1. Introduction
Chloronitrobenzenes are widely used as chemical intermediates in the production of dyes, pesticides, drugs, and other commercial products [
1,
2,
3,
4,
5]. Exposure to these compounds occurs primarily in an occupational setting, but chloronitrobenzenes are also present in the environment from wastewater at industrial sites and from accidental spills [
6,
7,
8].
Information concerning the potential toxicity induced by the chloronitrobenzenes comes primarily from studies on monochloronitrobenzenes (MCNBs). These compounds induce hematotoxicity, hepatotoxicity, immunotoxicity, splenotoxicity, and nephrotoxicity [
9,
10,
11,
12,
13,
14,
15]. Hematotoxicity is generally the most common toxicity induced by MCNBs and is present as methemoglobinemia and anemia, and these conditions may contribute to toxicity in other organs [
9,
10,
11]. Although the toxicity induced by MCNBs has been studied to some degree, few studies have examined the toxicity induced by higher chlorinated nitrobenzenes (dichloro-, trichloro-, etc.), and it is generally believed that dichloronitrobenzenes (DCNBs) and trichloronitrobenzenes (TCNBs) have the potential to induce the same kinds of toxicities as the MCNBs.
While there have been several studies examining chloronitrobenzene-induced hematoxicity, few studies have explored the nephrotoxicity induced by these compounds. Yoshida et al. found that 4-chloronitrobenzene (1.0 mmol/kg, intraperitoneally) induced diuresis, increased
N-acetyl-β-
d-glucosaminidase excretion, and caused swelling of the kidney 48 h after treatment [
14]. However, urinary creatinine and blood urea nitrogen (BUN) concentrations were not altered in the 4-chloronitrobenzene treatment group, suggesting that only a mild nephrotoxicity was induced. Matsumoto et al. [
13] found that feeding 2-chloronitrobenzene to Fisher 344 rats increased the number of renal cell adenomas in females and renal carcinomas in males and exacerbated age-related chronic progressive nephropathy in males, leading to the death of 47 of 50 rats treated at the 2000 ppm level. Hong et al. found that pyruvate-stimulated gluconeogenesis was significantly reduced at 120 min in renal cortical slices from male Fischer 344 rats incubated with 1.0 mM of any of the three MCNBs [
12]. However, lactate dehydrogenase (LDH) release, a marker of cell death, was not increased until very high MCNB concentrations (≥3 mM) were used. Thus, MCNBs appear to be mild nephrotoxicants in vitro as well.
Studies on the effects of polychlorinated nitrobenzenes on renal function are limited to the work of Hong et al. who also determined the in vitro nephrotoxicity induced by the six DCNBs and two of the TCNBs [
12]. The DCNBs were generally more potent nephrotoxicants than the MCNBs with only 3,4-dichloronitrobenzene (3,4-DCNB) reducing pyruvate-stimulated gluconeogenesis and increasing LDH release at a concentration as low as 0.5 mM. The two TCNBs tested (2,3,4- and 3,4,5-TCNB) were both potent nephrotoxicants and similar in their cytotoxic profile to 3,4-DCNB [
12].
The role of biotransformation in chloronitrobenzene-induced nephrotoxicity is unknown. Chloronitrobenzenes are primarily metabolized by reduction of the nitro group to form the corresponding chloroaniline, followed by oxidation, acetylation, and conjugation reactions [
16,
17,
18]. In addition, a chloro group in the para position to the nitro group can be displaced enzymatically by glutathione, followed by metabolism of the conjugate to a mercapturate metabolite [
18,
19]. The formation of mercapturate metabolites from a chloronitrobenzene is considered a biomarker of chloronitrobenzene exposure [
20]. While chloroanilines are nephrotoxicants in vivo and in vitro [
21,
22,
23,
24,
25], it is not clear if these metabolites contribute to chloronitrobenzene nephrotoxicity.
The purpose of this study was to extend earlier studies of TCNB in vitro nephrotoxicity by determining the nephrotoxic potential of four TCNB compounds (2,3,4-, 2,4,5-, 2,4,6-, and 3,4,5-TCNB;
Figure 1) using isolated renal cortical cells (IRCC) as the animal model. The role of free radicals and/or biotransformation in TCNB in vitro nephrotoxicity was also examined. The IRCC model was chosen for study because many of our recent studies on chloroaniline nephrotoxicity have used this model [
25]. The Fischer 344 rat was chosen because this rat strain responds to nephrotoxicants more like humans than other rat strains [
26].
3. Discussion
There is little toxicological information available on the potential harmful effects of the TCNBs in mammals. Hong et al. [
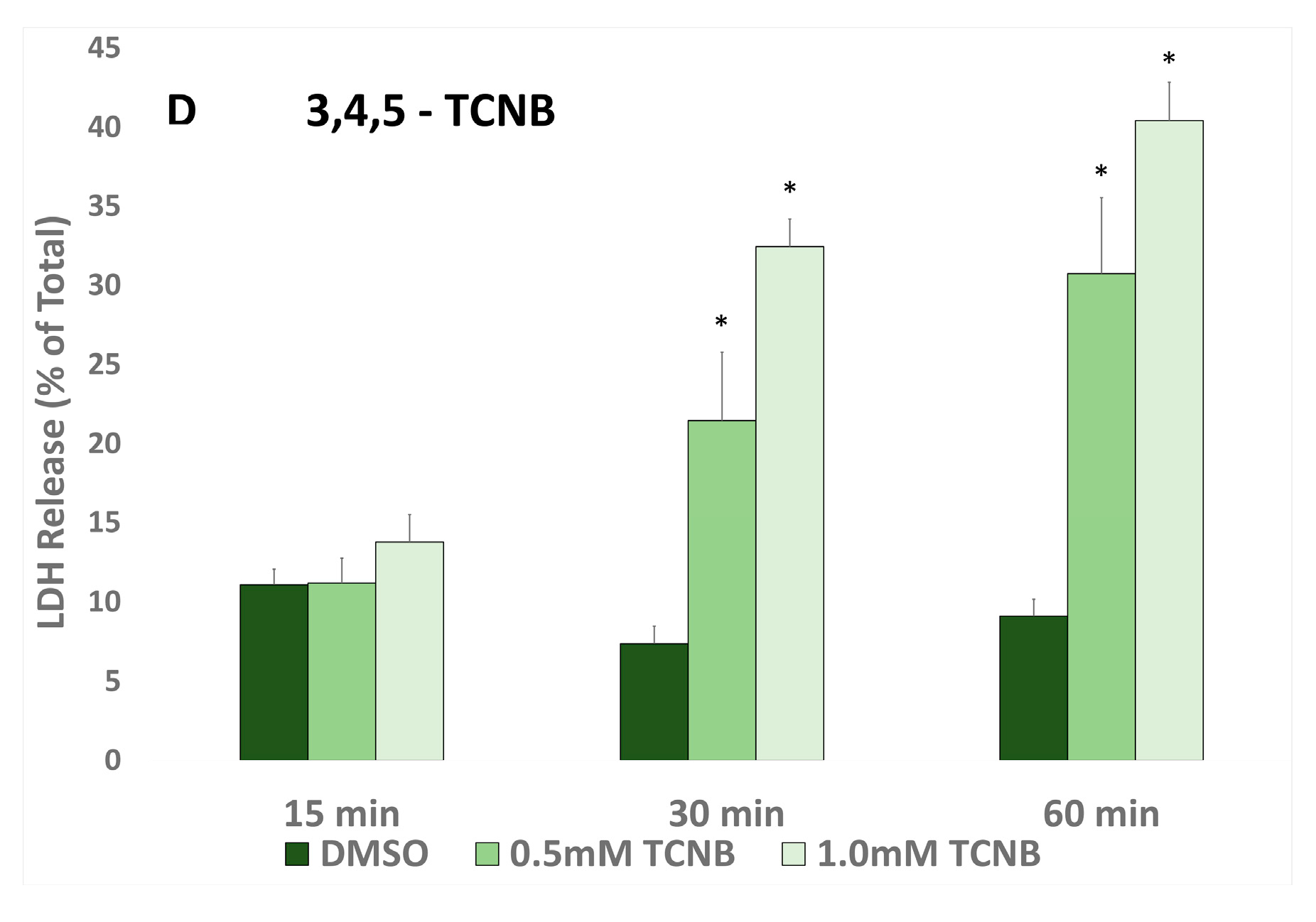
24] demonstrated that two TCNBs (2,3,4- and 3,4,5-TCNB) induced in vitro nephrotoxicity (increased LDH release and decreased pyruvate-stimulated gluconeogenesis) at concentrations as low as 0.5 mM after 120 min incubations in renal cortical slices from male Fischer 344 rats. In the present study using IRCC from male Fischer 344 rats, it was determined that TCNBs are also directly toxic to IRCC, and the order of decreasing nephrotoxic potential of the four TCNBs tested was approximately 3,4,5- > 2,4,6- > 2,3,4- > 2,4,5-TCNB. Cytotoxicity was evident at 0.5 mM and as early as 30 min with both 3,4,5- and 2,4,6- TCNB in IRCC, suggesting that IRCC might be a more sensitive model for measuring the nephrotoxic effects of the TCNBs than renal cortical slices.
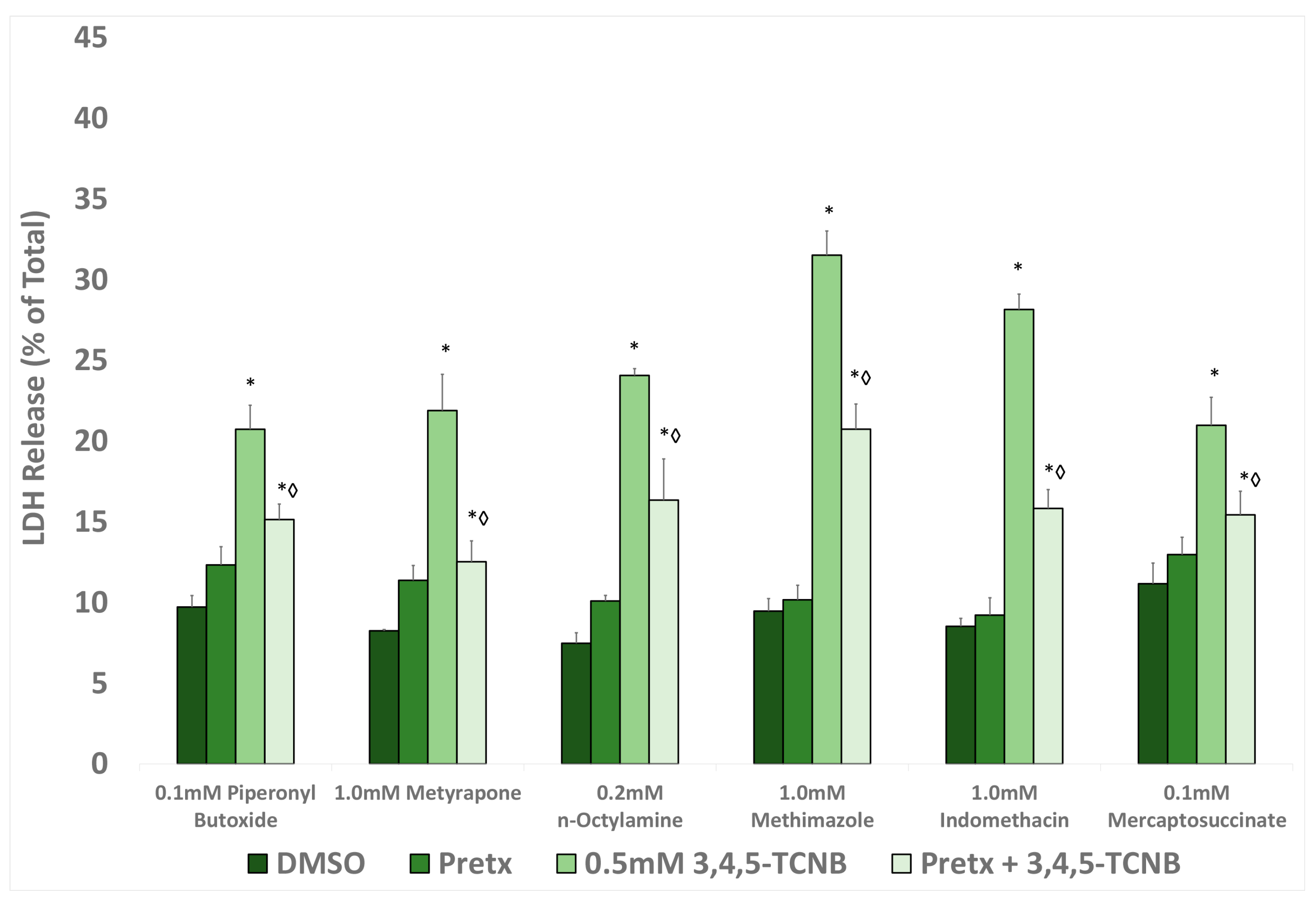
The ability of many of the inhibitors used in this study to attenuate TCNB cytotoxicity suggests that toxic metabolites of TCNBs contribute to TCNB in vitro nephrotoxicity. The biotransformation of chloronitrobenzenes has been studied in a number of species, with most of the studies to date having focused on the metabolism of MCNBs in rats [
27,
28,
29], rabbits [
30], or humans [
17,
18,
20]. In general, a MCNB undergoes three types of initial biotransformation reactions; (1) oxidation of the aromatic ring in a ring position ortho to a chloro group and meta to the nitro group, (2) displacement of a chloro group ortho or para to the nitro group by glutathione conjugation and (3) reduction of the nitro group to form a chloroaniline. The glutathione conjugates are further metabolized to mercapturate metabolites, which can be used as biomarkers of exposure to a chloronitrobenzene [
17,
20]. Reduction of the nitro group to form an aniline appears to be a major metabolic pathway in all species studied. The chloroanilines can be further metabolized to aminophenols, acetanilides, and oxanilic acids, plus glucuronide and sulfate conjugates [
18,
27,
29]. At least with 4-chloronitrobenzene, acetanilide and aminophenol formation was encountered less frequently in humans than in rats [
17].
In the one study of TCNB metabolism, Betts et al. examined the biotransformation of the six TCNBs in rabbits following oral administration (0.2–0.8 g/kg) of a TCNB by determining the identity of urinary metabolites at 24 h intervals for three days [
16]. In general, the metabolic pathways determined for the TCNBs in rabbit were very similar to the pathways identified for MCNBs (nitro reduction, glutathione conjugation of the TCNB, and aminochlorophenol formation), with the addition of hexachloroazoxybenzenes being formed as minor metabolites from 2,4,5- and 3,4,5-TCNB. Although TCNB metabolism by the kidney in rats has not been determined, a putative metabolic pathway for the TCNBs in rat kidney could be proposed (
Figure 8) based on the results of these previous studies.
It is unlikely that glutathione conjugation of a TCNB alone leads to toxic metabolites. Glutathione conjugation is normally a detoxifying mechanism that reduces the ability of electrophiles to react with cellular macromolecules. In cases where glutathione conjugates are nephrotoxiciants, the conjugates are produced extrarenally, and the glutathione or resultant mercapturate conjugate provides a means for renal accumulation and/or bioactivation [
31]. In this study, the TCNB would be expected to enter the IRCC via passive diffusion prior to metabolism and not require a glutathione derived conjugate to promote uptake. Thus, it is unlikely that the ultimate toxic metabolite results from glutathione conjugation.
N-Acetylation of an aniline metabolite to form a chloroacetanilide is also not a likely bioactivation mechanism.
N-Acetylation is catalyzed in kidney cells by cytosolic
N-acetyltransferase enzymes, and it is very unlikely that the pretreatments used in this study that attenuated TCNB cytotoxicity would significantly alter this metabolism reaction. In addition, chloroacetanilides have greatly reduced nephrotoxic potential in vivo when compared to the parent chloroanilines or aminochlorophenol metabolites [
32,
33]. Thus, it is unlikely that acetylation of any aniline derived metabolites would be a mechanism for bioactivation.
Reduction of chloronitrobenzenes to chloroanilines is a common biotransformation pathway for the MCNBs and TCNBs [
16,
20,
27]. Reduction of the nitro group in chloronitrobenzenes is catalyzed by microsomal enzymes (CYPs and nitroreductases) and is inhibited by cytochrome P450 (CYP) inhibitors [
27]. As the nitro group is reduced, chloronitrosobenzenes and then chlorophenylhydroxylamines are formed before the final reduction to the chloroaniline metabolite (
Figure 6). During the reduction, electrons can be transferred to oxygen to form the superoxide anion radical, which could lead to oxidative damage and cell death. The chloroaniline metabolites can also be oxidized back to the corresponding chloronitrobenzene through a reverse set of metabolic steps. Oxidation of the chloroaniline metabolite to an
N-hydroxyl metabolite (
Figure 6.) can be catalyzed by several enzyme systems including CYPs, flavin monooxygenases (FMOs), prostaglandin H synthase, and other peroxidases and contribute to chloroaniline toxicity [
34,
35,
36,
37,
38,
39,
40,
41]. Further oxidation of the chlorophenylhydroxylamine metabolites can occur via auto-oxidation. The result is that both chloroanilines and chloronitrobenzenes have the potential to set up a redox situation involving the production of chloronitrosobenzenes and chlorophenylhydroxylamines that could produce free radicals that could lead to potential oxidative damage and cellular toxicity. In erythrocytes, this redox cycling leads to methemoglobin formation via oxidation of the ferrous iron in hemoglobin as the various metabolites are reduced, and oxidative and reactive metabolites can eventually lead to the destruction of the erythrocytes and anemia [
9,
42,
43]. In addition, chloronitrosobenzenes are reactive compounds that can bind to cellular macromolecules and contribute to the cellular toxicity of these compounds [
44,
45,
46,
47]. Thus, the reversible metabolism of the TCNBs to trichloroanilines is a potential pathway for generating the nephrotoxic metabolites responsible for TCNB cytotoxicity observed in this study.
The results of this study demonstrated that cytotoxicity induced by 2,3,4-, 2,4,6-, and 3,4,5-TCNB was inhibited by pretreatment with at least one CYP inhibitor, FMO inhibitors, and peroxidase inhibitors, suggesting that the nitro reduction-amino oxidation pathway was contributing to the production of nephrotoxic metabolites. The placement of the three chloro groups on the TCNB appears to affect the ability of the TCNB to serve as a substrate for particular oxidation/reduction metabolizing enzymes, although no clear correlation pattern of substitution emerged. The nitro and resulting amino group in the aniline metabolite of 3,4,5-TCNB are the least sterically hindered and most accessible to metabolizing enzymes, and 3,4,5-TCNB is the most nephrotoxic of the four compound studies. However, although 2,4,6-TCNB was close to 3,4,5-TCNB in cytotoxic potential at 30 min (
Figure 2), two of the three chloro groups are ortho to the nitro group and provide the most steric hindrance for metabolizing the nitro group. Given the contributions of multiple enzyme systems contributing to TCNB metabolism, it is possible that the chloro groups, depending on ring location, direct the TCNBs and/or their metabolites to only certain metabolizing enzymes. It also appears that 3,4,5-TCNB may be metabolized by multiple CYPs to the ultimate toxic metabolite(s), as all of the specific CYP inhibitors used in this study offered some protection, or that multiple metabolic steps, catalyzed by different CYPs are required to produce the ultimate toxicant. However, more work needs to be done to establish the effect that the chloro group position on the aromatic ring has on the biotransformation and nephrotoxic potential of TCNBs.
The contributions of aminochlorophenol metabolites to TCNB nephrotoxicity must also be considered. Aminochlorophenols are known nephrotoxicants in vivo and in vitro in Fischer 344 rat models [
48,
49,
50,
51], and these metabolites are known to be produced in rats [
18], rabbits [
16], and humans [
17]. However, studies with 3,5-dichloroaniline and 3,4,5-trichloroaniline, the most potent nephrotoxicants among the di- and trichloroanilines isomers, in Fischer 344 rats have shown that in vitro,
N-oxidation pathways are more important than the formation of aminophenol metabolites for inducing nephrotoxicity [
25,
52]. Thus, while TCNBs may be metabolized to trichloroanilines in the rat kidney,
N-oxidation is the most likely pathway leading to nephrotoxic metabolites. Nonetheless, the contribution of aminophenol metabolites to TCNB nephrotoxicity needs to be studied further.
The ability of the four antioxidants (α-tocopherol, ascorbate, glutathione, and NAC) to attenuate the cytotoxicity of the four TCNBs provides insight into the role of free radicals in the toxicity induced by these compounds. Ascorbate is a water soluble radical scavenger that can quench radicals in solution. α-Tocopherol is a fat soluble antioxidant that primarily works by helping neutralize lipid peroxide formation and the generation of reactive oxygen species from oxidized lipids. NAC serves as an antioxidant primarily following the conversion to glutathione, but can, to a lesser extent, form disulfides (N,N-diacetylcystine) in the presence of free radicals to scavenge those radicals. Glutathione can neutralize free radicals (via glutathione peroxidase) or electrophiles (via glutathione S-transferase). Occasionally, NAC is not an effective protectant, when glutathione is protective. This observation could be due to the time required to effectively convert NAC to glutathione and/or to the fact that the cytotoxic chemical species is a reactive metabolite rather than a free radical and more easily neutralized by enzymatic conjugation with glutathione.
All four antioxidants attenuated the cytotoxicity caused by 2,3,4-, 2,4,5-, and 3,4,5-TCNB at the concentrations and times studied. Free radicals may be produced during the reduction of the nitro group or during the oxidation of trichloroaniline metabolites [
53], or may be the result of redox cycling of aminochlorophenol or chlorophenylhydroxylamine/chloronitrosobenzene metabolites to produce reactive oxygen species (ROS) and oxidative damage [
54,
55]. While some degree of oxidative stress may be induced by these compounds, the current work suggests that, at least for 3,4,5-TCNB, oxidative stress-induced lipid peroxidation is not the mechanism of cytotoxicity. The absence of an increase in 4-HNE or protein carbonylation, even at a time when 3,4,5-TCNB induced cytotoxicity, indicates that cells are dying by other mechanisms and antioxidants are attenuating TCNB cytotoxicity via mechanisms not involving quenching ROS or ROS-derived intermediates. Similarly, the finding that ascorbate but not α-tocopherol is protective against 2,4,6-TCNB could suggest that radical TCNB metabolites are formed but these metabolites are not leading to lipid peroxidation. A similar observation was noted for 3,5-dichloroaniline [
52], suggesting that chloroanilines and chloronitrobenzenes may share a common metabolic pathway, such as the generation of chloronitrosobenzenes, to induce nephrotoxicity. Additional work is needed to clarify if either or both of these potential pathways of free radical generation can explain the mechanism of protection by antioxidants on TCNB cytotoxicity and provide insights into the cellular targets and mechanism of TCNB nephrotoxicity.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}