The Effect of N-Terminal Cyclization on the Function of the HIV Entry Inhibitor 5P12-RANTES

Abstract
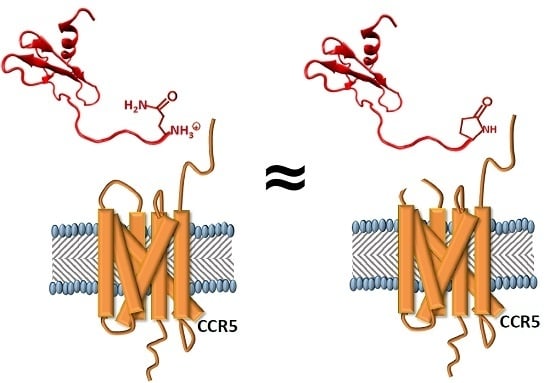
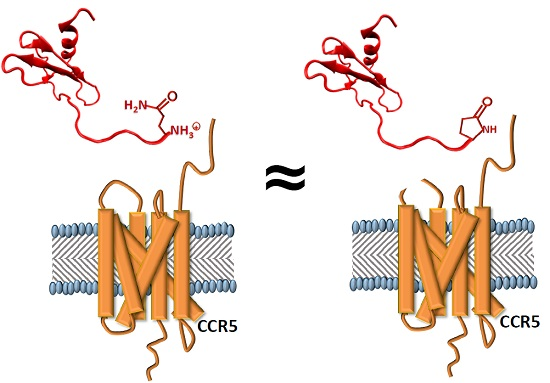
:
1. Introduction
2. Results
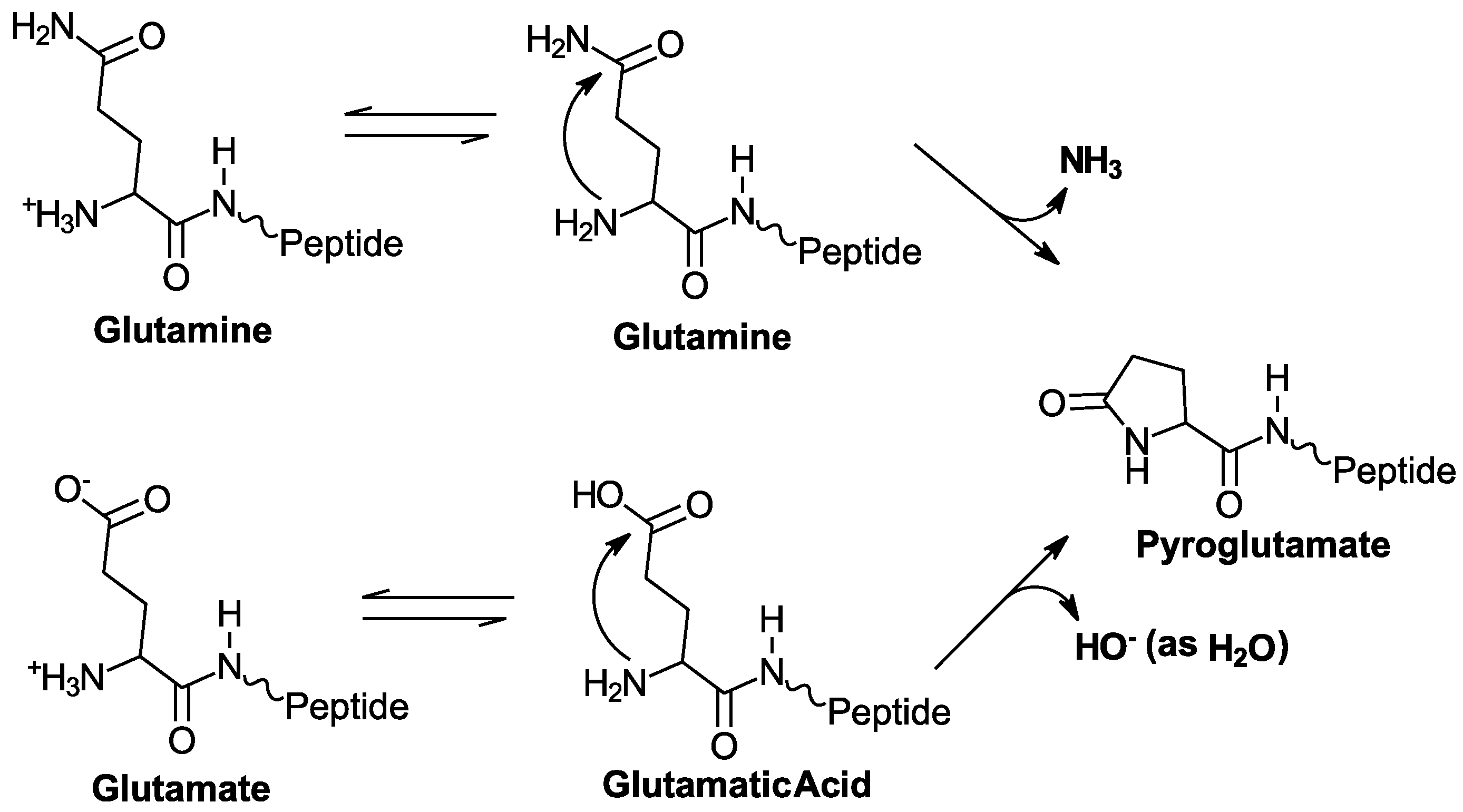
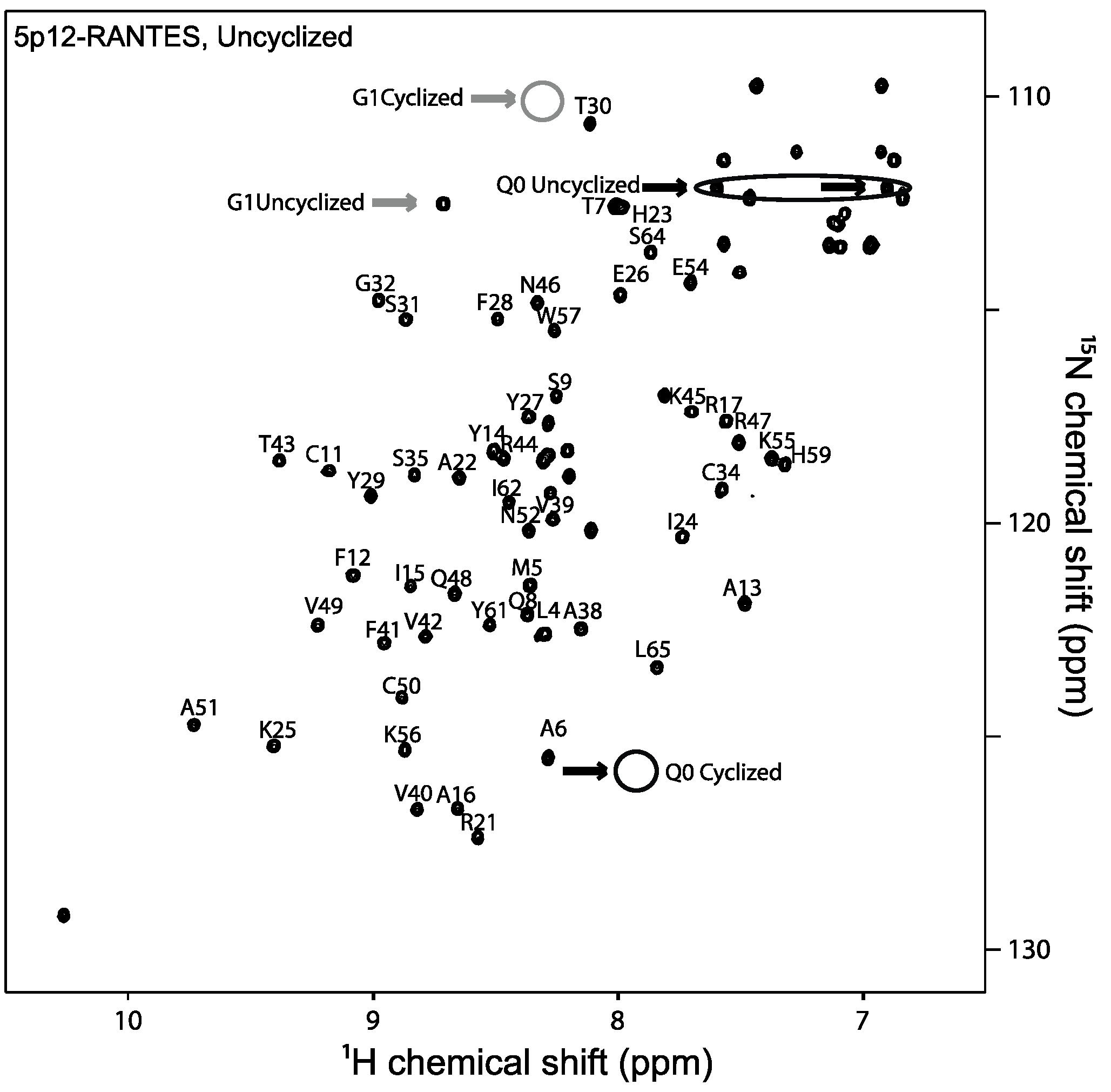
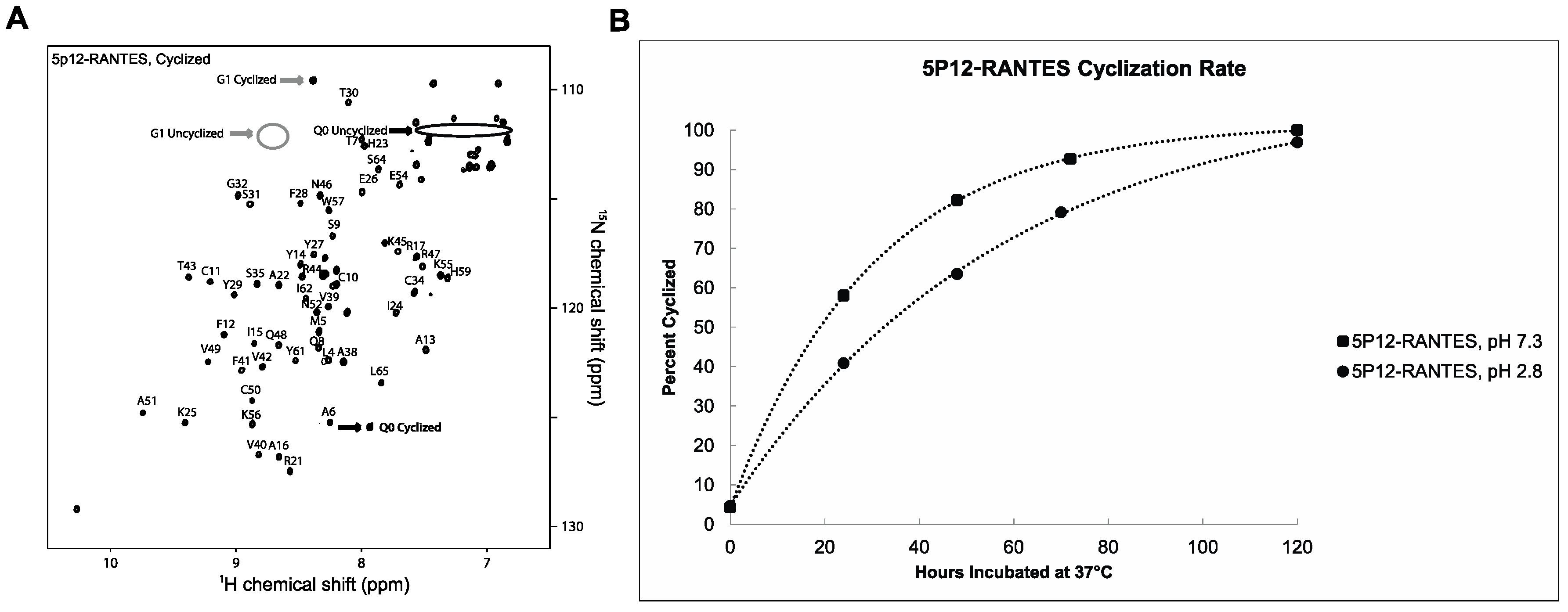
2.1. 5P12-RANTES N-Terminal Cyclization
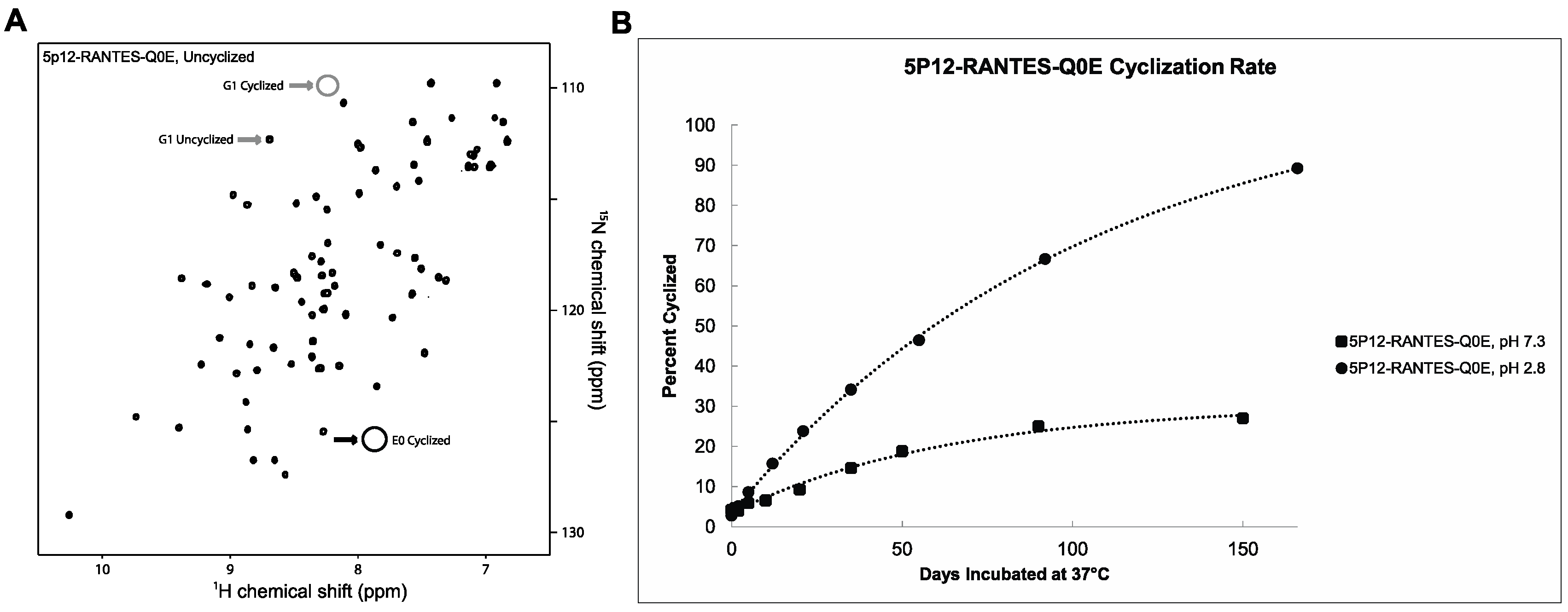
2.2. 5P12-Q0E Also Cyclizes to Produce Mature 5P12-RANTES
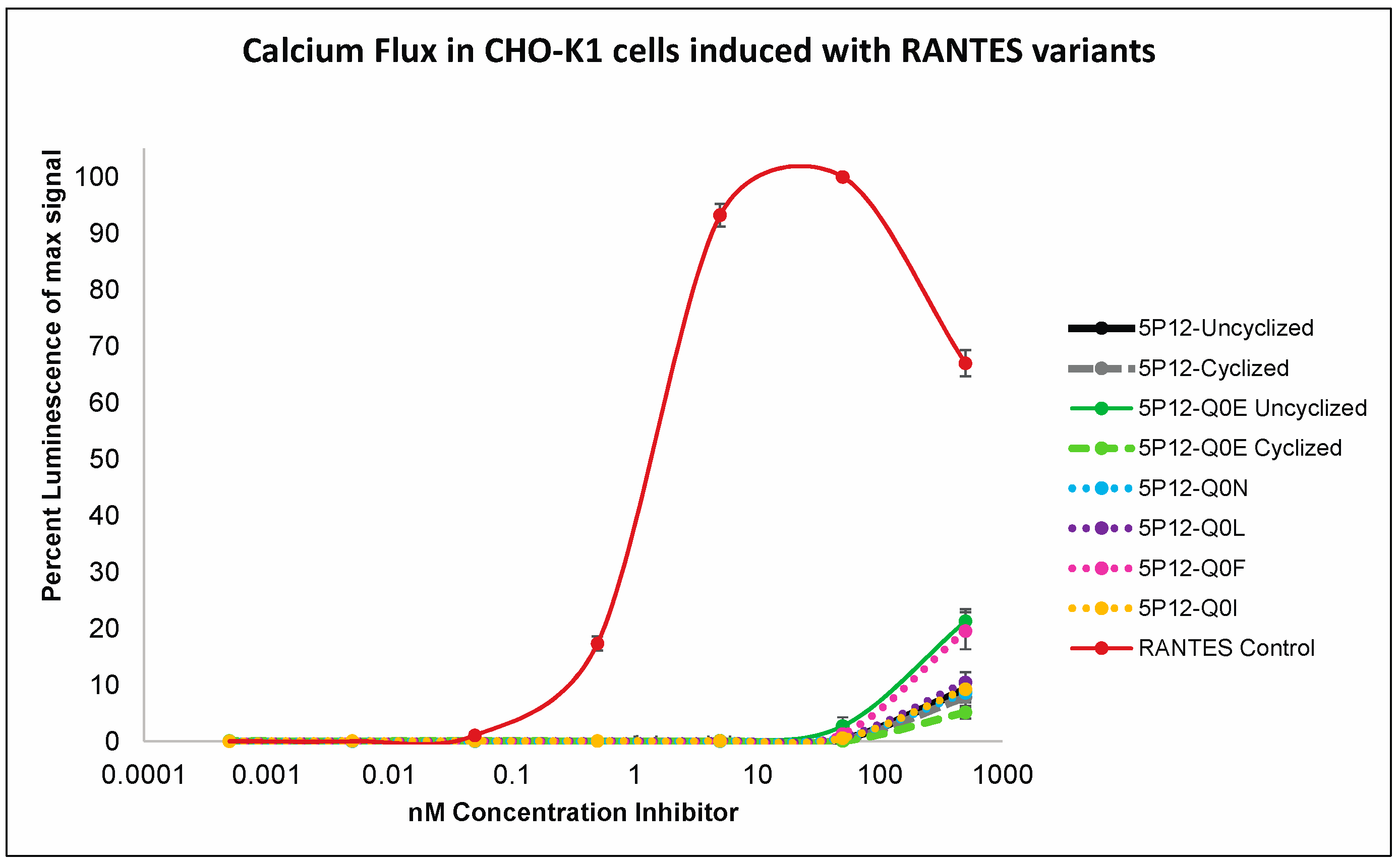
2.3. High Potency HIV Inhibition Does Not Require N-terminal Cyclization or N-Terminal Glutamine
2.4. The Effect of the N-Terminal Amino Acid and Its State of Cyclization on Activation of CC Chemokine Receptor 5 (CCR5)
3. Discussion
4. Materials and Methods
4.1. Protein Purification
4.2. Obtaining Rates of 5P12-RANTES and 5P12-RANTES-Q0E Cyclization
4.3. NMR Spectroscopy
4.4. Quantifying N-Terminal Cyclization
4.5. Cell Lines and Viruses
4.6. Single Round Pseudovirus Production
4.7. Single-Round Pseudoviral Assay
4.8. CCR5 Functional Assays in CHO-K1 Cells
4.9. CCR5 Activity in HeLa-P5L
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
Abbreviations
| HIV | Human Immunodeficiency Virus |
| AIDS | Acquired Immune Deficiency Syndrome |
| CCL5 | Chemokine (C-C Motif) Ligand 5; also called RANTES |
| RANTES | Regulated on Activation, Normal T-cell Expressed and Secreted; also called CCL5 |
| Gln/Q | Glutamine |
| Glu/E | Glutamate |
| Asn/N | Asparagine |
| Phe/F | Phenylalanine |
| Ile/I | Isoleucine |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| NMR | Nuclear Magnetic Resonance |
| HBSS | Hank’s Buffered Salt Solution |
| FBS | Fetal Bovine Serum |
| HSQC | Heteronuclear Single Quantum Coherence |
| CHO | Chinese Hamster Ovary |
| IPTG | Isopropyl β-d-1-thiogalactopyranoside |
| TFA | Trifluoroacetic Acid |
References
- Global Report: Unaids UNAIDS Report on the Global AIDS Epidemic 2013. Available online: http://www.unaids.org/sites/default/files/media_asset/UNAIDS_Global_Report_2013_en_1.pdf (accessed on 17 July 2017).
- Boskey, E.R.; Telsch, K.M.; Whaley, K.J.; Moench, T.R.; Cone, R.A. Acid production by vaginal flora in vitro is consistent with the rate and extent of vaginal acidification. Infect. Immun. 1999, 67, 5170–5175. [Google Scholar] [PubMed]
- Evans, D.F.; Pye, G.; Bramley, R.; Clark, A.G.; Dyson, T.J.; Hardcastle, J.D. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut 1988, 29, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, H.; Cerini, F.; Escola, J.-M.; Kuenzi, G.; Melotti, A.; Offord, R.; Ne Rossitto-Borlat, I.; Nedellec, R.; Salkowitz, J.; Gorochov, G.; et al. Highly potent, fully recombinant anti-HIV chemokines: Reengineering a low-cost microbicide. Proc. Natl. Acad. Sci. USA 2008, 105, 17706–17711. [Google Scholar] [CrossRef] [PubMed]
- Suresh, P.; Wanchu, A. Chemokines and chemokine receptors in HIV infection: Role in pathogenesis and therapeutics. J. Postgrad. Med. 2006, 52, 210–217. [Google Scholar] [PubMed]
- Dragic, T.; Litwin, V.; Allaway, G.P.; Martin, S.R.; Huang, Y.; Nagashima, K.A.; Cayanan, C.; Maddon, P.J.; Koup, R.A.; Moore, J.P.; et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Cerini, F.; Landay, A.; Gichinga, C.; Lederman, M.M.; Flyckt, R.; Starks, D.; Offord, R.E.; Xois, F.; Gal, L.; Hartley, O. Chemokine Analogues Show Suitable Stability for Development as Microbicides. J. Acquir. Immune Defic. Syndr. 2008, 49, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Veazey, R.S.; Ling, B.; Green, L.C.; Ribka, E.P.; Lifson, J.D.; Piatak, M.; Lederman, M.M.; Mosier, D.; Offord, R.; Hartley, O. Topically Applied Recombinant Chemokine Analogues Fully Protect Macaques from Vaginal Simian- Human Immunodeficiency Virus Challenge. J. Infect. Dis. 2009, 199, 1525–1527. [Google Scholar] [CrossRef] [PubMed]
- Wiktor, M.; Hartley, O.; Grzesiek, S. Characterization of structure, dynamics, and detergent interactions of the anti-HIV chemokine variant 5P12-RANTES. Biophys. J. 2013. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I.; Power, C.A.; Hoogewerf, A.J.; Montjovent, M.O.; Borlat, F.; Offord, R.E.; Wells, T.N.C. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J. Biol. Chem. 1996, 271, 2599–2603. [Google Scholar] [CrossRef] [PubMed]
- Laurence, J.S.; Blanpain, C.; De Leener, A.; Parmentier, M.; LiWang, P.J. Importance of basic residues and quaternary structure in the function of MIP-1β: CCR5 binding and cell surface sugar interactions. Biochemistry 2001, 40, 4990–4999. [Google Scholar] [CrossRef] [PubMed]
- Laurence, J.S.; LiWang, A.C.; LiWang, P.J. Effect of N-terminal truncation and solution conditions on chemokine dimer stability: Nuclear magnetic resonance structural analysis of macrophage inflammatory protein 1β mutants. Biochemistry 1998, 37, 9346–9354. [Google Scholar] [CrossRef] [PubMed]
- Hartley, O.; Dorgham, K.; Perez-Bercoff, D.; Cerini, F.; Heimann, A.; Gaertner, H.; Offord, R.E.; Pancino, G.; Debré, P.; Gorochov, G. Human Immunodeficiency Virus Type 1 Entry Inhibitors Selected on Living Cells from a Library of Phage Chemokines. J. Virol. 2003, 77, 6637–6644. [Google Scholar] [CrossRef] [PubMed]
- Arenzana-Seisdedos, F.; Virelizier, J.-L.; Rousset, D.; Clark-Lewis, I.; Loetscher, P.; Moser, B.; Baggiolini, M. HIV blocked by chemokine antagonist. Nature 1996, 383, 400. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Clapham, P.R.; Picard, L.; Offord, R.E.; Rosenkilde, M.M.; Schwartz, T.W.; Buser, R.; Wells, T.N.; Proudfoot, A.E.; Cocchi, F.; et al. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science 1997, 276, 276–279. [Google Scholar] [CrossRef] [PubMed]
- White, G.E.; Iqbal, A.J.; Greaves, D.R. CC chemokine receptors and chronic inflammation--therapeutic opportunities and pharmacological challenges. Pharmacol. Rev. 2013, 65, 47–89. [Google Scholar] [CrossRef] [PubMed]
- Pastore, C.; Picchio, G.R.; Galimi, F.; Fish, R.; Hartley, O.; Offord, R.E.; Mosier, D.E. Two mechanisms for human immunodeficiency virus type 1 inhibition by N-terminal modifications of RANTES. Antimicrob. Agents Chemother. 2003, 47, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Hartley, O.; Gaertner, H.; Wilken, J.; Thompson, D.; Fish, R.; Ramos, A.; Pastore, C.; Dufour, B.; Cerini, F.; Melotti, A.; et al. Medicinal chemistry applied to a synthetic protein: Development of highly potent HIV entry inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 16460–16465. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Chatterjee, A.; Khedkar, A.P.; Kusumanchi, M.; Adhikary, L. Mass spectrometric distinction of in-source and in-solution pyroglutamate and succinimide in proteins: A case study on rhG-CSF. J. Am. Soc. Mass Spectrom. 2013, 24, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.D.; Goetze, A.M.; Bass, R.B.; Flynn, G.C. N-terminal glutamate to pyroglutamate conversion in vivo for human IgG2 antibodies. J. Biol. Chem. 2011, 286, 11211–11217. [Google Scholar] [CrossRef] [PubMed]
- Tritsch, G.L.; Moore, G.E. Spontaneous decomposition of glutamine in cell culture media. Exp. Cell Res. 1962, 28, 360–364. [Google Scholar] [CrossRef]
- Yu, L.; Vizel, A.; Huff, M.B.; Young, M.; Remmele, R.L.; He, B. Investigation of N-terminal glutamate cyclization of recombinant monoclonal antibody in formulation development. J. Pharm. Biomed. Anal. 2006, 42, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Chelius, D.; Jing, K.; Lueras, A.; Rehder, D.S.; Dillon, T.M.; Vizel, A.; Rajan, R.S.; Li, T.; Treuheit, M.J.; Bondarenko, P.V. Formation of pyroglutamic acid from N-terminal glutamic acid in immunoglobulin gamma antibodies. Anal. Chem. 2006, 78, 2370–2376. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Valliere-Douglass, J.; Brady, L.; Steen, S.; Han, M.; Pace, D.; Elliott, S.; Yates, Z.; Han, Y.; Balland, A.; et al. Analysis of post-translational modifications in recombinant monoclonal antibody IgG1 by reversed-phase liquid chromatography/mass spectrometry. J. Chromatogr. A 2007, 1164, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bachhawat, A.K. Pyroglutamic acid: Throwing light on a lightly studied metabolite. Curr. Sci. 2012, 102, 288–297. [Google Scholar]
- Dick, L.W.; Kim, C.; Qiu, D.; Cheng, K.-C. Determination of the origin of the N-terminal pyro-glutamate variation in monoclonal antibodies using model peptides. Biotechnol. Bioeng. 2007, 97, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Cerini, F.; Gaertner, H.; Madden, K.; Tolstorukov, I.; Brown, S.; Laukens, B.; Callewaert, N.; Harner, J.C.; Oommen, A.M.; Harms, J.T.; et al. A scalable low-cost cGMP process for clinical grade production of the HIV inhibitor 5P12-RANTES in Pichia pastoris. Protein Expr. Purif. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towards NMR Analysis of the HIV-1 Coreceptor CCR5 and Its Interaction with RANTES. Available online: http://edoc.unibas.ch/30282/ (accessed on 17 July 2017).
- Grewe, C.; Beck, A.; Gelderblom, H.R. HIV: Early virus-cell interactions. J. Acquir. Immune Defic. Syndr. 1990, 3, 965–974. [Google Scholar] [PubMed]
- Allen, S.J.; Crown, S.E.; Handel, T.M. Chemokine:Receptor Structure, Interactions, and Antagonism. Annu. Rev. Immunol. 2007, 25, 787–820. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Brown, A.; Cribbes, S.; Randle, E.; Czaplewski, L.G. Aggregation of RANTES Is Responsible for Its Inflammatory Properties: Characterization of Nonaggregating, Noninflammatory RANTES Mutants. J. Biol. Chem. 1999, 274, 27505–27512. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Hangartner, L. Broadly Neutralizing Antibodies to HIV and Their Role in Vaccine Design. Annu. Rev. Immunol. 2016, 34, 635–659. [Google Scholar] [CrossRef] [PubMed]
- Klein, F.; Halper-Stromberg, A.; Horwitz, J.A.; Gruell, H.; Scheid, J.F.; Bournazos, S.; Mouquet, H.; Spatz, L.A.; Diskin, R.; Abadir, A.; et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature 2012, 492, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Gindin, T.; Zhang, B.; Wang, L.; Abel, R.; Murret, C.S.; Xu, F.; Bao, A.; Lu, N.J.; Zhou, T.; et al. Free Energy Perturbation Calculation of Relative Binding Free Energy between Broadly Neutralizing Antibodies and the gp120 Glycoprotein of HIV-1. J. Mol. Biol. 2016, 429, 930–947. [Google Scholar] [CrossRef] [PubMed]
- Pancera, M.; Shahzad-ul-Hussan, S.; Doria-Rose, N.A.; McLellan, J.S.; Bailer, R.T.; Dai, K.; Loesgen, S.; Louder, M.K.; Staupe, R.P.; Yang, Y.; et al. Structural basis for diverse N-glycan recognition by HIV-1–neutralizing V1–V2–directed antibody PG16. Nat. Struct. Mol. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, K.B.; Gray, E.S.; Lambson, B.E.; Moore, P.L.; Choge, I.A.; Mlisana, K.; Karim, S.S.A.; McMahon, J.; O’Keefe, B.; Chikwamba, R.; et al. Mannose-rich glycosylation patterns on HIV-1 subtype C gp120 and sensitivity to the lectins, Griffithsin, Cyanovirin-N and Scytovirin. Virology 2010, 402, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Akkouh, O.; Ng, T.B.; Singh, S.S.; Yin, C.; Dan, X.; Chan, Y.S.; Pan, W.; Cheung, R.C.F. Lectins with anti-HIV activity: A review. Molecules 2015, 20, 648–668. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.; Tam, S.-C.; Zheng, Y.-T. Current peptide HIV type-1 fusion inhibitors. Antivir. Chem. Chemother. 2009, 20, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Mankowski, M.K.; Snyder, B.A.; Ptak, R.G.; Liwang, P.J. Highly potent chimeric inhibitors targeting two steps of HIV cell entry. J. Biol. Chem. 2011, 286, 28370–28381. [Google Scholar] [CrossRef] [PubMed]
- Kagiampakis, I.; Gharibi, A.; Mankowski, M.K.; Snyder, B.A.; Ptak, R.G.; Alatas, K.; LiWang, P.J. Potent Strategy To Inhibit HIV-1 by Binding both gp120 and gp41. Antimicrob. Agents Chemother. 2011, 55, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Kufareva, I.; Holden, L.G.; Wang, C.; Zheng, Y.; Zhao, C.; Fenalti, G.; Wu, H.; Han, G.W.; Cherezov, V.; et al. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 2015, 347, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Herrera, C.; Coburn, J.; Olejniczak, N.; Ziprin, P.; Kaplan, D.L.; LiWang, P.J. Stabilization and sustained release of HIV inhibitors by encapsulation in silk fibroin disks. ACS Biomater. Sci. Eng. 2017. [Google Scholar] [CrossRef]
- Nedellec, R.; Coetzer, M.; Lederman, M.M.; Offord, R.E.; Hartley, O.; Mosier, D.E. Resistance to the CCR5 inhibitor 5P12-RANTES requires a difficult evolution from CCR5 to CXCR4 Coreceptor use. PLoS ONE 2011, 6, e22020. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Boyer, E.G.; Krebs, D.S.S. The Enzymes, 3rd ed.; Academic Press: New York, NY, USA, 1970; pp. 127–130. [Google Scholar]
- Chang, Y.-G.; Kuo, N.-W.; Tseng, R.; LiWang, A. Flexibility of the C-terminal, or CII, ring of KaiC governs the rhythm of the circadian clock of cyanobacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 14431–14436. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1 H, 13 C and 15 N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Garrett, D.S.; Powers, R.; Gronenborn, A.M.; Clore, G.M. A common sense approach to peak picking in two-, three-, and four-dimensional spectra using automatic computer analysis of contour diagrams. J. Magn. Reson. 1991, 95, 214–220. [Google Scholar] [CrossRef]
- Laurence, J.S.; Blanpain, C.; Burgner, J.W.; Parmentier, M.; LiWang, P.J. CC chemokine MIP-1β can function as a monomer and depends on Phe13 for receptor binding. Biochemistry 2000, 39, 3401–3409. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gao, F.; Mascola, J.R.; Stamatatos, L.; Polonis, V.R.; Koutsoukos, M.; Voss, G.; Goepfert, P.; Gilbert, P.; Greene, K.M.; et al. Human Immunodeficiency Virus Type 1 env Clones from Acute and Early Subtype B Infections for Standardized Assessments of Vaccine-Elicited Neutralizing Antibodies. J. Virol. 2005, 79, 10108–10125. [Google Scholar] [CrossRef] [PubMed]
- Derdeyn, C.A.; Decker, J.M.; Bibollet-Ruche, F.; Mokili, J.L.; Muldoon, M.; Denham, S.A.; Heil, M.L.; Kasolo, F.; Musonda, R.; Hahn, B.H.; et al. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 2004, 303, 2019–2022. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Shaw, G.M.; Kappes, J.C.; Wu, X. Emergence of Resistant Human Immunodeficiency Virus Type 1 in Patients Receiving Fusion Inhibitor (T-20) Monotherapy Emergence of Resistant Human Immunodeficiency Virus Type 1 in Patients Receiving Fusion Inhibitor (T-20) Monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Decker, J.M.; Wang, S.; Hui, H.; Kappes, J.C.; Wu, X.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Kilby, J.M.; Saag, M.S.; et al. Antibody neutralization and escape by HIV-1. Nature 2003, 422, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Shen, X.; Baggett, B.R.; Kong, X.; LiWang, P.J. The human CC chemokine MIP-1β dimer is not competent to bind to the CCR5 receptor. J. Biol. Chem. 2007, 282, 27976–27983. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 Values of 5P12 Variants | ||
|---|---|---|
| 5P12 Variant | PVO.4 (nM) | ZM53 (nM) |
| 5P12-RANTES Uncyclized | 1.51 ± 0.10 | 1.61 ± 0.12 |
| 5P12-RANTES Cyclized | 1.27 ± 0.10 | 1.46 ± 0.15 |
| 5P12-RANTES-Q0E Uncyclized | 6.30 ± 0.14 | 4.04 ± 0.29 |
| 5P12-RANTES-Q0E Cyclized | 1.09 ± 0.06 | 1.23 ± 0.09 |
| 5P12-RANTES-Q0N | 2.20 ± 0.33 | 1.31 ± 0.19 |
| 5P12-RANTES-Q0I | 1.21 ± 0.24 | 1.43 ± 0.11 |
| 5P12-RANTES-Q0F | 1.26 ± 0.19 | 1.84 ± 0.12 |
| 5P12-RANTES-Q0L | 1.31 ± 0.12 | 1.45 ± 0.15 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
F. Nguyen, A.; S. Schill, M.; Jian, M.; J. LiWang, P. The Effect of N-Terminal Cyclization on the Function of the HIV Entry Inhibitor 5P12-RANTES. Int. J. Mol. Sci. 2017, 18, 1575. https://doi.org/10.3390/ijms18071575
F. Nguyen A, S. Schill M, Jian M, J. LiWang P. The Effect of N-Terminal Cyclization on the Function of the HIV Entry Inhibitor 5P12-RANTES. International Journal of Molecular Sciences. 2017; 18(7):1575. https://doi.org/10.3390/ijms18071575
Chicago/Turabian StyleF. Nguyen, Anna, Megan S. Schill, Mike Jian, and Patricia J. LiWang. 2017. "The Effect of N-Terminal Cyclization on the Function of the HIV Entry Inhibitor 5P12-RANTES" International Journal of Molecular Sciences 18, no. 7: 1575. https://doi.org/10.3390/ijms18071575
APA StyleF. Nguyen, A., S. Schill, M., Jian, M., & J. LiWang, P. (2017). The Effect of N-Terminal Cyclization on the Function of the HIV Entry Inhibitor 5P12-RANTES. International Journal of Molecular Sciences, 18(7), 1575. https://doi.org/10.3390/ijms18071575