TGF-?-Dependent Growth Arrest and Cell Migration in Benign and Malignant Breast Epithelial Cells Are Antagonistically Controlled by Rac1 and Rac1b




Abstract
:1. Introduction
2. Results
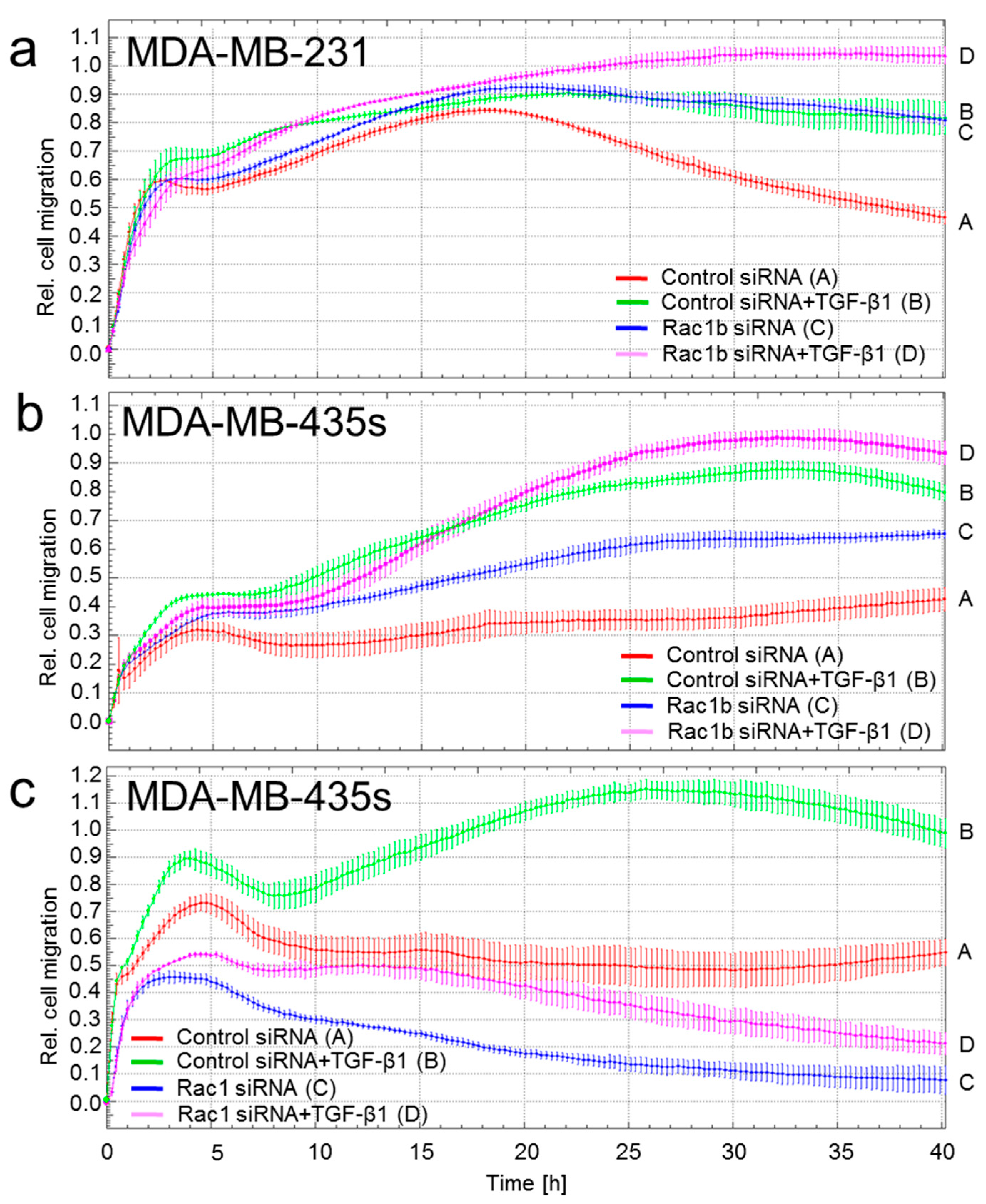
2.1. Depletion of Rac1 Suppresses While Rac1b Enhances Basal and TGF-β1-Induced Migration of Breast Cancer Cells
2.2. Depletion of Rac1b in MDA-MB-231 Cells Increases TGF-β-Induced p21WAF1 Expression and ERK1/2 Phosphorylation
2.3. Differential Expression of Metastasis-Associated Genes in HMEC of Low and High Passage Number
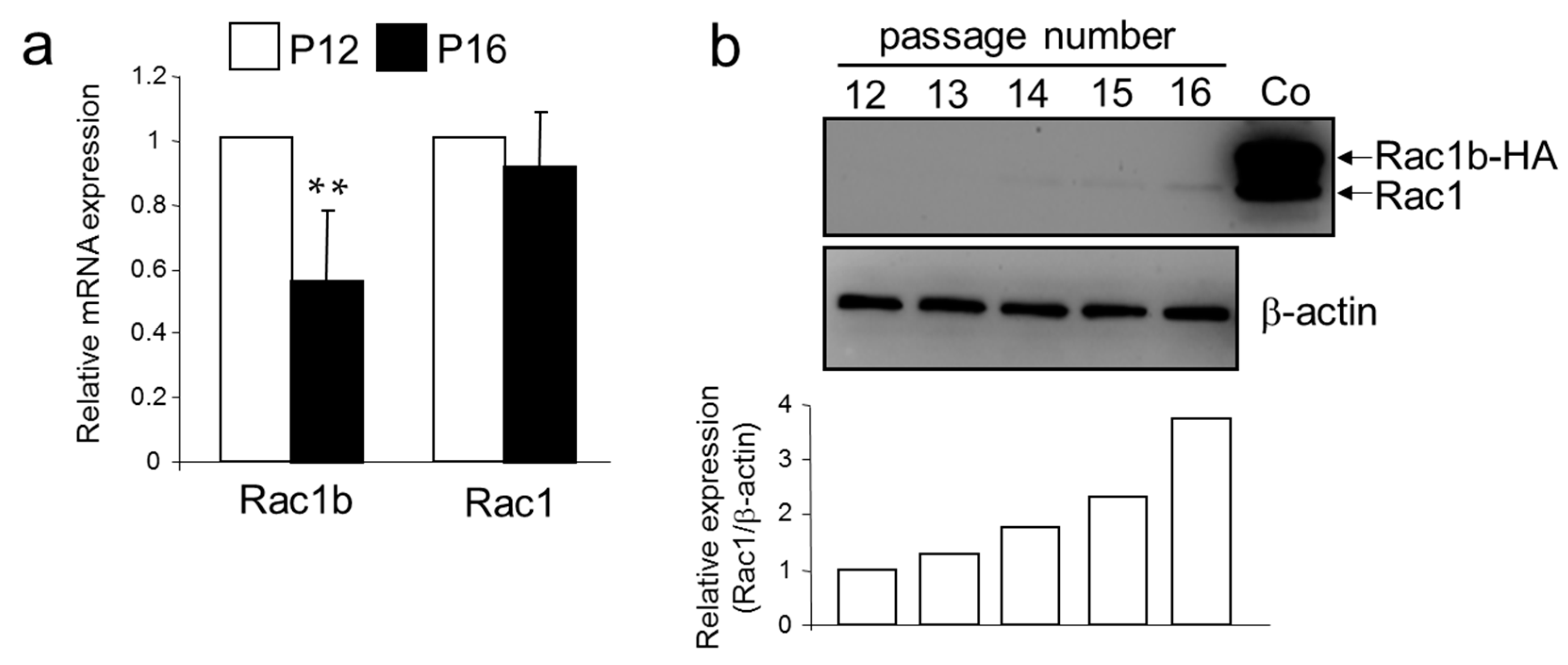
2.4. A Senescent Phenotype of HMEC was Associated with an Increase in Rac1 Expression
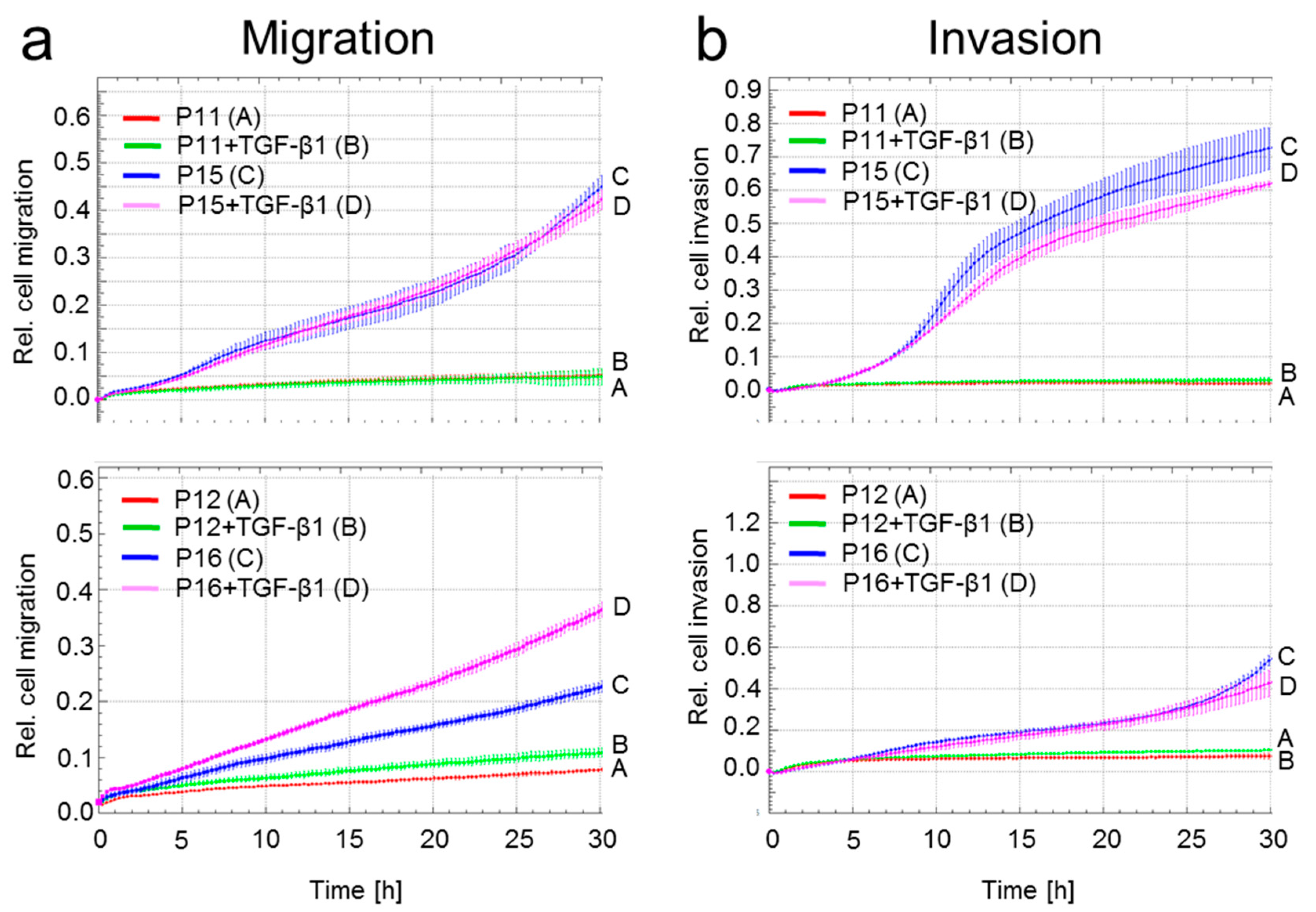
2.5. A Senescent Phenotype is Associated with Increased Migratory Activity in HMEC
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Cultures of Human Mammary Epithelial Cells
4.3. Culture of Established Breast Cancer Cell Lines
4.4. Transient Transfection of siRNAs
4.5. Human Tumor Metastasis PCR Array and qPCR Analysis
4.6. Immunoblotting
4.7. Flow Cytometry Analysis of HMEC
4.8. Migration and Invasion Assays
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HMEC | Human mammary epithelial cells |
| MnSOD | Manganese-dependent superoxide dismutase |
| ERK | Extracellular signal-regulated kinase |
| TGF-β | Transforming growth factor-β |
References
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Isayeva, T.; Siegal, G.P.; Ponnazhagan, S. Silencing of transforming growth factor-β1 in situ by RNA interference for breast cancer: Implications for proliferation and migration in vitro and metastasis in vivo. Clin. Cancer Res. 2008, 14, 4961–4970. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. TGF-β and cancer. Cytokine Growth Factor Rev. 2006, 17, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Bild, A.H.; Yao, G.; Chang, J.T.; Wang, Q.; Potti, A.; Chasse, D.; Joshi, M.B.; Harpole, D.; Lancaster, J.M.; Berchuck, A.; et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006, 439, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Tremain, R.; Marko, M.; Kinnimulki, V.; Ueno, H.; Bottinger, E.; Glick, A. Defects in TGF-β signaling overcome senescence of mouse keratinocytes expressing v-Ha-ras. Oncogene 2000, 19, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yang, J.; Elkahloun, A.G.; Bandyopadhyay, A.; Wang, L.; Cornell, J.E.; Yeh, I.T.; Agyin, J.; Tomlinson, G.; Sun, L.Z. Attenuation of TGF-β signaling suppresses premature senescence in a p21-dependent manner and promotes oncogenic Ras-mediated metastatic transformation in human mammary epithelial cells. Mol. Biol. Cell 2012, 23, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Bertram, C.; Hass, R. MMP-7 is involved in the aging of primary human mammary epithelial cells (HMEC). Exp. Gerontol. 2008, 43, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Bertram, C.; Hass, R. Cellular senescence of human mammary epithelial cells (HMEC) is associated with an altered MMP-7/HB-EGF signaling and increased formation of elastin-like structures. Mech. Ageing Dev. 2009, 130, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Bertram, C.; Hass, R. Cellular responses to reactive oxygen species-induced DNA damage and aging. Biol. Chem. 2008, 389, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Hass, R. Retrodifferentiation and cell death. Crit. Rev. Oncog. 1994, 5, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Konzack, A.; Kietzmann, T. Manganese superoxide dismutase in carcinogenesis: Friend or foe? Biochem. Soc. Trans. 2014, 42, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Yaswen, P.; Stampfer, M.R. Molecular changes accompanying senescence and immortalization of cultured human mammary epithelial cells. Int. J. Biochem. Cell Biol. 2002, 34, 1382–1394. [Google Scholar] [CrossRef]
- Bottinger, E.P.; Jakubczak, J.L.; Haines, D.C.; Bagnall, K.; Wakefield, M.L. Transgenic mice overexpressing a dominant-negative mutant type II transforming growth factor β receptor show enhanced tumorigenesis in the mammary gland and lung in response to the carcinogen 7,12-dimethylbenz-[a]-anthracene. Cancer Res. 1997, 57, 5564–5570. [Google Scholar] [PubMed]
- Wang, X.J.; Greenhalgh, D.A.; Bickenbach, J.R.; Jiang, A.; Bundman, D.S.; Krieg, T.; Derynck, R.; Roop, D.R. Expression of a dominant-negative type II transforming growth factor β (TGF-β) receptor in the epidermis of transgenic mice blocks TGF-β-mediated growth inhibition. Proc. Natl. Acad. Sci. USA 1997, 94, 2386–2391. [Google Scholar] [CrossRef] [PubMed]
- Schnelzer, A.; Prechtel, D.; Knaus, U.; Dehne, K.; Gerhard, M.; Graeff, H.; Harbeck, N.; Schmitt, M.; Lengyel, E. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000, 19, 3013–3020. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Sebens, S.; Giehl, K.; Helm, O.; Groth, S.; Fandrich, F.; Rocken, C.; Sipos, B.; Lehnert, H.; Gieseler, F. Rac1b negatively regulates TGF-β1-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signalling. Oncotarget 2014, 5, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, P.L. Regulation of NADPH oxidases: The role of Rac proteins. Circ. Res. 2006, 98, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.F.; Barros, P.; Moyer, M.P.; Matos, P.; Jordan, P. Expression of tumor-related Rac1b antagonizes B-Raf-induced senescence in colorectal cells. Cancer Lett. 2015, 369, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Hass, R. Extracellular signals in young and aging breast epithelial cells and possible connections to age-associated breast cancer development. Mech. Ageing Dev. 2011, 132, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Baek, G.Y.; Park, H.R.; Jo, S.K.; Jung, U. Smad2/3-regulated expression of DLX2 is associated with radiation-induced epithelial-mesenchymal transition and radioresistance of A549 and MDA-MB-231 human cancer cell lines. PLoS ONE 2016, 11, e0147343. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Malfettone, A.; Soukupova, J. New insights into the crossroads between EMT and stemness in the context of cancer. J. Clin. Med. 2016, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayachandra, K.; Higgins, W.; Lee, J.; Glick, A. Induction of p16ink4a and p19ARF by TGF-β1 contributes to growth arrest and senescence response in mouse keratinocytes. Mol. Carcinog. 2009, 48, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.W.; Nikpay, M.; Silva, A.; Lau, P.; Martinuk, A.; Linseman, T.A.; Soubeyrand, S.; McPherson, R. Functional interaction between COL4A1/COL4A2 and Smad3 risk loci for coronary artery disease. Atherosclerosis 2015, 242, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Integrative genomic analyses of CXCR4: Transcriptional regulation of CXCR4 based on TGFβ, Nodal, Activin signaling and POU5F1, FOXA2, FOXC2, FOXH1, SOX17, and GFI1 transcription factors. Int. J. Oncol. 2010, 36, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Shinriki, S.; Jono, H.; Guo, J.; Ueda, M.; Hayashi, M.; Yamashita, S.; Zijlstra, A.; Nakayama, H.; Hiraki, A.; et al. Intrinsic TGF-β2-triggered SDF-1-CXCR4 signaling axis is crucial for drug resistance and a slow-cycling state in bone marrow-disseminated tumor cells. Oncotarget 2015, 6, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Wiercinska, E.; Naber, H.P.; Pardali, E.; van der Pluijm, G.; van Dam, H.; ten Dijke, P. The TGF-β/Smad pathway induces breast cancer cell invasion through the up-regulation of matrix metalloproteinase 2 and 9 in a spheroid invasion model system. Breast Cancer Res. Treat. 2011, 128, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Gogineni, V.R.; Gupta, R.; Nalla, A.K.; Velpula, K.K.; Rao, J.S. uPAR and cathepsin B shRNA impedes TGF-β1-driven proliferation and invasion of meningioma cells in a XIAP-dependent pathway. Cell Death Dis. 2012, 3, e439. [Google Scholar] [CrossRef] [PubMed]
- Sugioka, K.; Kodama, A.; Okada, K.; Iwata, M.; Yoshida, K.; Kusaka, S.; Matsumoto, C.; Kaji, H.; Shimomura, Y. TGF-β2 promotes RPE cell invasion into a collagen gel by mediating urokinase-type plasminogen activator (uPA) expression. Exp. Eye Res. 2013, 115, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Luga, V.; McLean, S.; Le Roy, C.; O’Connor-McCourt, M.; Wrana, J.L.; di Guglielmo, G.M. The extracellular domain of the TGFβ type II receptor regulates membrane raft partitioning. Biochem. J. 2009, 421, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Mandel, K.; Seidl, D.; Rades, D.; Lehnert, H.; Gieseler, F.; Hass, R.; Ungefroren, H. Characterization of spontaneous and TGF-β-induced cell motility of primary human normal and neoplastic mammary cells in vitro using novel real-time technology. PLoS ONE 2013, 8, e56591. [Google Scholar] [CrossRef]
- Carl, C.; Flindt, A.; Hartmann, J.; Dahlke, M.; Rades, D.; Dunst, J.; Lehnert, H.; Gieseler, F.; Ungefroren, H. Ionizing radiation induces a motile phenotype in human carcinoma cells in vitro through hyperactivation of the TGF-β signaling pathway. Cell Mol. Life Sci. 2016, 73, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Wang, S.; Feng, R. STIM1 plays an important role in TGF-β-induced suppression of breast cancer cell proliferation. Oncotarget 2016, 7, 16866–16878. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Hass, R.; von der Ohe, J.; Lehnert, H.; Ungefroren, H. The role of TGF-β and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun. Signal. 2017, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.; Bakin, A.V.; Arteaga, C.L. Autocrine transforming growth factor-β signaling mediates Smad-independent motility in human cancer cells. J. Biol. Chem. 2003, 278, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Northey, J.J.; Primeau, M.; Machado, R.D.; Trembath, R.; Siegel, P.M.; Lamarche-Vane, N. CdGAP is required for transforming growth factor β- and Neu/ErbB-2-induced breast cancer cell motility and invasion. Oncogene 2011, 30, 1032–1045. [Google Scholar] [CrossRef] [PubMed]
- Rosman, D.S.; Phukan, S.; Huang, C.C.; Pasche, B. TGFBR1*6A enhances the migration and invasion of MCF-7 breast cancer cells through RhoA activation. Cancer Res. 2008, 68, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Bae, G.U.; Kang, J.K.; Park, J.W.; Lee, E.K.; Lee, H.Y.; Choi, W.S.; Lee, H.W.; Han, J.W. Cooperation of H2O2-mediated ERK activation with Smad pathway in TGF-β1 induction of p21WAF1/Cip1. Cell Signal. 2006, 18, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Senturk, S.; Mumcuoglu, M.; Gursoy-Yuzugullu, O.; Cingoz, B.; Akcali, K.C.; Ozturk, M. Transforming growth factor-β induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology 2010, 52, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Kucerova, A.; Michlits, G.; Kyjacova, L.; Reinis, M.; Korolov, O.; Bartek, J.; Hodny, Z. IFNγ induces oxidative stress, DNA damage and tumor cell senescence via TGFβ/Smad signaling-dependent induction of Nox4 and suppression of ANT2. Oncogene 2016, 35, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Gao, Y.; Hoyle, D.L.; Cheng, T.; Wang, Z.Z. Suppression of transforming growth factor-β signaling delays cellular senescence and preserves the function of endothelial cells derived from human pluripotent stem cells. Stem Cells Transl. Med. 2016, 6, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.J.; Der, C.J. Aberrant function of the Ras signal transduction pathway in human breast cancer. Breast Cancer Res. Treat. 1995, 35, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Suo, Z.; Risberg, B.; Karlsson, M.G.; Villman, K.; Nesland, J.M. Expression of EphB2 and EphB4 in breast carcinoma. Pathol. Oncol. Res. 2004, 10, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.V.; Shinohara, M.; Porchia, L.M.; Chung, Y.J.; McCarty, S.; Saji, M.; Ringel, M.D. Regulator of calcineurin 1 modulates cancer cell migration in vitro. Clin. Exp. Metastasis 2009, 26, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, V.V.; Sakthivel, K.M.; Guruvayoorappan, C. Kisspeptins (KiSS-1): Essential players in suppressing tumor metastasis. Asian Pac. J. Cancer Prev. 2013, 14, 6215–6220. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.G.; Yi, Z.; Pang, X.; Yi, T.; Wang, Y.; Luo, J.; Wu, Z.; Li, D.; Liu, M. Kisspeptin-10, a KISS1-derived decapeptide, inhibits tumor angiogenesis by suppressing Sp1-mediated VEGF expression and FAK/Rho GTPase activation. Cancer Res. 2009, 69, 7062–7070. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Groth, S.; Sebens, S.; Lehnert, H.; Gieseler, F.; Fandrich, F. Differential roles of Smad2 and Smad3 in the regulation of TGF-β1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: Control by Rac1. Mol. Cancer 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, R.; Kan, C.E.; Graham, J.; Danielpour, D.; Stampfer, M.; Jackson, M.W. TGF-β signaling engages an ATM-CHK2-p53-independent RAS-induced senescence and prevents malignant transformation in human mammary epithelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8668–8673. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Su, J.; Shi, L.; Liao, Q.; Su, Q. DADS downregulates the Rac1-ROCK1/PAK1-LIMK1-ADF/cofilin signaling pathway, inhibiting cell migration and invasion. Oncol. Rep. 2013, 29, 605–612. [Google Scholar] [PubMed]
- Hass, R.; Bertram, C. Characterization of human breast cancer epithelial cells (HBCEC) derived from long term cultured biopsies. J. Exp. Clin. Cancer Res. 2009, 28. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HMEC | P11/P12 | P15/P16 | |
|---|---|---|---|
| Gene | Protein | ||
| APC | Apc | 34.5 | 30.8 |
| CD44 | CD44/hyaluron receptor | 29.3 | 26.9 |
| CDKN2A | p16INK4A | 32.7 | 30.4 |
| COL4A2 | collagen 4A2 | 37.2 | 35.2 |
| CXCR2 | CXCR2 | 32.3 | 30.1 |
| CXCR4 | CXCR4 | 39 | 35.5 |
| EPHB2 | Ephrin B2 receptor | 28.1 | 26 |
| ETV4 | ETS variant 4 | 28.8 | 27.2 |
| FXYD5 | FXYD domain-containing ion transport regulator 5 | 36.2 | 33.5 |
| IL18 | IL18 | 27 | 24 |
| KRAS | Ki-Ras | 34.3 | 30.8 |
| MET | Hepatocyte growth factor receptor | 33.4 | 30.9 |
| MGAT5 | N-acetylglucosaminyltransferase V | 31.8 | 28.7 |
| MMP2 | MMP-2 | 31.6 | 29.5 |
| NME4 | non-metastatic cells 4 | 27.6 | 25.5 |
| NR4A3 | nuclear receptor subfamily 4 group A member 3 | 23.8 | 21.8 |
| PLAUR | urokinase-type plasminogen activator receptor | 31.2 | 28 |
| RORB | retinoid-related orphan receptor β | 29.3 | 26.7 |
| SMAD2 | SMAD2 | 31 | 28.3 |
| SMAD4 | SMAD4 | 33.2 | 30.9 |
| KISS1 | Kisspeptin-10 | 35.6 | 37 |
| KISS1R | GPR54 | 35.5 | 37 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melzer, C.; Von der Ohe, J.; Hass, R.; Ungefroren, H. TGF-?-Dependent Growth Arrest and Cell Migration in Benign and Malignant Breast Epithelial Cells Are Antagonistically Controlled by Rac1 and Rac1b. Int. J. Mol. Sci. 2017, 18, 1574. https://doi.org/10.3390/ijms18071574
Melzer C, Von der Ohe J, Hass R, Ungefroren H. TGF-?-Dependent Growth Arrest and Cell Migration in Benign and Malignant Breast Epithelial Cells Are Antagonistically Controlled by Rac1 and Rac1b. International Journal of Molecular Sciences. 2017; 18(7):1574. https://doi.org/10.3390/ijms18071574
Chicago/Turabian StyleMelzer, Catharina, Juliane Von der Ohe, Ralf Hass, and Hendrik Ungefroren. 2017. "TGF-?-Dependent Growth Arrest and Cell Migration in Benign and Malignant Breast Epithelial Cells Are Antagonistically Controlled by Rac1 and Rac1b" International Journal of Molecular Sciences 18, no. 7: 1574. https://doi.org/10.3390/ijms18071574
APA StyleMelzer, C., Von der Ohe, J., Hass, R., & Ungefroren, H. (2017). TGF-?-Dependent Growth Arrest and Cell Migration in Benign and Malignant Breast Epithelial Cells Are Antagonistically Controlled by Rac1 and Rac1b. International Journal of Molecular Sciences, 18(7), 1574. https://doi.org/10.3390/ijms18071574