The Application of REDOR NMR to Understand the Conformation of Epothilone B

Abstract
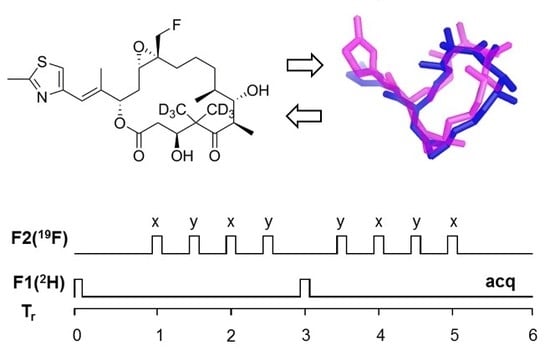
:
1. Introduction
2. Results
2.1. Design of Epothilone B for Rotational-Echo Double-Resonance (REDOR)
2.2. Cell Cytotocicity of Synthetic Epothilone B
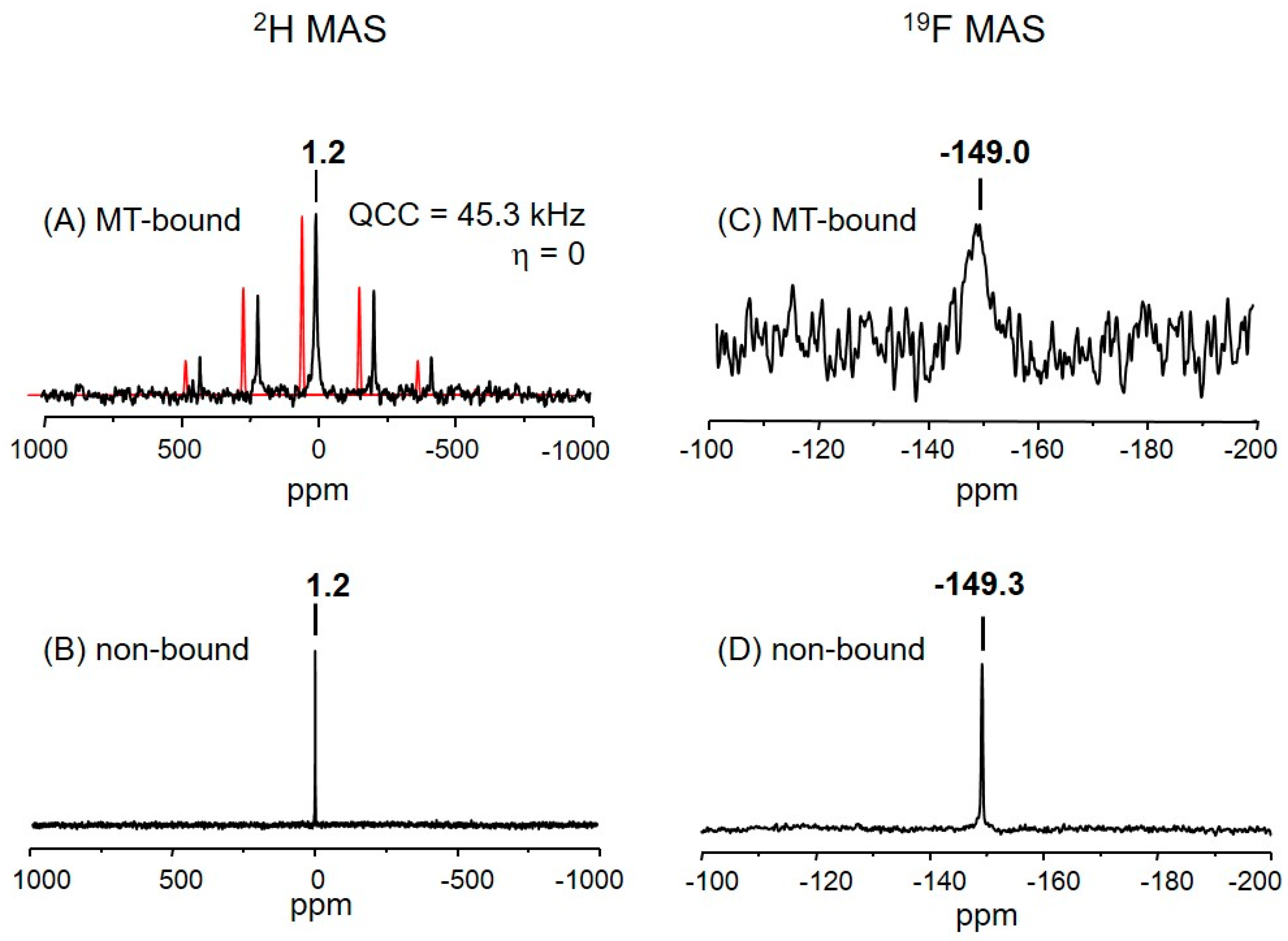
2.3. The Deuterium Lineshape of Synthetic Epothilone B, Microtubule-Bound
2.4. The Fluorine Signal of Synthetic Epothilone B, Microtubule-Bound
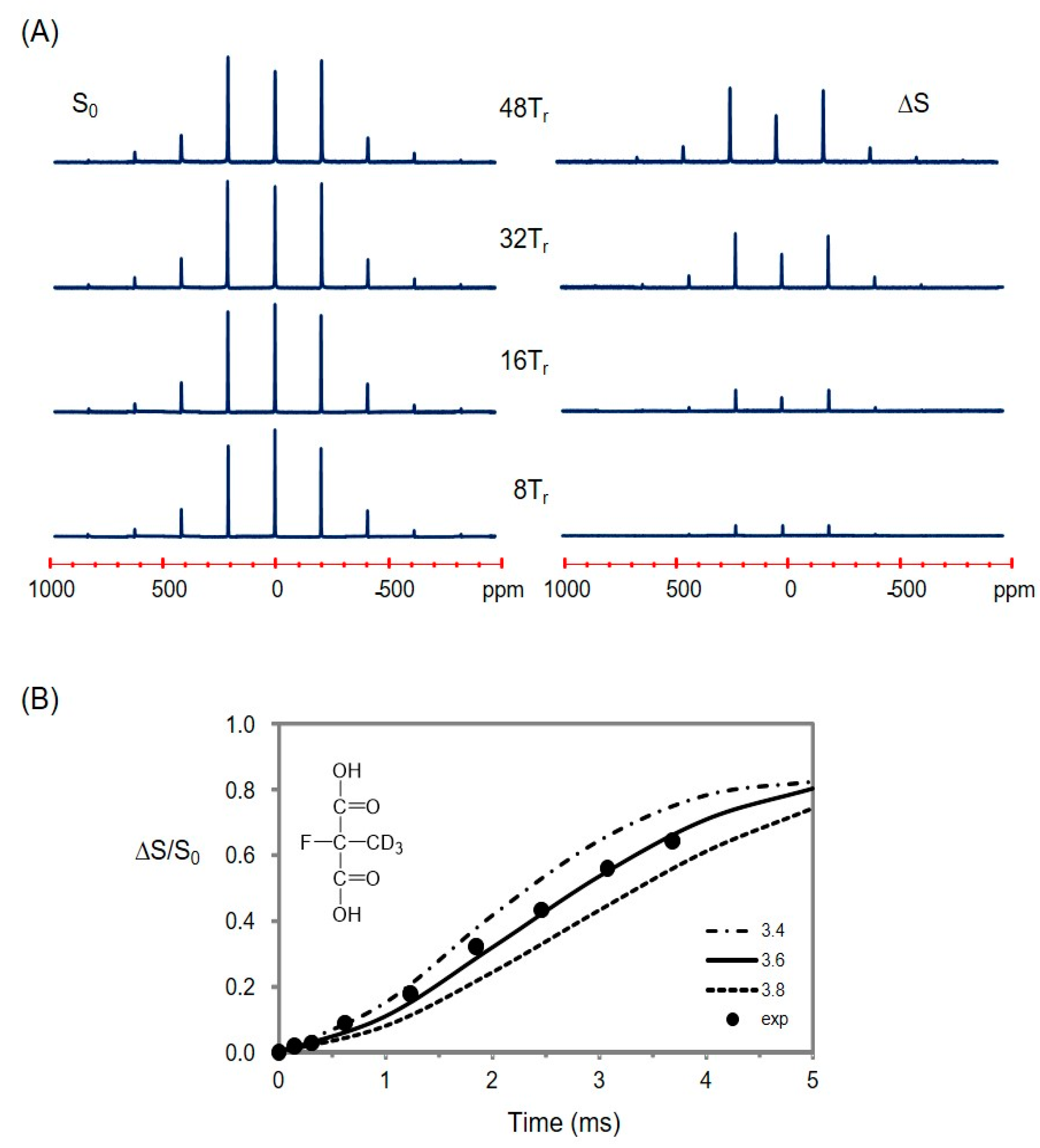
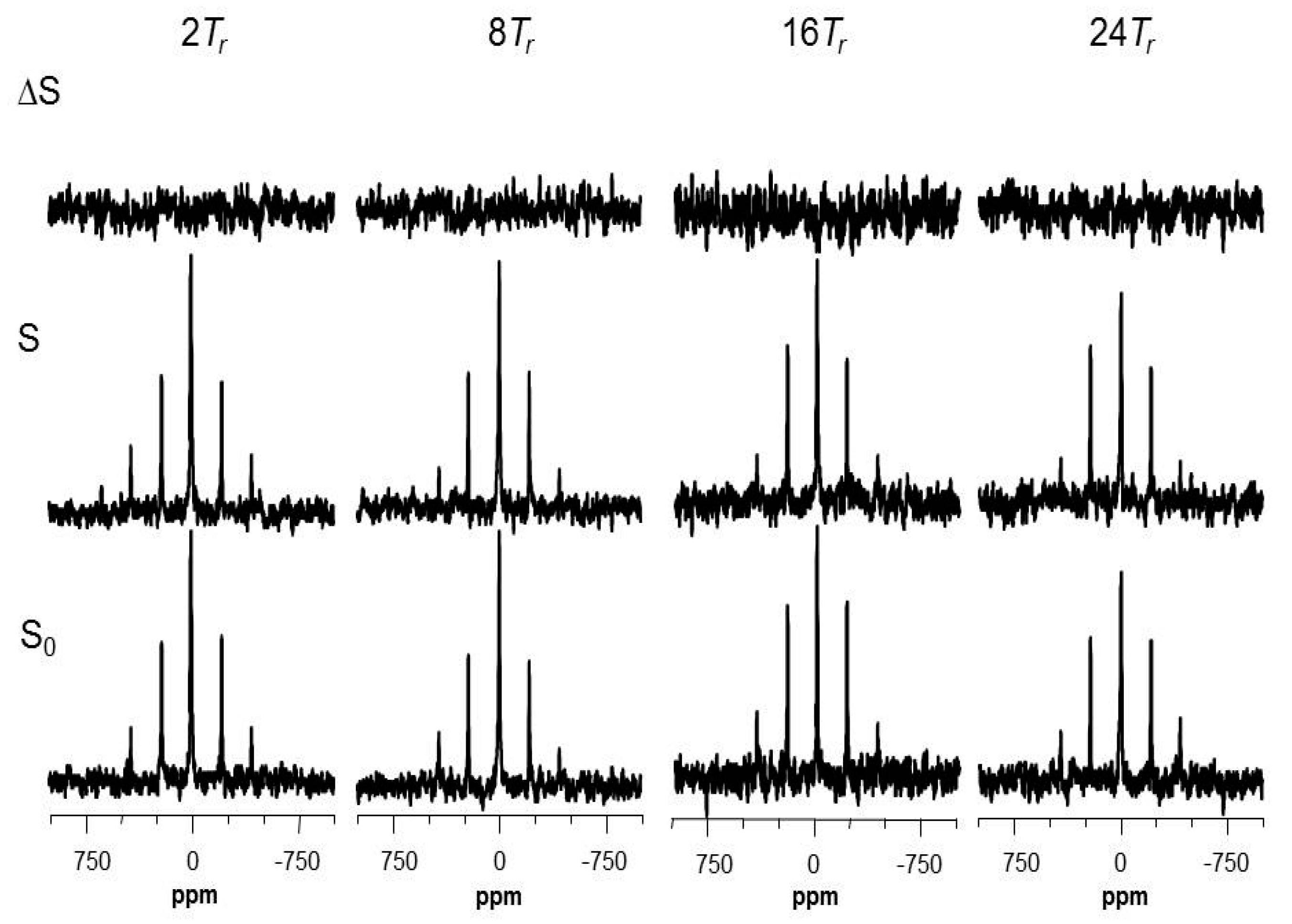
2.5. Using a Double Resonance Spectrometer for 2H{19F} REDOR
2.6. Information for the Microtubule-Bound Epothilone B Conformation
3. Discussion
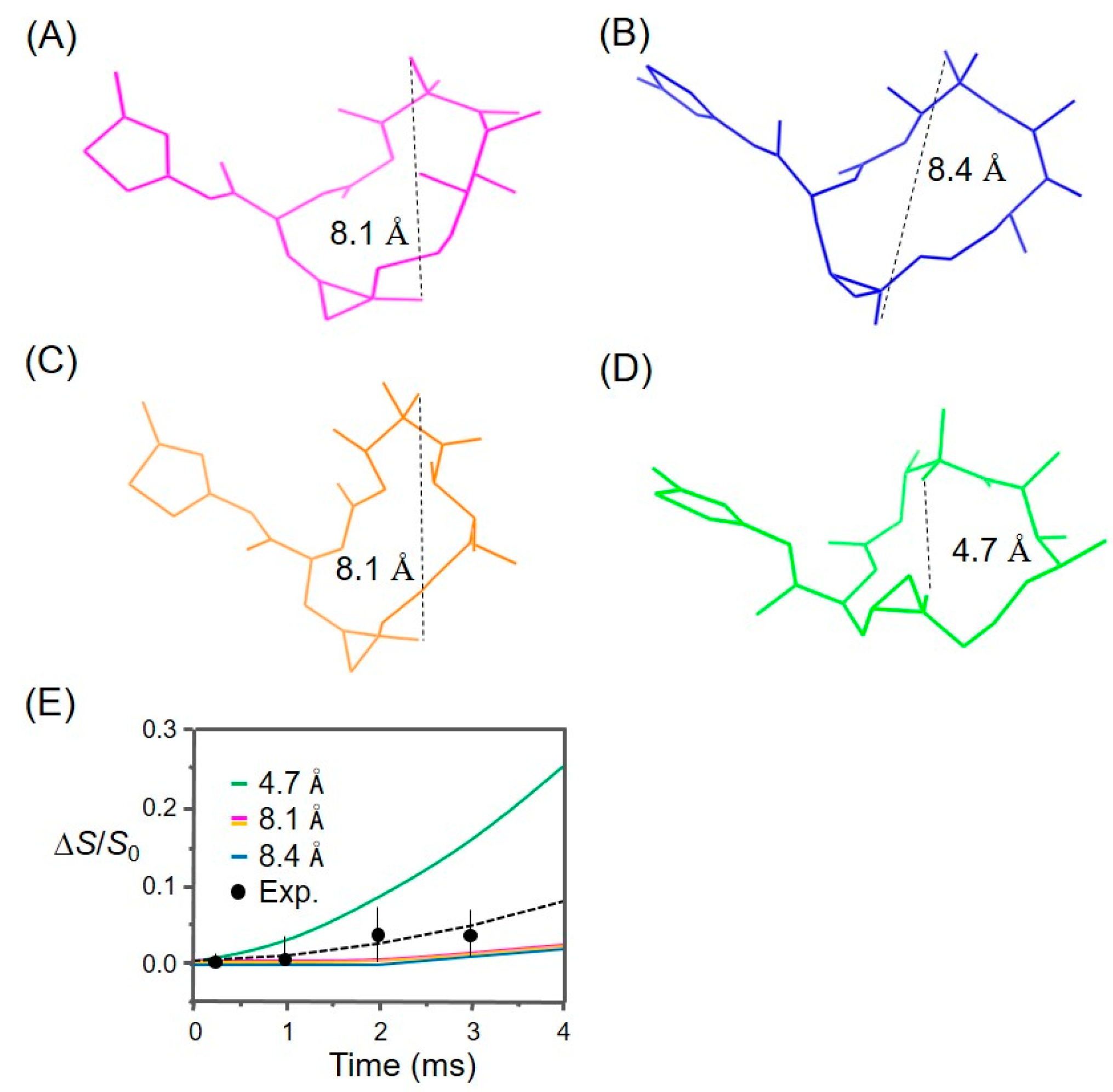
3.1. Analysis of the REDOR Data for Epothilone B Conformation
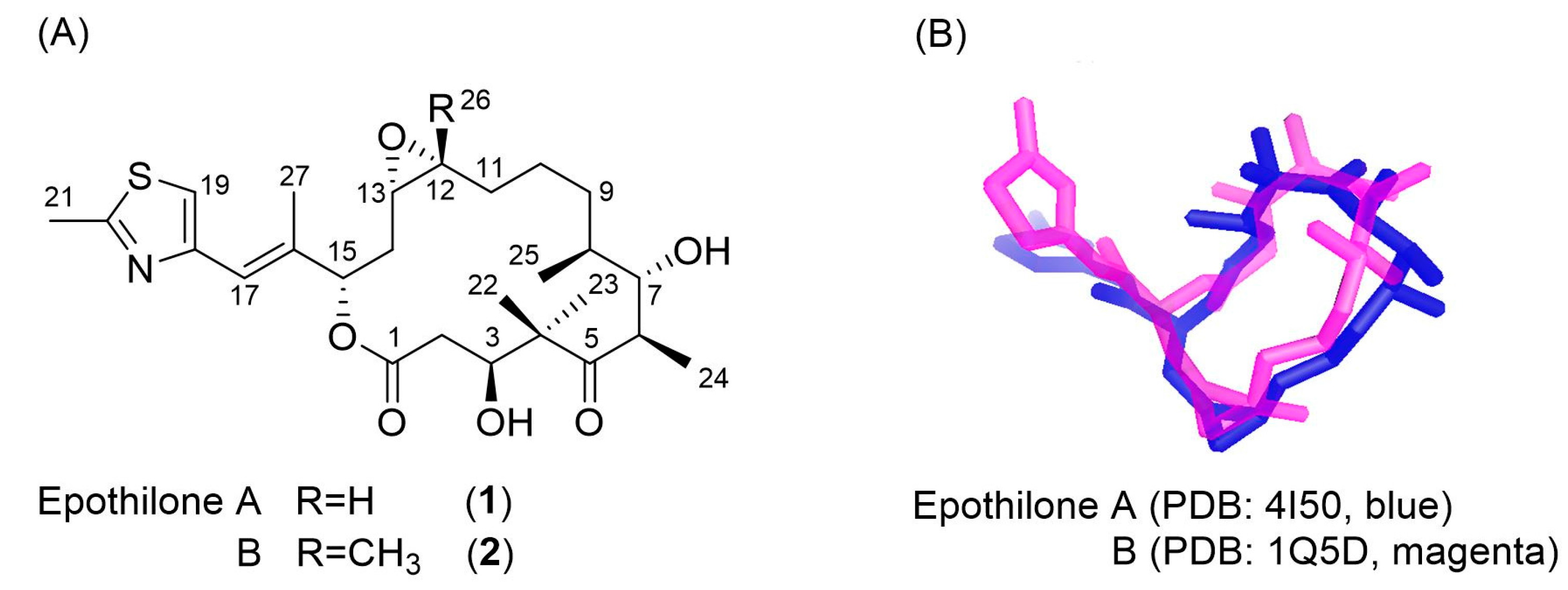
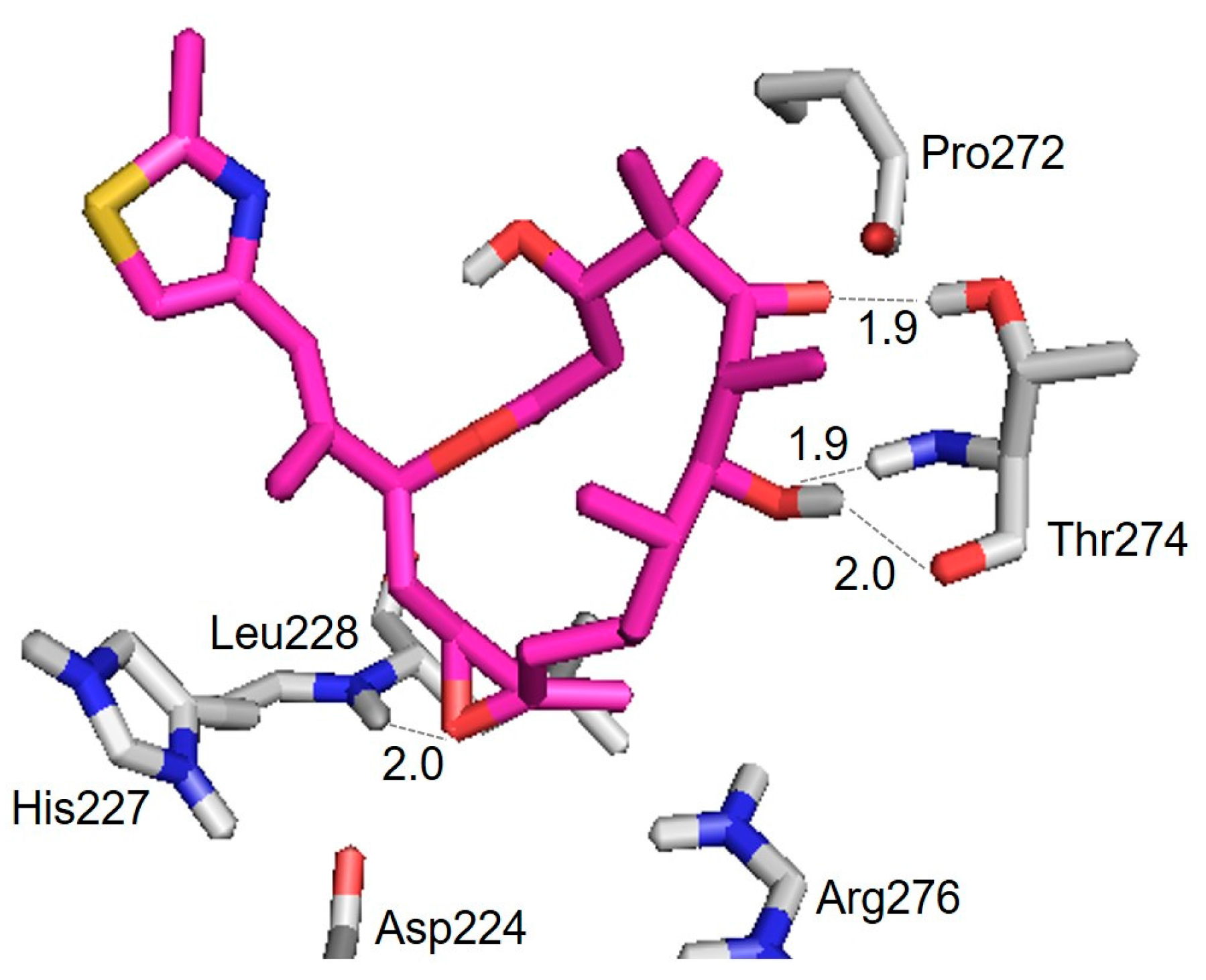
3.2. Evaluation of an X-ray Crystal Structure (PDB 1Q5D) for the Epothilone B Conformation
3.3. Applicability to Other Epothilone Derivatives
3.4. Molecular Motions of the Microtubule-Bound Epothilone B
3.4.1. Indications for the Binding
3.4.2. The Fraction of Non-Bound Analog 3
3.4.3. The Fluorine Linewidth
4. Materials and Methods
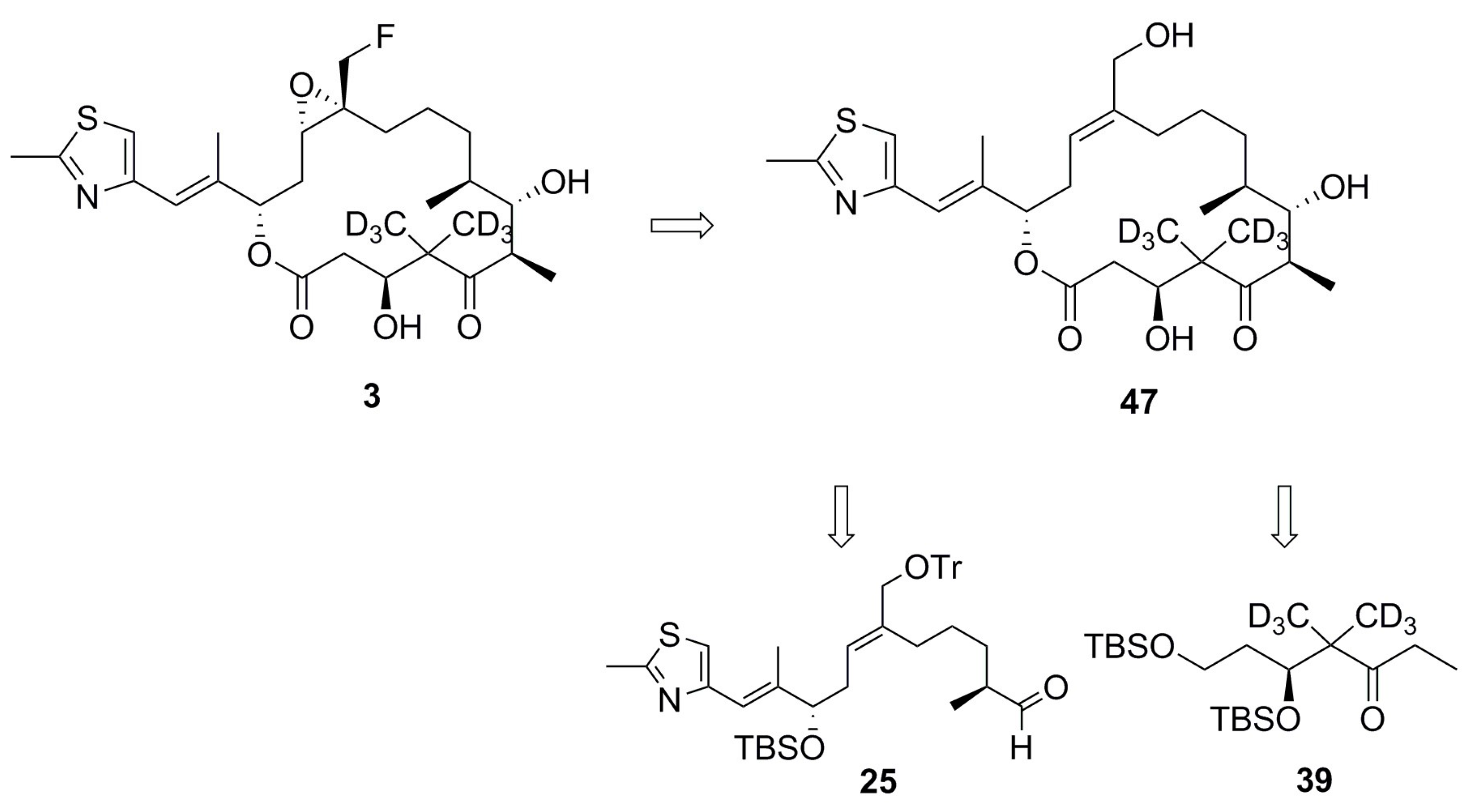
4.1. Synthesis of Analog 3
4.2. Cell Cytotoxicity (IC50) Test
4.3. Sample Preparations
4.4. REDOR NMR Experiments
4.5. AutoDock Calculations
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| [2-F,2-Me-d3]MA | 2-fluroro-2-methyl-d3-malonic acid |
| MAS | Magic angle spinning |
| MT | Microtubule |
| NSCL | Non-small cell lung |
| QCC | Quadrupole coupling constant |
| REDOR | Rotational-echo double-resonance |
| SAR | Structure-activity relationship |
References
- Matsuoka, S.; Inoue, M. Application of REDOR NMR in natural product chemistry. Chem. Commun. 2009, 14, 5664–5675. [Google Scholar] [CrossRef] [PubMed]
- Cegelski, L. REDOR NMR for drug discovery. Bioorg. Med. Chem. Lett. 2013, 23, 5767–5775. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, J. “Development of REDOR rotational-echo double-resonance NMR” by Terry Gullion and Jacob Schaefer [J. Magn. Reson. 81 (1989) 196–200]. J. Magn. Reson. 2011, 213, 421–422. [Google Scholar] [CrossRef] [PubMed]
- Grage, S.L.; Watts, J.A.; Watts, A. 2H{19F} REDOR for distance measurements in biological solids using a double resonance spectrometer. J. Magn. Reson. 2004, 166, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cady, S.; Wang, T.; Hong, M. Membrane-dependent effects of a cytoplasmic helix on the structure and drug binding of the influenza virus M2 protein. J. Am. Chem. Soc. 2011, 133, 11572–11579. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Poliks, B.; Cegelski, L.; Poliks, M.; Gryczynski, Z.; Piszczek, G.; Jagtap, P.G.; Studelska, D.R.; Kingston, D.G.I.; Schaefer, J.; et al. Conformation of microtubule-bound paclitaxel determined by fluorescence spectroscopy and REDOR NMR. Biochemistry 2000, 39, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.; Yang, C.; Metaferia, B.; Tang, S.; Bane, S.; Ravindra, R.; Shanker, N.; Alcaraz, A.A.; Johnson, S.A.; Schaefer, J.; et al. Rotational-echo double-resonance NMR distance measurements for the tubulin-bound paclitaxel conformation. J. Am. Chem. Soc. 2007, 129, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.P.; Nettles, J.H.; Conett, B.; Downing, K.H.; Nogales, E. The binding conformation of taxol in β-tubulin: A model based on electron crystallographic density. Proc. Natl. Acad. Sci. USA 2001, 98, 5312–5316. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Veith, J.M.; Pera, P.; Bernacki, R.J.; Ojima, I. Design and synthesis of de novo cytotoxic alkaloids by mimicking the bioactive conformation of paclitaxel. Bioorg. Med. Chem. 2010, 18, 7101–7112. [Google Scholar] [CrossRef] [PubMed]
- Gee, C.T.; Arntson, K.E.; Urick, A.K.; Mishra, N.K.; Hawk, L.M.; Wisniewski, A.J.; Pomerantz, W.C.K. Protein-observed 19F-NMR for fragment screening, affinity quantification and druggability assessment. Nat. Protoc. 2016, 11, 1414–1427. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration Website. Available online: http//www.fda.gov/ (accessed on 16 October 2007).
- Hofle, G.; Bedorf, N.; Steinmetz, H.; Schomburg, D.; Gerth, K.; Reichenbach, H. Epothilone A and B-novel 16-membered macrolides with cytotoxic activity: Isolation, crystal structure, and conforamtion in solution. Angew. Chem. Int. Ed. 1996, 35, 1567–1569. [Google Scholar] [CrossRef]
- Bollag, D.M.; McQueney, P.A.; Hensens, O.; Zhu, J.; Koupal, L.; Liesch, J.; Goetz, M.; Lazarides, E.; Woods, C.M. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995, 55, 2325–2333. [Google Scholar] [PubMed]
- Oehler, C.; Frei, K.; Rushing, E.J.; Mcsheehy, P.M.J.; Weber, D.; Allergrini, P.R.; Weniger, D.; Lutolf, U.M.; Knuth, A.; Yonekawa, Y.; et al. Patupilone (epothilone B) for recurrent glioblastoma: Clinical outcome and translational analysis of a single-institution phase I/II trial. Oncology 2012, 83, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gerth, K.; Steinmetz, H.; Hofle, G.; Reichenbach, H. Studies on the biosynthesis of epothilones: The biosynthetic origin of the carbon skeleton. J. Antibiot. 2000, 53, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Kingston, D.G.I. Taxol and its analogs. In Anticancer Agents from Natural Products, 1st ed.; Crag, G.M., Kingston, D.G.I., Newman, D.J., Eds.; Taylor & Francis Group: Boca Raton, FL, USA, 2005; Chapter 6; pp. 89–135. [Google Scholar]
- Carlomagno, T.; Blommers, M.J.J.; Meiler, J.; Jahnke, W.; Schupp, T.; Petersen, F.; Schinzer, D.; Altmann, K.-H.; Griesinger, C. The high-resolution solution structure of epothilone A bound to tubulin: An understanding of the structure-activity relationships for a powerful class of antitumor agents. Angew. Chem. Int. Ed. 2003, 42, 2511–2515. [Google Scholar] [CrossRef] [PubMed]
- Nettles, J.H.; Li, H.; Cornett, B.; Krahn, J.M.; Snyder, J.P.; Downing, K.H. The binding mode of Epothilone A on α,ß-tubulin by electron crystallography. Science 2004, 305, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Bargsten, K.; Zurwerra, D.; Field, J.J.; Diaz, J.F.; Altmann, K.-H.; Steinmetz, M.O. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013, 339, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Canales, A.; Nieto, L.; Rodríguez-Salarichs, J.; Sánchez-Murcia, P.A.; Cordrech, C.; Cortes-Cabrera, A.; Paterson, I.; Carlomagno, T.; Federico, G.; Andreu, J.; et al. Molecular recognition of epothilones by microtubules and tubulin dimers revealed by biochemical and NMR approaches. ACS Chem. Biol. 2014, 9, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Coderch, C.; Klett, J.; Morreale, A.; Diaz, J.F.; Gago, F. Comparative binding energy (COMBINE) analysis supports a proposal for the binding mode of epothilones to β-tubulin. ChemMedChem 2012, 7, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Giannakakou, P.; Gussio, R.; Nogales, E.; Downing, K.H.; Zaharevitz, D.; Bollbuck, B.; Poy, G.; Sackett, D.; Nicolaou, K.C.; Fojo, T. A common pharmacophore for epothilone and taxanes: Molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2904–2909. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Jagtap, P.G.; Kingston, D.G.I.; Shen, H.-J.; Orr, G.A.; Horwitz, S.B. A common pharmacophore for taxol and the epothilones based on the biological activity of a taxane molecule lacking a C-13 side chain. Biochemistry 2000, 39, 3972–3978. [Google Scholar] [CrossRef] [PubMed]
- Narvi, E.; Jaakkola, K.; Oetken-Lindholm, C.; Halonen, P.; Kallio, L.; Kallio, M.J. Altered TUBB3 expression contributes to the epothilone response of mitotic cells. Br. J. Cancer 2013, 108, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Brizuela, M. The microtubule-stabilizing drug epothilone D increases axonal sprouting following transection injury in vitro. Mol. Cell. Neurosci. 2015, 66, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Kitagawa, K.; Mera, K.; Sasaki, Y. First clinical pharmacokinetic dose-escalation study of sagopilone, a novel, fully synthetic epothilone, in Japanese patients with refractory solid tumors. Investig. New Drugs 2012, 30, 2327–2333. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.-H.; Wartmann, M.; O’Reilly, T. Epothilones and related structures—A new class of microtubule inhibitors with potent in vivo antitumor activity. Biochim. Biophys. Acta 2000, 1470, M79–M91. [Google Scholar] [CrossRef]
- Chou, T.-C.; Zhang, X.-G.; Balog, D.-S.; Meng, D.; Savin, K.; Bertino, J.R.; Danishefsky, S.J. Desoxyepothilone B: An efficacious microtubule-targeted antitumor agent with a promising in vivo profile relative to epothilone B. Proc. Natl. Acad. Sci. USA 1998, 95, 9642–9647. [Google Scholar] [CrossRef] [PubMed]
- Heinz, D.W.; Schubert, W.-D.; Hofle, G. Much anticipated-the bioactive conformation of epothilone and its binding to tubulin. Angew. Chem. Int. Ed. 2005, 44, 1298–1301. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Heise, H.; Blommers, M.J.J.; Krastel, P.; Schmitt, E.; Petersen, F.; Jeganathan, S.; Mandelkow, E.-M.; Carlomagno, T.; Griesinger, C.; et al. Interaction of epothilone B (patupilone) with microtubules as detected by two-dimensional solid-state NMR spectroscopy. Angew. Chem. Int. Ed. 2010, 49, 7504–7507. [Google Scholar] [CrossRef] [PubMed]
- Nagano, S.; Li, H.; Shimizu, H.; Nishida, C.; Ogura, H.; de Montellano, P.R.O.; Poulos, T.L. Crystal structures of epothilone D-bound, epothilone B-bound, and substrate-free forms of cytochrome P450epoK. J. Biol. Chem. 2003, 278, 44886–44893. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.A.; Yang, J.; Finlay, M.R.V.; Cabral, F.; Vourloumis, D.; Stephens, L.C.; Troncoso, P.; Wu, X.; Logothetis, C.J.; Nicolaou, K.C.; et al. Antitumor efficacy of 26-fluoroepothilone B against human prostate cancer xenografts. Cancer Chemother. Pharmacol. 2001, 48, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Guido, K.; Olivier, L.; Karl-Heinz, A. Total synthesis of 26-fluoro-epothilone B. Synlett 2004, 4, 693–697. [Google Scholar]
- Bak, M.; Rasmussen, J.T.; Nielsen, N.C. SIMPSON: A general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 2000, 147, 296–330. [Google Scholar] [CrossRef] [PubMed]
- Goetz, J.M.; Schaefer, J. REDOR dephasing by multiple spins in the presence of molecular motion. J. Magn. Reson. 1997, 127, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Grage, S.L.; Durr, U.H.N.; Afonin, S.; Mikhailiuk, P.K.; Komarov, I.V.; Ulrich, A.S. Solid-state 19F NMR parameters of fluorine-labeled amino acids. Part II: Aliphatic substituents. J. Magn. Reson. 2008, 191, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Grage, S.L.; Ulrich, A.S. Orientation-dependent 19F dipolar couplings within a trifluoromethyl group are revealed by static multipulse NMR in the solid state. J. Magn. Reson. 2000, 146, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Toke, O.; Maloy, W.L.; Kim, S.J.; Blazyk, J.; Schaefer, J. Secondary structure and lipid contact of a peptide antibiotic in phospholipid bilayers by REDOR. Biophys. J. 2004, 87, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Ninkovic, S.; Finlay, M.R.V.; Sarabia, F.; Li, T. Total synthesis of 26-hydroxyepothilone B and related analogues. Chem. Commun. 1997, 24, 2343–2344. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Finlay, M.R.V.; Ninkovic, S.; Sarabia, F. Total synthesis of 26-hydroxy-epothilone B and related analogs via a macrolactonization based strategy. Tetrahedron 1998, 54, 7127–7166. [Google Scholar] [CrossRef]
- Lee, H.W.; Lee, I.-Y.C.; Hong, Y.D. Synthesis of the C11–C21 and C13–C21 fragments of epothilones from D-glucose. Bull. Korean Chem. Soc. 2000, 21, 1177–1178. [Google Scholar]
- Opella, S.J. [17]Protein dynamics by solid state nuclear magnetic resonance. Methods Enzymol. 1986, 131, 327–361. [Google Scholar] [PubMed]
- Altmann, K.-H.; Hofle, G.; Muller, R.; Mulzer, J.; Prantz, K. The Epothilones: An Outstanding Family of Anti-Tumor Agents; Springer: Wien, Austria, 2009; pp. 1–260. [Google Scholar]
- He, L.; Yang, C.-P.H.; Horwitz, S.B. Mutations in β-tubulin map to domains involved in regulation of microtubule stability in epothilone-resistant cell lines. Mol. Cancer Ther. 2001, 1, 3–10. [Google Scholar] [PubMed]
- Erdelyi, M.; Pfeiffer, B.; Hauenstein, K.; Fohrer, J.; Gertsch, J.; Altmann, K.-H.; Carlomagno, T. Conformational preferences of natural and C3-modified epothilones in aqueous solution. J. Med. Chem. 2008, 51, 1469–1473. [Google Scholar] [CrossRef] [PubMed]
- Kamel, K.; Kolinski, A. Computational study of binding of epothilone A to β-tubulin. Acta Biochim. Pol. 2011, 58, 255–260. [Google Scholar] [PubMed]
- Diaz-Padilla, I.; Oza, A.M. Epothilones in the treatment of ovarian cancer. Future Oncol. 2011, 7, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Lopus, M.; Smiyun, G.; Miller, H.; Oroudjev, E.; Wilson, L.; Jordan, M.A. Mechanism of action of ixabepilone and its interactions with the βIII-tubulin isotype. Cancer Chemother. Pharmacol. 2015, 76, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Lozynski, M. Patupilone and ixabepilone: The effect of a point structural change on the exo-endo conformational profile. J. Phys. Chem. B 2012, 116, 7605–7617. [Google Scholar] [CrossRef] [PubMed]
- Kelusky, E.C.; Smith, I.C.P.; Elliger, C.A.; Cameron, D.G. Molecular motions in the solid phases of n-heneicosane: A 2H NMR study. J. Am. Chem. Soc. 1984, 106, 2267–2270. [Google Scholar] [CrossRef]
- O’Connor, R.D.; Poliks, B.; Bolton, D.H.; Goetz, J.M.; Byers, J.A.; Wooley, K.L.; Schaefer, J. Chain packing in linear phenol-polycarbonate by 13C{2H} REDOR. Macromolecules 2002, 35, 2608–2617. [Google Scholar] [CrossRef]
- Lee, G.H.; Oh, S.Y.; Yeo, K.J.Y.; Ferdous, T.; Cho, M.; Paik, Y. Solid-state 31P NMR investigation on the status of guanine nucleotides in paclitaxel-stabilized microtubules. Magn. Reson. Chem. 2015, 53, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H. Synthesis of an Epothilone B Derivative with Fluorine and Deuterium Substituents. Master’s Thesis, Chungbuk National University, Cheong Ju, Korea, February 2014. [Google Scholar]
- Paik, Y.; Lee, S.; Kim, H.S.; Lee, G.H. Sampling Tool and Sample Analysis Method Using the Same. KR Patent 2016-0172469, 16 December 2016. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | A549 | HeLa |
|---|---|---|
| Epo B (2) | 127 | 41 |
| Analog (3) | 163 | 67 |
| Hydrogen Bond | Distance (Å) |
|---|---|
| Thr274(CO) ⋯ HO(7) | 2.0 |
| Thr274(NH) ⋯ O(7) | 1.9 |
| Thr274(OH) ⋯ O(5) | 1.9 |
| Leu228(NH) ⋯ O | 2.0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-H.; Kim, M.-S.; Lee, H.W.; Lee, I.-Y.C.; Kim, H.K.; Kim, N.D.; Lee, S.; Seo, H.; Paik, Y. The Application of REDOR NMR to Understand the Conformation of Epothilone B. Int. J. Mol. Sci. 2017, 18, 1472. https://doi.org/10.3390/ijms18071472
Lee J-H, Kim M-S, Lee HW, Lee I-YC, Kim HK, Kim ND, Lee S, Seo H, Paik Y. The Application of REDOR NMR to Understand the Conformation of Epothilone B. International Journal of Molecular Sciences. 2017; 18(7):1472. https://doi.org/10.3390/ijms18071472
Chicago/Turabian StyleLee, Jae-Ho, Moon-Su Kim, Hyo Won Lee, Ihl-Young C. Lee, Hyun Kyoung Kim, Nam Doo Kim, SangGap Lee, Hwajeong Seo, and Younkee Paik. 2017. "The Application of REDOR NMR to Understand the Conformation of Epothilone B" International Journal of Molecular Sciences 18, no. 7: 1472. https://doi.org/10.3390/ijms18071472
APA StyleLee, J.-H., Kim, M.-S., Lee, H. W., Lee, I.-Y. C., Kim, H. K., Kim, N. D., Lee, S., Seo, H., & Paik, Y. (2017). The Application of REDOR NMR to Understand the Conformation of Epothilone B. International Journal of Molecular Sciences, 18(7), 1472. https://doi.org/10.3390/ijms18071472