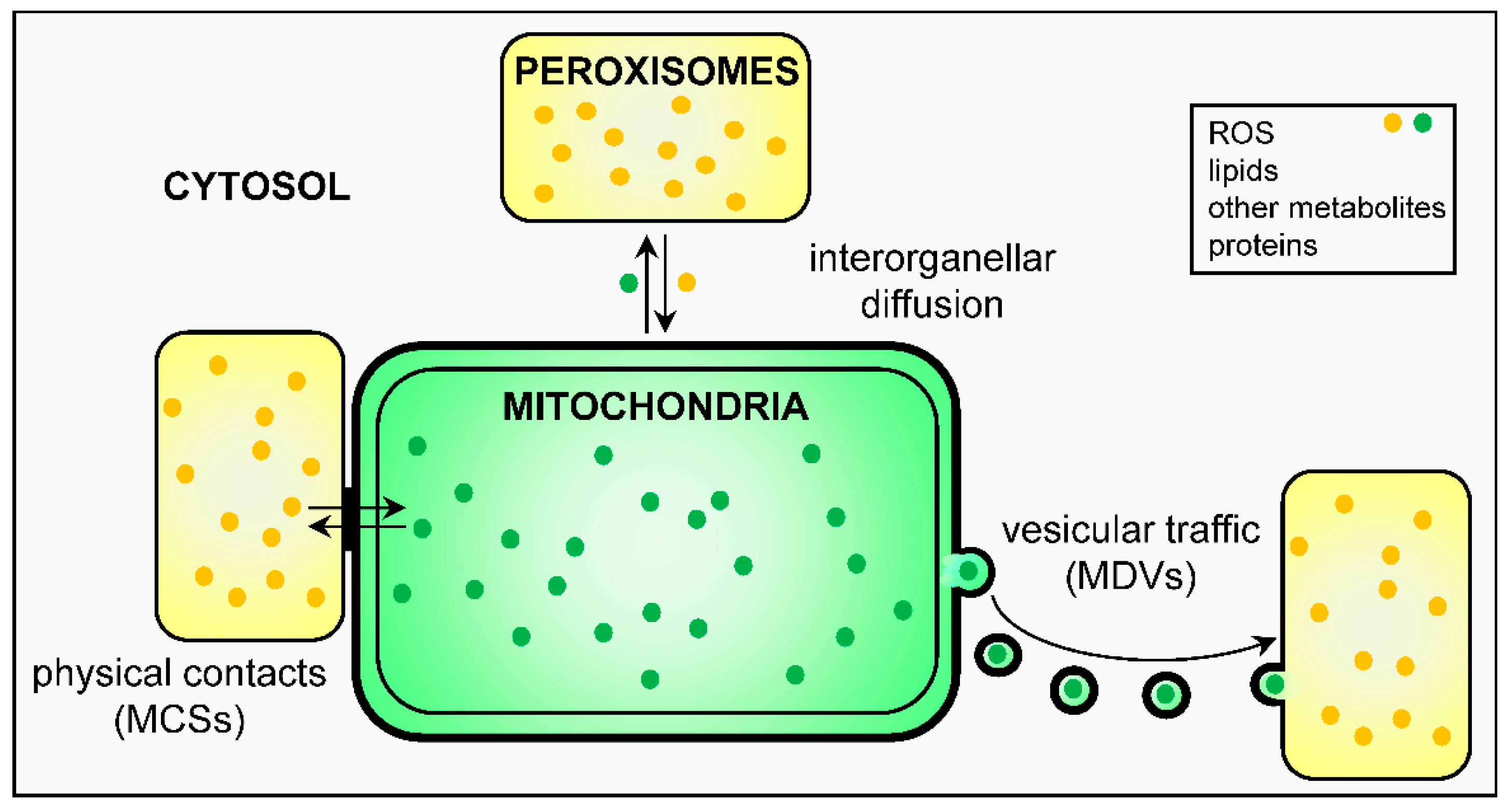
3. Peroxisome-Mitochondria Communication
3.1. Physical Contact Sites and Tethers
3.2. Vesicular Transport
3.3. Biological Messengers
4. Metabolic Interplay
4.1. β-Oxidation of Fatty Acids
4.2. Reactive Oxygen Species (ROS) Metabolism
4.2.1. ROS/RNS Producing and Scavenging Enzymes
4.2.2. Peroxisome-Mitochondria Redox Interplay
5. Interorganelle Signaling
5.1. Release of Messenger Molecules
5.1.1. ROS
5.1.2. Lipids
5.1.3. Other Metabolites
5.1.4. Proteins
5.2. Membrane-Associated Signaling Scaffolds
6. Peroxisome-Mitochondria Communication: Physiological Importance in Health and Disease
6.1. Organelle Function Deficiencies
6.2. Defects in Shared Components of the Fission Machinery
6.3. Dysregulation of Pexophagy
6.4. Viral Infections
6.5. Cellular Aging and Age-Related Diseases
7. Conclusions, Challenges and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ABCD | ATP-binding cassette transporters of subfamily D |
| ACAD | Acyl-CoA dehydrogenase |
| ACOX | Acyl-CoA oxidase |
| ADP | Adenine dinucleotide phosphate |
| ATP | Adenosine triphosphate |
| BAK | BCL2-antagonist/killer |
| BCL | B-cell lymphoma |
| BRCFA | Branched-chain fatty acid |
| CAC | Carnitine-acylcarnitine carrier |
| cAMP | Cyclic adenosine monophosphate |
| CoA | Coenzyme A |
| DNM1L | Dynamin 1-like protein |
| ECI | Enoyl-CoA δ isomerase |
| EMPF | Encephalopathy due to defective mitochondrial and peroxisomal fission |
| ER | Endoplasmic reticulum |
| ETF | Electron transfer flavoprotein |
| ETFDH | ETF dehydrogenase |
| FA | Fatty acid |
| FAD | Flavin adenine dinucleotide |
| FADH2 | Reduced FAD |
| FIS | Mitochondrial fission protein |
| FOXO | Forkhead box O |
| GDAP | Ganglioside-induced differentiation-associated protein |
| IFN | Interferon |
| IMM | Inner mitochondrial membrane |
| LCFA | Long-chain fatty acid |
| MAM | Mitochondria-associated ER membrane |
| MAVS | Mitochondrial antiviral-signaling protein |
| MCFA | Medium-chain fatty acid |
| MCT | Monocarboxylate transporter |
| MDV | Mitochondria-derived vesicle |
| MIEF | Mitochondrial elongation factor |
| MFF | Mitochondrial fission factor |
| MT | Mitochondria/mitochondrial |
| MUL | Mitochondrial E3 ubiquitin protein ligase |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH | Reduced NAD+ |
| NCOR | Nuclear receptor corepressor |
| NOS | Nitric oxide synthase |
| NRF | Nuclear respiratory factor |
| NRS | NAD(H) redox shuttle |
| OMM | Outer mitochondrial membrane |
| OXPHOS | Oxidative phosphorylation |
| PEX | peroxin |
| PGC | PPAR-γ co-activator |
| PO | Peroxisome/peroxisomal |
| PPAR | Peroxisome proliferator-activated receptor |
| PRDX | Peroxiredoxin |
| PXMP2 | Peroxisomal membrane protein 2 |
| RIG-I | Retinoic acid-inducible gene 1 |
| RLR | RIG-I-like receptor |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RXR | Retinoid X receptor |
| SOD | Superoxide dismutase |
| TCA | Tricarboxylic acid |
| TF | Transcription factor |
| VDAC | Voltage-dependent anion-selective channel |
| VLCFA | Very-long-chain fatty acid |
| X-ALD | X-linked adrenoleukodystrophy |
| XDH | Xanthine oxidoreductase |
| ZSS | Zellweger syndrome spectrum |
References
- Chen, A.H.; Silver, P.A. Designing biological compartmentalization. Trends Cell Biol. 2012, 22, 662–670. [Google Scholar] [CrossRef]
- Fransen, M. Peroxisome dynamics: Molecular players, mechanisms, and (dys) functions. ISRN Cell Biol. 2012, 2012, 714192. [Google Scholar] [CrossRef]
- Islinger, M.; Cardoso, M.J.; Schrader, M. Be different—The diversity of peroxisomes in the animal kingdom. Biochim. Biophys. Acta 2010, 1803, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Antonenkov, V.D.; Grunau, S.; Ohlmeier, S.; Hiltunen, J.K. Peroxisomes are oxidative organelles. Antioxid. Redox Signal. 2010, 13, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 2016, 1863, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Apanasets, O.; Nordgren, M.; Fransen, M. Dissecting peroxisome-mediated signaling pathways: A new and exciting research field. In Molecular Machines Involved in Peroxisome Biogenesis and Maintenance, 1st ed.; Brocard, C., Hartig, A., Eds.; Springer: Wien, Austria, 2014; pp. 255–273. [Google Scholar]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O.; Van Veldhoven, P.P. Aging, age-related diseases and peroxisomes. Subcell. Biochem. 2013, 69, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal dysfunction in age-related diseases. Trends. Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P. Interfaces between mitochondrial dynamics and disease. Cell Calcium 2016, 60, 190–198. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Kauppila, T.E.; Kauppila, J.H.; Larsson, N.G. Mammalian mitochondria and aging: An update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Geuze, H.J.; Murk, J.L.; Stroobants, A.K.; Griffith, J.M.; Kleijmeer, M.J.; Koster, A.J.; Verkleij, A.J.; Distel, B.; Tabak, H.F. Involvement of the endoplasmic reticulum in peroxisome formation. Mol. Biol. Cell 2003, 14, 2900–2907. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.K.; Mullen, R.T.; Schumann, U.; Lippincott-Schwartz, J. The origin and maintenance of mammalian peroxisomes involves a de novo PEX16-dependent pathway from the ER. J. Cell Biol. 2006, 173, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; Mattie, S.; Prudent, J.; McBride, H.M. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 2017, 542, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Huybrechts, S.J.; van Veldhoven, P.P.; Brees, C.; Mannaerts, G.P.; Los, G.V.; Fransen, M. Peroxisome dynamics in cultured mammalian cells. Traffic 2009, 10, 1722–1733. [Google Scholar] [CrossRef] [PubMed]
- Delille, H.K.; Agricola, B.; Guimaraes, S.C.; Borta, H.; Lüers, G.H.; Fransen, M.; Schrader, M. Pex11pβ-mediated growth and division of mammalian peroxisomes follows a maturation pathway. J. Cell Sci. 2010, 123, 2750–2762. [Google Scholar] [CrossRef] [PubMed]
- Nordgren, M.; Wang, B.; Apanasets, O.; Fransen, M. Peroxisome degradation in mammals: Mechanisms of action, recent advances, and perspectives. Front. Physiol. 2013, 4, 145. [Google Scholar] [CrossRef] [PubMed]
- Honsho, M.; Yamashita, S.; Fujiki, Y. Peroxisome homeostasis: Mechanisms of division and selective degradation of peroxisomes in mammals. Biochim. Biophys. Acta 2016, 1863, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta 2015, 1853, 2784–2790. [Google Scholar] [CrossRef] [PubMed]
- Bonekamp, N.A.; Sampaio, P.; de Abreu, F.V.; Lüers, G.H.; Schrader, M. Transient complex interactions of mammalian peroxisomes without exchange of matrix or membrane marker proteins. Traffic 2012, 13, 960–978. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Costello, J.; Godinho, L.F.; Islinger, M. Peroxisome-mitochondria interplay and disease. J. Inherit. Metab. Dis. 2015, 38, 681–702. [Google Scholar] [CrossRef] [PubMed]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor α target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef] [PubMed]
- Lodhi, I.J.; Semenkovich, C.F. Peroxisomes: A nexus for lipid metabolism and cellular signaling. Cell Metab. 2014, 19, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Song, J.; Park, K.W. The multifaceted factor peroxisome proliferator-activated receptor γ (PPARγ) in metabolism, immunity, and cancer. Arch. Pharm. Res. 2015, 38, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Barlaka, E.; Galatou, E.; Mellidis, K.; Ravingerova, T.; Lazou, A. Role of pleiotropic properties of peroxisome proliferator-activated receptors in the heart: Focus on the nonmetabolic effects in cardiac protection. Cardiovasc. Ther. 2016, 34, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Gutierrez, M.P.; Roszer, T.; Ricote, M. Biology and therapeutic applications of peroxisome proliferator- activated receptors. Curr. Top. Med. Chem. 2012, 12, 548–584. [Google Scholar] [CrossRef] [PubMed]
- Viswakarma, N.; Jia, Y.; Bai, L.; Vluggens, A.; Borensztajn, J.; Xu, J.; Reddy, J.K. Coactivators in PPAR-regulated gene expression. PPAR Res. 2010, 2010, 250126. [Google Scholar] [CrossRef] [PubMed]
- Cook, W.S.; Yeldandi, A.V.; Rao, M.S.; Hashimoto, T.; Reddy, J.K. Less extrahepatic induction of fatty acid β-oxidation enzymes by PPAR α. Biochem. Biophys. Res. Commun. 2000, 278, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Hoivik, D.J.; Qualls, C.W.; Mirabile, R.C.; Cariello, N.F.; Kimbrough, C.L.; Colton, H.M.; Anderson, S.P.; Santostefano, M.J.; Morgan, R.J.; Dahl, R.R.; et al. Fibrates induce hepatic peroxisome and mitochondrial proliferation without overt evidence of cellular proliferation and oxidative stress in cynomolgus monkeys. Carcinogenesis 2004, 25, 1757–1769. [Google Scholar] [CrossRef] [PubMed]
- Corona, J.C.; de Souza, S.C.; Duchen, M.R. PPARγ activation rescues mitochondrial function from inhibition of complex I and loss of PINK1. Exp. Neurol. 2014, 253, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism—Emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef] [PubMed]
- Wenz, T. PGC-1α activation as a therapeutic approach in mitochondrial disease. IUBMB Life 2009, 61, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Bagattin, A.; Hugendubler, L.; Mueller, E. Transcriptional coactivator PGC-1α promotes peroxisomal remodeling and biogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 20376–20381. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Zheng, D.; Houmard, J.A.; Brault, J.J.; Hickner, R.C.; Cortright, R.N. Overexpression of PGC-1α increases peroxisomal activity and mitochondrial fatty acid oxidation in human primary myotubes. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E253–E263. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Costello, J.L.; Godinho, L.F.; Azadi, A.S.; Islinger, M. Proliferation and fission of peroxisomes—An update. Biochim. Biophys. Acta 2016, 1863, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Yoon, Y. Mitochondrial fission and fusion. Biochem. Soc. Trans. 2016, 44, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Yoon, Y.; Bonekamp, N.A.; McNiven, M.A.; Schrader, M. A role for Fis1 in both mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2005, 16, 5077–5086. [Google Scholar] [CrossRef] [PubMed]
- Gandre-Babbe, S.; van der Bliek, A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- Huber, N.; Guimaraes, S.; Schrader, M.; Suter, U.; Niemann, A. Charcot-Marie-Tooth disease-associated mutants of GDAP1 dissociate its roles in peroxisomal and mitochondrial fission. EMBO Rep. 2013, 14, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gould, S.J. The dynamin-like DLP1ase DLP1 is essential for peroxisome division and is recruited to peroxisomes in part by PEX11. J. Biol. Chem. 2003, 278, 17012–17020. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Thiemann, M.; Grabenbauer, M.; Yoon, Y.; McNiven, M.A.; Schrader, M. Dynamin-like protein 1 is involved in peroxisomal fission. J. Biol. Chem. 2003, 278, 8597–8605. [Google Scholar] [CrossRef] [PubMed]
- Antonny, B.; Burd, C.; De Camilli, P.; Chen, E.; Daumke, O.; Faelber, K.; Ford, M.; Frolov, V.A.; Frost, A.; Hinshaw, J.E.; et al. Membrane fission by dynamin: What we know and what we need to know. EMBO J. 2016, 35, 2270–2284. [Google Scholar] [CrossRef] [PubMed]
- Itoyama, A.; Michiyuki, S.; Honsho, M.; Yamamoto, T.; Moser, A.; Yoshida, Y.; Fujiki, Y. Mff functions with Pex11pβ and DLP1 in peroxisomal fission. Biol. Open 2013, 2, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Kobayashi, S.; Fujiki, Y. Peroxisome division is impaired in a CHO cell mutant with an inactivating point-mutation in dynamin-like protein 1 gene. Exp. Cell Res. 2006, 312, 1671–1684. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Liu, X.; Feng, Y.; Klionsky, D.J. The progression of peroxisomal degradation through autophagy requires peroxisomal division. Autophagy 2014, 10, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Kohno, H.; Ishihara, N. Physiological roles of mitochondrial fission in cultured cells and mouse development. Ann. N. Y. Acad. Sci. 2015, 1350, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Peeters, A.; Shinde, A.B.; Dirkx, R.; Smet, J.; de Bock, K.; Espeel, M.; Vanhorebeek, I.; Vanlander, A.; van Coster, R.; Carmeliet, P.; et al. Mitochondria in peroxisome-deficient hepatocytes exhibit impaired respiration, depleted DNA, and PGC-1α independent proliferation. Biochim. Biophys. Acta 2015, 1853, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, V.; Phadke, R.; Saggar, A.; Hargreaves, I.P.; Yates, R.; Fokoloros, C.; Mankad, K.; Hertecant, J.; Ruggieri, M.; McCormick, D.; et al. Zellweger syndrome and secondary mitochondrial myopathy. Eur. J. Pediatr. 2015, 174, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, R.; Vanhorebeek, I.; Martens, K.; Schad, A.; Grabenbauer, M.; Fahimi, D.; Declercq, P.; Van Veldhoven, P.P.; Baes, M. Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities. Hepatology 2005, 41, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Vickers, A.E.; Bentley, P.; Fisher, R.L. Consequences of mitochondrial injury induced by pharmaceutical fatty acid oxidation inhibitors is characterized in human and rat liver slices. Toxicol. Vitr. 2006, 20, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Wicks, S.E.; Vandanmagsar, B.; Haynie, K.R.; Fuller, S.E.; Warfel, J.D.; Stephens, J.M.; Wang, M.; Han, X.; Zhang, J.; Noland, R.C.; et al. Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, E3300–E3309. [Google Scholar] [CrossRef] [PubMed]
- Rahim, R.S.; Chen, M.; Nourse, C.C.; Meedeniya, A.C.; Crane, D.I. Mitochondrial changes and oxidative stress in a mouse model of Zellweger syndrome neuropathogenesis. Neuroscience 2016, 334, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Hicks, L.; Fahimi, H.D. Peroxisomes (microbodies) in the myocardium of rodents and primates. A comparative ultrastructural cytochemical study. Cell Tissue Res. 1977, 175, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Islinger, M.; Lüers, G.H.; Zischka, H.; Ueffing, M.; Völkl, A. Insights into the membrane proteome of rat liver peroxisomes: Microsomal glutathione-S-transferase is shared by both subcellular compartments. Proteomics 2006, 6, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Wilkins, C.; Badil, S.; Iskarpatyoti, J.; Gale, M. Proteomic analysis of mitochondrial-associated ER membranes (MAM) during RNA virus infection reveals dynamic changes in protein and organelle trafficking. PLoS ONE 2015, 10, e0117963. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, X.; Issop, L.; Culty, M.; Papadopoulos, V. ACBD2/ECI2-mediated peroxisome-mitochondria interactions in Leydig cell steroid biosynthesis. Mol. Endocrinol. 2016, 30, 763–782. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.; Klug, Y.A.; Dimitrov, L.; Erez, Z.; Chuartzman, S.G.; Elinger, D.; Yofe, I.; Soliman, K.; Gärtner, J.; Thoms, S.; et al. Peroxisomes are juxtaposed to strategic sites on mitochondria. Mol. Biosyst. 2014, 10, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi Ušaj, M.; Brložnik, M.; Kaferle, P.; Žitnik, M.; Wolinski, H.; Leitner, F.; Kohlwein, S.D.; Zupan, B.; Petrovič, U. Genome-wide localization study of yeast Pex11 identifies peroxisome-mitochondria interactions through the ERMES complex. J. Mol. Biol. 2015, 427, 2072–2087. [Google Scholar] [CrossRef] [PubMed]
- Klecker, T.; Böckler, S.; Westermann, B. Making connections: Interorganelle contacts orchestrate mitochondrial behavior. Trends. Cell Biol. 2014, 24, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Navarro, M.A.; Sanchez-Pulido, L.; McBride, H.M. Mitochondrial vesicles: An ancient process providing new links to peroxisomes. Curr. Opin. Cell Biol. 2009, 21, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Neuspiel, M.; Schauss, A.C.; Braschi, E.; Zunino, R.; Rippstein, P.; Rachubinski, R.A.; Andrade-Navarro, M.A.; McBride, H.M. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 2008, 18, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Braschi, E.; Goyon, V.; Zunino, R.; Mohanty, A.; Xu, L.; McBride, H.M. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 2010, 20, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M. VDAC in cancer. Biochim. Biophys. Acta 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Pierri, C.L. Mitochondrial metabolite transport. Essays Biochem. 2010, 47, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Rokka, A.; Antonenkov, V.D.; Soininen, R.; Immonen, H.L.; Pirilä, P.L.; Bergmann, U.; Sormunen, R.T.; Weckström, M.; Benz, R.; Hiltunen, J.K. Pxmp2 is a channel-forming protein in mammalian peroxisomal membrane. PLoS ONE 2009, 4, e5090. [Google Scholar] [CrossRef] [PubMed]
- Antonenkov, V.D.; Hiltunen, J.K. Transfer of metabolites across the peroxisomal membrane. Biochim. Biophys. Acta 2012, 1822, 1374–1386. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Carrier, D.J.; Schaedler, T.; Waterham, H.R.; van Roermund, C.W.; Theodoulou, F.L. Peroxisomal ABC transporters: Functions and mechanism. Biochem. Soc. Trans. 2015, 43, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Visser, W.F.; van Roermund, C.W.; Ijlst, L.; Waterham, H.R.; Wanders, R.J. Metabolite transport across the peroxisomal membrane. Biochem. J. 2007, 401, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Agrimi, G.; Russo, A.; Scarcia, P.; Palmieri, F. The human gene SLC25A17 encodes a peroxisomal transporter of coenzyme A, FAD and NAD+. Biochem. J. 2012, 443, 241–247. [Google Scholar] [CrossRef] [PubMed]
- McClelland, G.B.; Khanna, S.; González, G.F.; Butz, C.E.; Brooks, G.A. Peroxisomal membrane monocarboxylate transporters: Evidence for a redox shuttle system? Biochem. Biophys. Res. Commun. 2003, 304, 130–135. [Google Scholar] [CrossRef]
- Valença, I.; Pértega-Gomes, N.; Vizcaino, J.R.; Henrique, R.M.; Lopes, C.; Baltazar, F.; Ribeiro, D. Localization of MCT2 at peroxisomes is associated with malignant transformation in prostate cancer. J. Cell Mol. Med. 2015, 19, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Gee, R.; McGroarty, E.; Hsien, B.; Wied, D.M.; Tolbert, N.E. Glycerol phosphate dehydrogenase in mammalian peroxisomes. Arch. Biochem. Biophys. 1974, 161, 187–193. [Google Scholar] [CrossRef]
- Baumgart, E.; Fahimi, H.D.; Stich, A.; Völkl, A. L-lactate dehydrogenase A4- and A3B isoforms are bona fide peroxisomal enzymes in rat liver. Evidence for involvement in intraperoxisomal NADH reoxidation. J. Biol. Chem. 1996, 271, 3846–3855. [Google Scholar] [CrossRef] [PubMed]
- Satrústegui, J.; Pardo, B.; Del Arco, A. Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol. Rev. 2007, 87, 29–67. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front. Cell Dev. Biol. 2016, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Poirier, Y.; Antonenkov, V.D.; Glumoff, T.; Hiltunen, J.K. Peroxisomal β-oxidation—A metabolic pathway with multiple functions. Biochim. Biophys. Acta 2006, 1763, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Ghisla, S.; Thorpe, C. Acyl-CoA dehydrogenases. A mechanistic overview. Eur. J. Biochem. 2004, 271, 494–508. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Ruiter, J.P.; IJLst, L.; Waterham, H.R.; Houten, S.M. The enzymology of mitochondrial fatty acid β-oxidation and its application to follow-up analysis of positive neonatal screening results. J. Inherit. Metab. Dis. 2010, 33, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Mitochondrial ROS metabolism: 10 years later. Biochemistry 2015, 80, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, E.; López-Bernardo, E.; Cadenas, S. Functional evidence for nitric oxide production by skeletal-muscle mitochondria from lipopolysaccharide-treated mice. Mitochondrion 2012, 12, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 23, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Sparacino-Watkins, C.E.; Tejero, J.; Sun, B.; Gauthier, M.C.; Thomas, J.; Ragireddy, V.; Merchant, B.A.; Wang, J.; Azarov, I.; Basu, P.; et al. Nitrite reductase and nitric-oxide synthase activity of the mitochondrial molybdopterin enzymes mARC1 and mARC2. J. Biol. Chem. 2014, 289, 10345–10358. [Google Scholar] [CrossRef] [PubMed]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Van Broekhoven, A.; Peeters, M.C.; Debeer, L.J.; Mannaerts, G.P. Subcellular distribution of coenzyme A: Evidence for a separate coenzyme A pool in peroxisomes. Biochem. Biophys. Res. Commun. 1981, 100, 305–312. [Google Scholar] [CrossRef]
- Horie, S.; Isobe, M.; Suga, T. Changes in CoA pools in hepatic peroxisomes of the rat under various conditions. J. Biochem. 1986, 99, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Maroz, A.; Anderson, R.F.; Smith, R.A.; Murphy, M.P. Reactivity of ubiquinone and ubiquinol with superoxide and the hydroperoxyl radical: Implications for in vivo antioxidant activity. Free Radic. Biol. Med. 2009, 46, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Sagun, K.C.; Cárcamo, J.M.; Golde, D.W. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J. 2005, 19, 1657–1667. [Google Scholar] [CrossRef]
- Lauridsen, C.; Jensen, S.K. α-Tocopherol incorporation in mitochondria and microsomes upon supranutritional vitamin E supplementation. Genes Nutr. 2012, 7, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Schlüter, T.; Fischer, K.D.; Reiser, G. Non-esterified polyunsaturated fatty acids distinctly modulate the mitochondrial and cellular ROS production in normoxia and hypoxia. J. Neurochem. 2011, 118, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, M.; Maack, C. Calcium release microdomains and mitochondria. Cardiovasc. Res. 2013, 98, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Koepke, J.I.; Wood, C.S.; Terlecky, L.J.; Walton, P.A.; Terlecky, S.R. Progeric effects of catalase inactivation in human cells. Toxicol. Appl. Pharmacol. 2008, 232, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 2011, 22, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Lee, J.; Huh, J.Y.; Park, J.; Lee, H.B.; Ho, Y.S.; Ha, H. Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes 2012, 61, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Walton, P.A.; Pizzitelli, M. Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 2012, 3, 108. [Google Scholar] [CrossRef] [PubMed]
- López-Erauskin, J.; Galino, J.; Ruiz, M.; Cuezva, J.M.; Fabregat, I.; Cacabelos, D.; Boada, J.; Martínez, J.; Ferrer, I.; Pamplona, R.; et al. Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X-linked adrenoleukodystrophy. Hum. Mol. Genet. 2013, 22, 3296–3305. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, S.; Ferrer, I.; Pujol, A. Oxidative stress, mitochondrial and proteostasis malfunction in adrenoleukodystrophy: A paradigm for axonal degeneration. Free Radic. Biol. Med. 2015, 88, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, E.; Vanhorebeek, I.; Grabenbauer, M.; Borgers, M.; Declercq, P.E.; Fahimi, H.D.; Baes, M. Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse). Am. J. Pathol. 2001, 159, 1477–1494. [Google Scholar] [CrossRef]
- Koepke, J.I.; Nakrieko, K.A.; Wood, C.S.; Boucher, K.K.; Terlecky, L.J.; Walton, P.A.; Terlecky, S.R. Restoration of peroxisomal catalase import in a model of human cellular aging. Traffic 2007, 8, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Liepinsh, E.; Skapare, E.; Kuka, J.; Makrecka, M.; Cirule, H.; Vavers, E.; Sevostjanovs, E.; Grinberga, S.; Pugovics, O.; Dambrova, M. Activated peroxisomal fatty acid metabolism improves cardiac recovery in ischemia-reperfusion. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Van Zutphen, T.; Ciapaite, J.; Bloks, V.W.; Ackereley, C.; Gerding, A.; Jurdzinski, A.; de Moraes, R.A.; Zhang, L.; Wolters, J.C.; Bischoff, R.; et al. Malnutrition-associated liver steatosis and ATP depletion is caused by peroxisomal and mitochondrial dysfunction. J. Hepatol. 2016, 65, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox. Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Khan, H.; Shahab, U.; Rehman, S.; Rafi, Z.; Khan, M.Y.; Ansari, A.; Siddiqui, Z.; Ashraf, J.M.; Abdullah, S.M.; et al. Protein oxidation: An overview of metabolism of sulphur containing amino acid, cysteine. Front. Biosci. 2017, 9, 71–87. [Google Scholar] [CrossRef]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.R.; Sampaio, I.H.; Teodoro, B.G.; Sousa, T.A.; Zoppi, C.C.; Queiroz, A.L.; Passos, M.A.; Alberici, L.C.; Teixeira, F.R.; Manfiolli, A.O.; et al. Hydrogen peroxide production regulates the mitochondrial function in insulin resistant muscle cells: Effect of catalase overexpression. Biochim. Biophys. Acta 2013, 1832, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Behring, J.B.; Shao, D.; Sverdlov, A.L.; Whelan, S.A.; Elezaby, A.; Yin, X.; Siwik, D.A.; Seta, F.; Costello, C.E.; et al. Overexpression of catalase diminishes oxidative cysteine modifications of cardiac proteins. PLoS ONE 2015, 10, e0144025. [Google Scholar] [CrossRef] [PubMed]
- Netto, L.E.; Antunes, F. The roles of peroxiredoxin and thioredoxin in hydrogen peroxide sensing and in signal transduction. Mol. Cells 2016, 39, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Wadley, A.J.; Aldred, S.; Coles, S.J. An unexplored role for peroxiredoxin in exercise-induced redox signalling? Redox Biol. 2016, 8, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Epstein, C.B.; Waddle, J.A.; Hale, W., 4th; Davé, V.; Thornton, J.; Macatee, T.L.; Garner, H.R.; Butow, R.A. Genome-wide responses to mitochondrial dysfunction. Mol. Biol. Cell 2001, 12, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Bulina, M.E.; Chudakov, D.M.; Britanova, O.V.; Yanushevich, Y.G.; Staroverov, D.B.; Chepurnykh, T.V.; Merzlyak, E.M.; Shkrob, M.A.; Lukyanov, S.; Lukyanov, K.A. A genetically encoded photosensitizer. Nat. Biotechnol. 2006, 24, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; van Veldhoven, P.P.; Brees, C.; Rubio, N.; Nordgren, M.; Apanasets, O.; Kunze, M.; Baes, M.; Agostinis, P.; Fransen, M. Mitochondria are targets for peroxisome-derived oxidative stress in cultured mammalian cells. Free Radic. Biol. Med. 2013, 65, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Walbrecq, G.; Wang, B.; Becker, S.; Hannotiau, A.; Fransen, M.; Knoops, B. Antioxidant cytoprotection by peroxisomal peroxiredoxin-5. Free Radic. Biol. Med. 2015, 84, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Gross, R.W.; Han, X. Lipidomics at the interface of structure and function in systems biology. Chem. Biol. 2011, 18, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Reiser, G.; Schönfeld, P.; Kahlert, S. Mechanism of toxicity of the branched-chain fatty acid phytanic acid, a marker of Refsum disease, in astrocytes involves mitochondrial impairment. Int. J. Dev. Neurosci. 2006, 24, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Rönicke, S.; Kruska, N.; Kahlert, S.; Reiser, G. The influence of the branched-chain fatty acids pristanic acid and Refsum disease-associated phytanic acid on mitochondrial functions and calcium regulation of hippocampal neurons, astrocytes, and oligodendrocytes. Neurobiol. Dis. 2009, 36, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Baarine, M.; Andréoletti, P.; Athias, A.; Nury, T.; Zarrouk, A.; Ragot, K.; Vejux, A.; Riedinger, J.M.; Kattan, Z.; Bessede, G.; et al. Evidence of oxidative stress in very long chain fatty acid-treated oligodendrocytes and potentialization of ROS production using RNA interference-directed knockdown of ABCD1 and ACOX1 peroxisomal proteins. Neuroscience 2012, 213, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Busanello, E.N.; Amaral, A.U.; Tonin, A.M.; Grings, M.; Moura, A.P.; Eichler, P.; Vargas, C.R.; Wajner, M. Experimental evidence that pristanic acid disrupts mitochondrial homeostasis in brain of young rats. J. Neurosci. Res. 2012, 90, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Farr, R.L.; Lismont, C.; Terlecky, S.R.; Fransen, M. Peroxisome biogenesis in mammalian cells: The impact of genes and environment. Biochim. Biophys. Acta 2016, 1863, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and epigenetics—Crosstalk in homeostasis and stress. Trends. Cell Biol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, K.I.; Miyata, N.; Mukai, S.; Furuki, S.; Okumoto, K.; Cheng, E.H.; Fujiki, Y. The VDAC2-BAK axis regulates peroxisomal membrane permeability. J. Cell Biol. 2017, 216, 709–722. [Google Scholar] [CrossRef] [PubMed]
- De Pinto, V.; Reina, S.; Gupta, A.; Messina, A.; Mahalakshmi, R. Role of cysteines in mammalian VDAC isoforms’ function. Biochim. Biophys. Acta 2016, 1857, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, K.; García-Sáez, A.J. Bax and Bak pores: Are we closing the circle? Trends. Cell Biol. 2017, 27, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Walton, P.A.; Brees, C.; Lismont, C.; Apanasets, O.; Fransen, M. The peroxisomal import receptor PEX5 functions as a stress sensor, retaining catalase in the cytosol in times of oxidative stress. Biochem. Biophys. Acta 2017. in revision. [Google Scholar]
- Kagan, J.C.; Magupalli, V.G.; Wu, H. SMOCs: Supramolecular organizing centres that control innate immunity. Nat. Rev. Immunol. 2014, 14, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Jang, H. Dynamic multiprotein assemblies shape the spatial structure of cell signaling. Prog. Biophys. Mol. Biol. 2014, 116, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Odendall, C.; Kagan, J.C. Activation and pathogenic manipulation of the sensors of the innate immune system. Microbes Infect. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Durbin, A.F.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Reuter, A.; Eberle, F.; Einhorn, E.; Binder, M.; Bartenschlager, R. Activation of type I and III interferon response by mitochondrial and peroxisomal MAVS and inhibition by hepatitis C virus. PLoS Pathog. 2015, 11, e1005264. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.K.; Melchjorsen, J.; Rintahaka, J.; Diget, E.; Søby, S.; Horan, K.A.; Gorelick, R.J.; Matikainen, S.; Larsen, C.S.; Ostergaard, L.; et al. Genomic HIV RNA induces innate immune responses through RIG-I-dependent sensing of secondary-structured RNA. PLoS ONE 2012, 7, e29291. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem. 2004, 279, 32804–32812. [Google Scholar] [CrossRef] [PubMed]
- Baes, M.; Van Veldhoven, P.P. Mouse models for peroxisome biogenesis defects and β-oxidation enzyme deficiencies. Biochim. Biophys. Acta 2012, 1822, 1489–1500. [Google Scholar] [CrossRef] [PubMed]
- Torraco, A.; Peralta, S.; Iommarini, L.; Diaz, F. Mitochondrial diseases part I: Mouse models of OXPHOS deficiencies caused by defects in respiratory complex subunits or assembly factors. Mitochondrion 2015, 21, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Iommarini, L.; Peralta, S.; Torraco, A.; Diaz, F. Mitochondrial diseases part II: Mouse models of OXPHOS deficiencies caused by defects in regulatory factors and other components required for mitochondrial function. Mitochondrion 2015, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Fujita, T.; Usuda, N.; Cook, W.; Qi, C.; Peters, J.M.; Gonzalez, F.J.; Yeldandi, A.V.; Rao, M.S.; Reddy, J.K. Peroxisomal and mitochondrial fatty acid β-oxidation in mice nullizygous for both peroxisome proliferator-activated receptor α and peroxisomal fatty acyl-CoA oxidase. Genotype correlation with fatty liver phenotype. J. Biol. Chem. 1999, 274, 19228–19236. [Google Scholar] [CrossRef] [PubMed]
- Vluggens, A.; Andreoletti, P.; Viswakarma, N.; Jia, Y.; Matsumoto, K.; Kulik, W.; Khan, M.; Huang, J.; Guo, D.; Yu, S.; et al. Reversal of mouse Acyl-CoA oxidase 1 (ACOX1) null phenotype by human ACOX1b isoform. Lab. Investig. 2010, 90, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Góth, L.; Nagy, T. Inherited catalase deficiency: Is it benign or a factor in various age related disorders? Mutat. Res. 2013, 753, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Oettl, K.; Höfler, G.; Ness, G.C.; Sattler, W.; Malle, E. An apparent decrease in cholesterol biosynthesis in peroxisomal-defective Chinese hamster ovary cells is related to impaired mitochondrial oxidation. Biochem. Biophys. Res. Commun. 2003, 305, 957–963. [Google Scholar] [CrossRef]
- McGuinness, M.C.; Lu, J.F.; Zhang, H.P.; Dong, G.X.; Heinzer, A.K.; Watkins, P.A.; Powers, J.; Smith, K.D. Role of ALDP (ABCD1) and mitochondria in X-linked adrenoleukodystrophy. Mol. Cell Biol. 2003, 23, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Kapfhammer, J.P.; Hindelang, C.; Kemp, S.; Troffer-Charlier, N.; Broccoli, V.; Callyzot, N.; Mooyer, P.; Selhorst, J.; Vreken, P.; et al. Inactivation of the peroxisomal ABCD2 transporter in the mouse leads to late-onset ataxia involving mitochondria, Golgi and endoplasmic reticulum damage. Hum. Mol. Genet. 2005, 14, 3565–3577. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.Y.; Pan, J.; Chu, R.; Lee, D.; Kluckman, K.D.; Usuda, N.; Singh, I.; Yeldandi, A.V.; Rao, M.S.; Maeda, N.; et al. Hepatocellular and hepatic peroxisomal alterations in mice with a disrupted peroxisomal fattyacyl-coenzyme A oxidase gene. J. Biol. Chem. 1996, 271, 24698–24710. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.H.; Robak, L.A.; Xia, F.; Koenig, M.K.; Adesina, A.; Bacino, C.A.; Scaglia, F.; Bellen, H.J.; Wangler, M.F. Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila. Hum. Mol. Genet. 2016, 25, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Vanstone, J.R.; Smith, A.M.; McBride, S.; Naas, T.; Holcik, M.; Antoun, G.; Harper, M.E.; Michaud, J.; Sell, E.; Chakraborty, P.; et al. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur. J. Hum. Genet. 2016, 24, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Alshammari, M.; Al-Sheddi, T.; Salih, M.A.; Alkhalidi, H.; Kentab, A.; Repetto, G.M.; Hashem, M.; Alkuraya, F.S. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 2012, 49, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Feichtinger, R.G.; Freisinger, P.; Pies, M.; Schrödl, F.; Iuso, A.; Sperl, W.; Mayr, J.A.; Prokisch, H.; Haack, T.B. Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. Med. Genet. 2016, 53, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Cassereau, J.; Chevrollier, A.; Bonneau, D.; Verny, C.; Procaccio, V.; Reynier, P.; Ferré, M. A locus-specific database for mutations in GDAP1 allows analysis of genotype-phenotype correlations in Charcot-Marie-Tooth diseases type 4A and 2K. Orphanet J. Rare Dis. 2011, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.F.; Golden, M.H.; Brooks, S.E. Peroxisomes and the fatty liver of malnutrition: An hypothesis. Am. J. Clin. Nutr. 1991, 54, 674–677. [Google Scholar] [PubMed]
- Vasko, R.; Ratliff, B.B.; Bohr, S.; Nadel, E.; Chen, J.; Xavier, S.; Chander, P.; Goligorsky, M.S. Endothelial peroxisomal dysfunction and impaired pexophagy promotes oxidative damage in lipopolysaccharide-induced acute kidney injury. Antioxid. Redox Signal. 2013, 19, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.M.; Schönenberger, M.J.; Trötzmüller, M.; Horn, M.; Elsässer, H.P.; Moser, A.B.; Lucas, M.S.; Schwarz, T.; Gerber, P.A.; Faust, P.L.; et al. Hif-2α promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab. 2014, 20, 882–897. [Google Scholar] [CrossRef] [PubMed]
- Lazarow, P.B. Viruses exploiting peroxisomes. Curr. Opin. Microbiol. 2011, 14, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef] [PubMed]
- Tanner, L.B.; Chng, C.; Guan, X.L.; Lei, Z.; Rozen, S.G.; Wenk, M.R. Lipidomics identifies a requirement for peroxisomal function during influenza virus replication. J. Lipid Res. 2014, 55, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Hou, S.; Malik-Soni, N.; Xu, Z.; Kumar, A.; Rachubinski, R.A.; Frappier, L.; Hobman, T.C. Flavivirus infection impairs peroxisome biogenesis and early antiviral signaling. J. Virol. 2015, 89, 12349–12361. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sun, T.; Park, S.; Shen, G.; Liu, J. The role of hepatitis B virus X protein is related to its differential intracellular localization. Acta Biochim. Biophys. Sin. 2011, 43, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Kang, J.A.; Han, M.H.; Chung, K.H.; Lee, C.R.; Song, W.K.; Jun, Y.; Park, S.G. Peroxisome-localized hepatitis Bx protein increases the invasion property of hepatocellular carcinoma cells. Arch. Virol. 2014, 159, 2549–2557. [Google Scholar] [CrossRef] [PubMed]
- Titorenko, V.I.; Terlecky, S.R. Peroxisome metabolism and cellular aging. Traffic 2011, 12, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, D.; Palacín, M.; Zorzano, A. Mitochondrial dynamics: Coupling mitochondrial fitness with healthy aging. Trends. Mol. Med. 2017, 23, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.J.; Quintanilla, R.A.; Toro, A.; Grandy, R.; Dinamarca, M.C.; Godoy, J.A.; Inestrosa, N.C. Peroxisomal proliferation protects from β-amyloid neurodegeneration. J. Biol. Chem. 2005, 280, 41057–41068. [Google Scholar] [CrossRef] [PubMed]
- Nell, H.J.; Au, J.L.; Giordano, C.R.; Terlecky, S.R.; Walton, P.A.; Whitehead, S.N.; Cechetto, D.F. Targeted antioxidant, catalase-SKL, reduces β-amyloid toxicity in the rat brain. Brain Pathol. 2017, 27, 86–94. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18, 1126. https://doi.org/10.3390/ijms18061126
Fransen M, Lismont C, Walton P. The Peroxisome-Mitochondria Connection: How and Why? International Journal of Molecular Sciences. 2017; 18(6):1126. https://doi.org/10.3390/ijms18061126
Chicago/Turabian StyleFransen, Marc, Celien Lismont, and Paul Walton. 2017. "The Peroxisome-Mitochondria Connection: How and Why?" International Journal of Molecular Sciences 18, no. 6: 1126. https://doi.org/10.3390/ijms18061126
APA StyleFransen, M., Lismont, C., & Walton, P. (2017). The Peroxisome-Mitochondria Connection: How and Why? International Journal of Molecular Sciences, 18(6), 1126. https://doi.org/10.3390/ijms18061126