Oncogene-Induced Replication Stress Drives Genome Instability and Tumorigenesis

Abstract
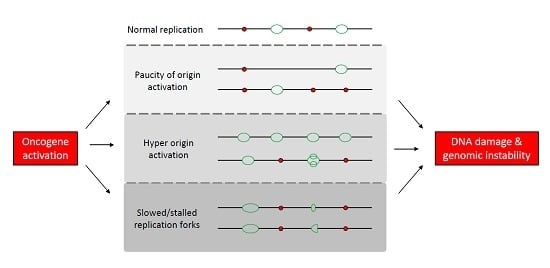
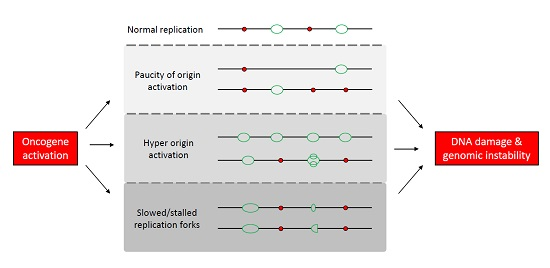
:
{kind=link}
{kind=link}
1. Introduction
1.1. DNA Replication Regulation—From Licensing to Termination
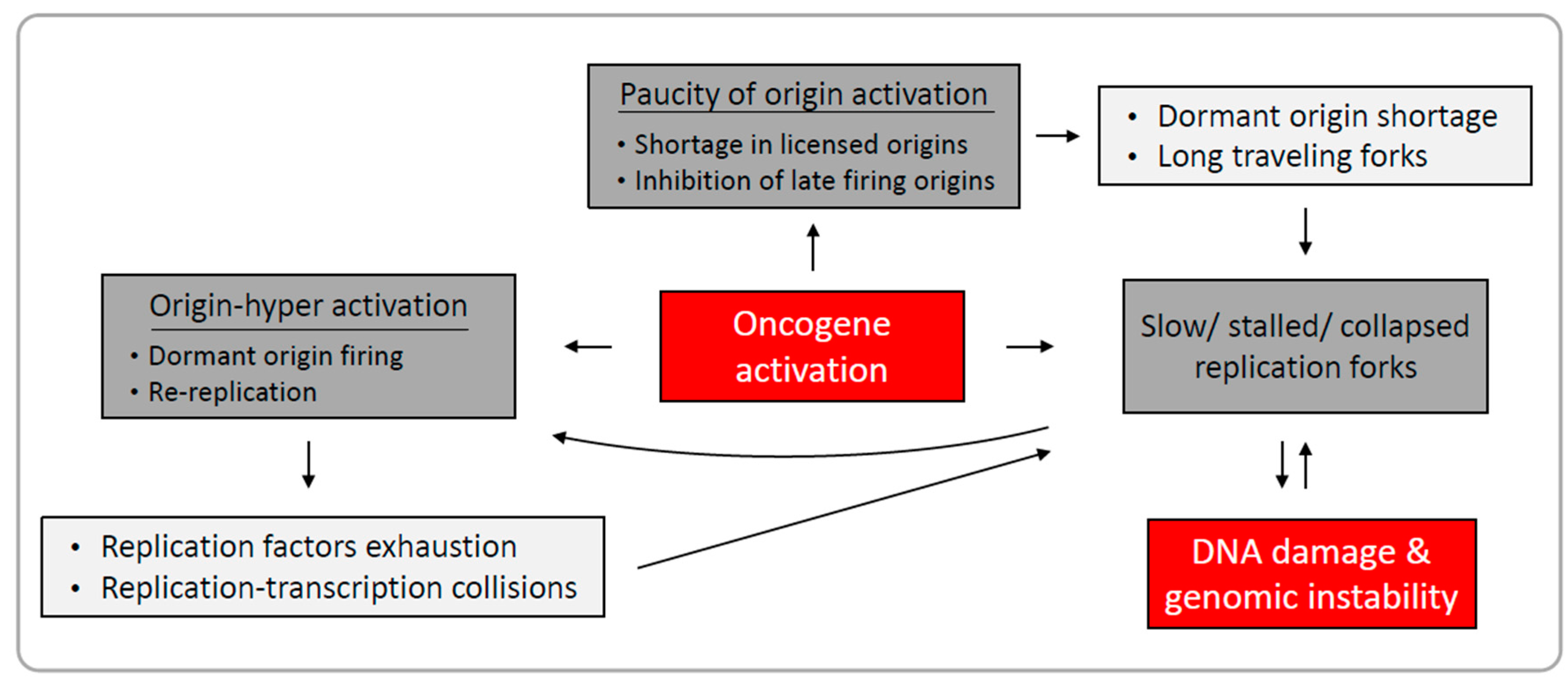
1.2. Mechanisms Underlying Oncogene-Induced Replication Stress
1.2.1. Paucity of Origin Activation
1.2.2. Origin Hyper-Activation
1.2.3. Perturbation of Replication Fork Elongation
1.3. Replication-Induced Genomic Instability in Cancer
2. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Plug-DeMaggio, A.W.; Sundsvold, T.; Wurscher, M.A.; Koop, J.I.; Klingelhutz, A.J.; McDougall, J.K. Telomere erosion and chromosomal instability in cells expressing the HPV oncogene 16E6. Oncogene 2004, 23, 3561–3571. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2013, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Ragland, R.L.; Arlt, M.F.; Mulle, J.G.; Warren, S.T.; Glover, T.W. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc. Natl. Acad. Sci. USA 2008, 105, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Bilousova, G.; Marusyk, A.; Porter, C.C.; Cardiff, R.D.; DeGregori, J. Impaired DNA replication within progenitor cell pools promotes leukemogenesis. PLoS Biol. 2005, 3, e401. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Miron, K.; Golan-Lev, T.; Dvir, R.; Ben-David, E.; Kerem, B. Oncogenes create a unique landscape of fragile sites. Nat. Commun. 2015, 6, 7094. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Evrin, C.; Clarke, P.; Zech, J.; Lurz, R.; Sun, J.; Uhle, S.; Li, H.; Stillman, B.; Speck, C. A double-hexameric MCM2–7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. USA 2009, 106, 20240–20245. [Google Scholar] [CrossRef] [PubMed]
- Gambus, A.; Khoudoli, G.A.; Jones, R.C.; Blow, J.J. MCM2–7 form double hexamers at licensed origins in xenopus egg extract. J. Biol. Chem. 2011, 286, 11855–11864. [Google Scholar] [CrossRef] [PubMed]
- Remus, D.; Beuron, F.; Tolun, G.; Griffith, J.D.; Morris, E.P.; Diffley, J.F.X. Concerted loading of MCM2–7 double hexamers around DNA during DNA replication origin licensing. Cell 2009, 139, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.H. Increase in DNA replication sites in cells held at the beginning of S phase. Chromosoma 1977, 62, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Santocanale, C.; Sharma, K.; Diffley, J.F.X. Activation of dormant origins of DNA replication in budding yeast. Genes Dev. 1999, 13, 2360–2364. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, D.; Blow, J.J. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb. Perspect. Biol. 2012, 4, a012955. [Google Scholar] [CrossRef] [PubMed]
- Moyer, S.E.; Lewis, P.W.; Botchan, M.R. Isolation of the Cdc45/Mcm2–7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. USA 2006, 103, 10236–10241. [Google Scholar] [CrossRef] [PubMed]
- Ilves, I.; Petojevic, T.; Pesavento, J.J.; Botchan, M.R. Activation of the MCM2–7 helicase by association with Cdc45 and GINS proteins. Mol. Cell 2010, 37, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Gambus, A.; Jones, R.C.; Sanchez-Diaz, A.; Kanemaki, M.; van Deursen, F.; Edmondson, R.D.; Labib, K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 2006, 8, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Dewar, J.M.; Budzowska, M.; Walter, J.C. The mechanism of DNA replication termination in vertebrates. Nature 2015, 525, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Hodgson, B. Replication licensing—Origin licensing: Defining the proliferative state? Trends Cell Biol. 2002, 12, 72–78. [Google Scholar] [CrossRef]
- Sugimoto, N.; Tatsumi, Y.; Tsurumi, T.; Matsukage, A.; Kiyono, T.; Nishitani, H.; Fujita, M. Cdt1 phosphorylation by cyclin A-dependent kinases negatively regulates its function without affecting geminin binding. J. Biol. Chem. 2004, 279, 19691–19697. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Li, X.; Yan, F.; Zhao, Q.; Wu, X. Cyclin-dependent kinases phosphorylate human Cdt1 and induce its degradation. J. Biol. Chem. 2004, 279, 17283–17288. [Google Scholar] [CrossRef] [PubMed]
- Méndez, J.; Zou-Yang, X.H.; Kim, S.Y.; Hidaka, M.; Tansey, W.P.; Stillman, B. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol. Cell 2002, 9, 481–491. [Google Scholar] [CrossRef]
- Wohlschlegel, J.A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 2000, 290, 2309–2312. [Google Scholar] [CrossRef] [PubMed]
- Rhind, N.; Gilbert, D.M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011, 36, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Schwob, E.; Méndez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef] [PubMed]
- Ekholm-Reed, S.; Méndez, J.; Tedesco, D.; Zetterberg, A.; Stillman, B.; Reed, S.I. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 2004, 165, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Slater, D.M.; Lenburg, M.; Nevis, K.; Cook, J.G.; Vaziri, C. Replication licensing promotes cyclin D1 expression and G1 progression in untransformed human cells. Cell Cycle 2009, 8, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Steckel, M.; Molina-Arcas, M.; Weigelt, B.; Marani, M.; Warne, P.H.; Kuznetsov, H.; Kelly, G.; Saunders, B.; Howell, M.; Downward, J.; et al. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 2012, 22, 1227–1245. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, K.M.; Jones, R.M.; Petermann, E.; Jeggo, P.A. Diminished origin-licensing capacity specifically sensitizes tumor cells to replication stress. Mol. Cancer Res. 2013, 11, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Frum, R.A.; Singh, S.; Vaughan, C.; Mukhopadhyay, N.D.; Grossman, S.R.; Windle, B.; Deb, S.; Deb, S.P. The human oncoprotein MDM2 induces replication stress eliciting early intra-S-phase checkpoint response and inhibition of DNA replication origin firing. Nucleic Acids Res. 2014, 42, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Liontos, M.; Koutsami, M.; Sideridou, M.; Evangelou, K.; Kletsas, D.; Levy, B.; Kotsinas, A.; Nahum, O.; Zoumpourlis, V.; Kouloukoussa, M.; et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007, 67, 10899–10909. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Gautier, J. MYC and the control of DNA replication. Cold Spring Harb. Perspect. Med. 2014, 4, a014423. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic Instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; García, P.; Helleday, T.; Petermann, E. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Anglana, M.; Apiou, F.; Bensimon, A.; Debatisse, M. Dynamics of DNA replication in mammalian somatic cells. Cell 2003, 114, 385–394. [Google Scholar] [CrossRef]
- Ge, X.Q.; Blow, J.J. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J. Cell Biol. 2010, 191, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, C.; Saxena, S.; Jeon, Y.; Lee, C.; Murata, K.; Machida, Y.; Wagle, N.; Hwang, D.S.; Dutta, A. A p53-dependent checkpoint pathway prevents rereplication. Mol. Cell 2003, 11, 997–1008. [Google Scholar] [CrossRef]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.P.; Poli, J.; Marians, K.J.; Pasero, P. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013, 5, a012815. [Google Scholar] [CrossRef] [PubMed]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Ozeri-Galai, E.; Lebofsky, R.; Rahat, A.; Bester, A.C.; Bensimon, A.; Kerem, B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell 2011, 43, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Lamm, N.; Maoz, K.; Bester, A.C.; Im, M.M.; Shewach, D.S.; Karni, R.; Kerem, B. Folate levels modulate oncogene-induced replication stress and tumorigenicity. EMBO Mol. Med. 2015, 7, 1138–1152. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Wei, X. Segregation of transcription and replication sites into higher order domains. Science 1998, 281, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Sánchez, M.S.; Barroso, S.; Gómez-González, B.; Luna, R.; Aguilera, A. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 2011, 7, e1002386. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Burrell, R.A.; Endesfelder, D.; Novelli, M.R.; Swanton, C. Cancer chromosomal instability: Therapeutic and diagnostic challenges. EMBO Rep. 2012, 13, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Magdalou, I.; Barascu, A.; Techer, H.; Debatisse, M.; Lopez, B.S. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 2014, 111, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Koundrioukoff, S.; Carignon, S.; Técher, H.; Letessier, A.; Brison, O.; Debatisse, M. Stepwise activation of the ATR signaling pathway upon increasing replication stress impacts fragile site integrity. PLoS Genet. 2013, 9, e1003643. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Sun, Z.; Liu, Z.; Guo, H.; Liu, Q.; Jiang, H.; Zou, Y.; Gong, Y.; Tischfield, J.A.; Shao, C. Replication stress induces micronuclei comprising of aggregated DNA double-strand breaks. PLoS ONE 2011, 6, e18618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of mutation and selection in the cancer genome. Nature 2010, 463, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Zlotorynski, E.; Scherer, S.W.; Cheung, J.; Vincent, J.B.; Smith, D.I.; Trakhtenbrot, L.; Kerem, B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell 2002, 1, 89–97. [Google Scholar] [CrossRef]
- Kotzot, D. Parental origin and mechanisms of formation of cytogenetically recognisable de novo direct and inverted duplications. J. Med. Genet. 2000, 37, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.T.; Lin, L.; Casper, A.M.; Lim, J.; Thomas, D.G.; Orringer, M.B.; Chang, A.C.; Chambers, A.F.; Giordano, T.J.; Glover, T.W.; et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene 2005, 1, 10. [Google Scholar] [CrossRef]
- Tsantoulis, P.K.; Kotsinas, A.; Sfikakis, P.P.; Evangelou, K.; Sideridou, M.; Levy, B.; Mo, L.; Kittas, C.; Wu, X.R.; Papavassiliou, A.G.; et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008, 27, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81–EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Minocherhomji, S.; Chan, K.L.; Palmai-Pallag, T.; Chu, W.K.; Wass, T.; Mankouri, H.W.; Liu, Y.; Hickson, I.D. MUS81 promotes common fragile site expression. Nat. Cell Biol. 2013, 15, 1001–1007. [Google Scholar] [CrossRef]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarni, D.; Kerem, B. Oncogene-Induced Replication Stress Drives Genome Instability and Tumorigenesis. Int. J. Mol. Sci. 2017, 18, 1339. https://doi.org/10.3390/ijms18071339
Sarni D, Kerem B. Oncogene-Induced Replication Stress Drives Genome Instability and Tumorigenesis. International Journal of Molecular Sciences. 2017; 18(7):1339. https://doi.org/10.3390/ijms18071339
Chicago/Turabian StyleSarni, Dan, and Batsheva Kerem. 2017. "Oncogene-Induced Replication Stress Drives Genome Instability and Tumorigenesis" International Journal of Molecular Sciences 18, no. 7: 1339. https://doi.org/10.3390/ijms18071339
APA StyleSarni, D., & Kerem, B. (2017). Oncogene-Induced Replication Stress Drives Genome Instability and Tumorigenesis. International Journal of Molecular Sciences, 18(7), 1339. https://doi.org/10.3390/ijms18071339