Growth Hormone’s Effect on Adipose Tissue: Quality versus Quantity

Abstract
:1. Introduction
2. Complexity of AT
2.1. Types of AT and Adipocytes
2.2. Endocrine Function
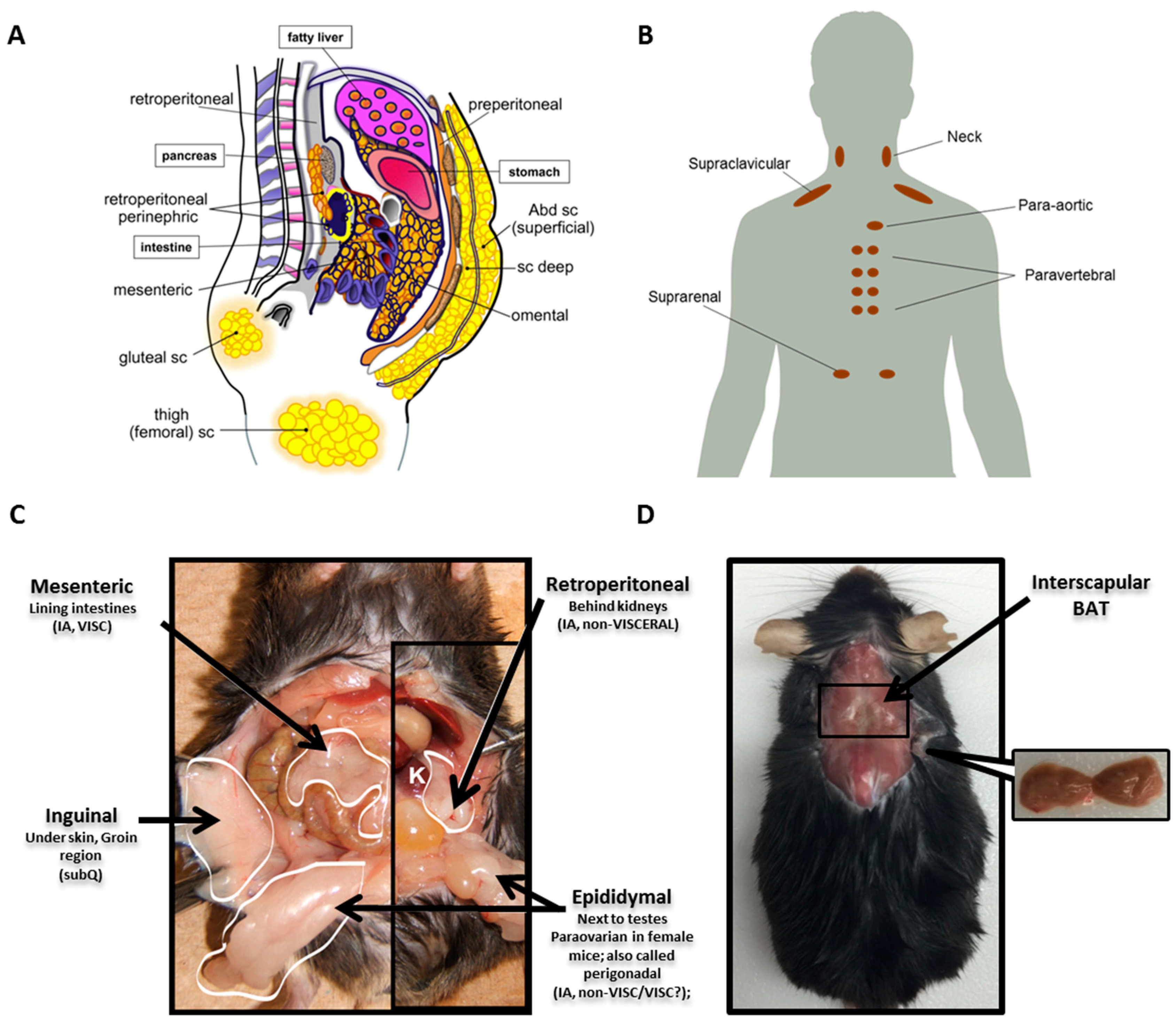
2.3. Depot Differences
2.4. Other Cellular and Non-Cellular Components of AT
3. Clinical Conditions and Mouse Lines with Alterations in GH
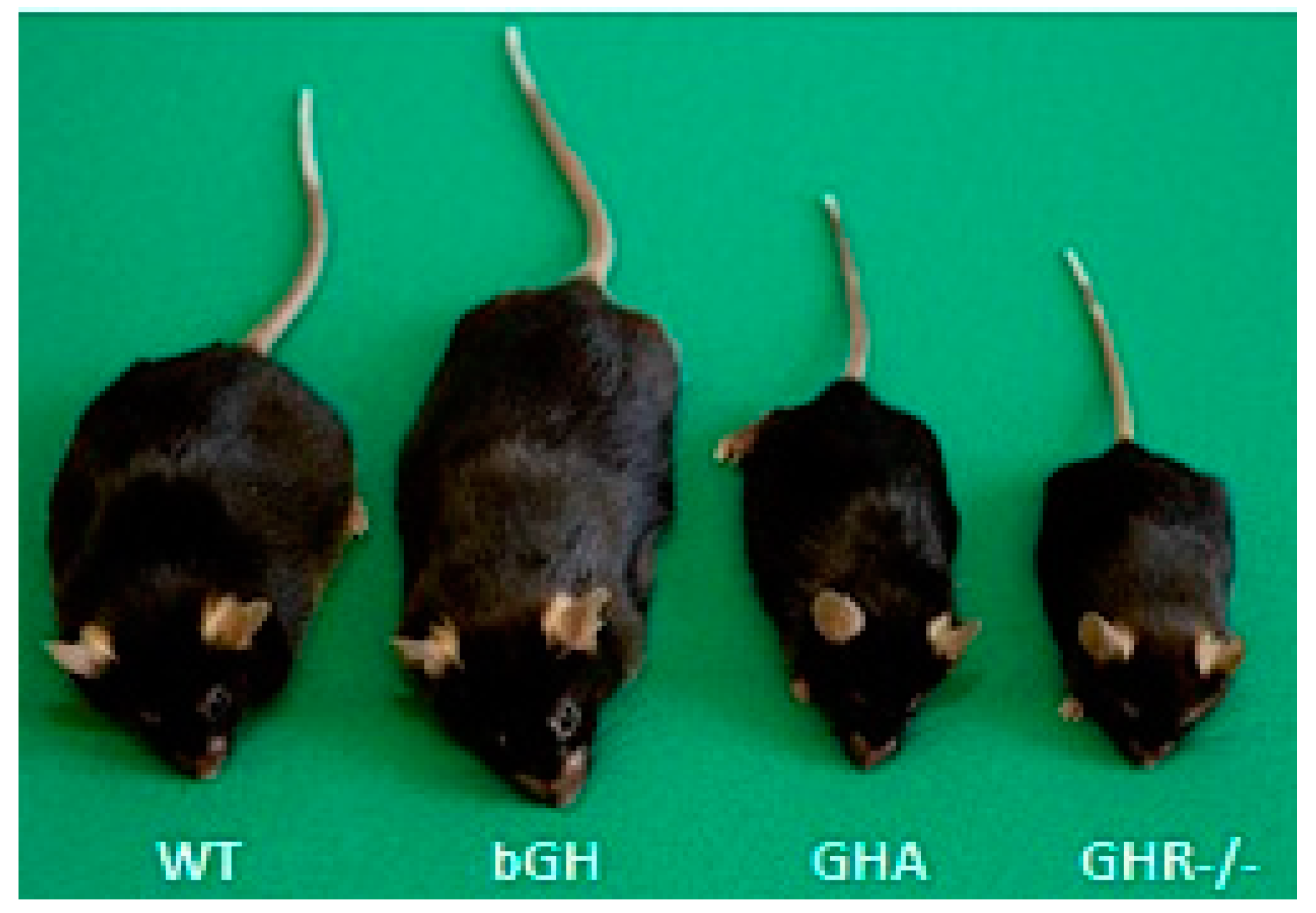
3.1. Elevated GH: Acromegaly and bGH Transgenic Mice
3.2. GH Deficiency: GHD and GHA and Ames Dwarf Mice
3.3. GH Insensitivity: Laron Syndrome, GHR-/- Mice and aGHRKO Mice
4. Adipose Tissue and GH
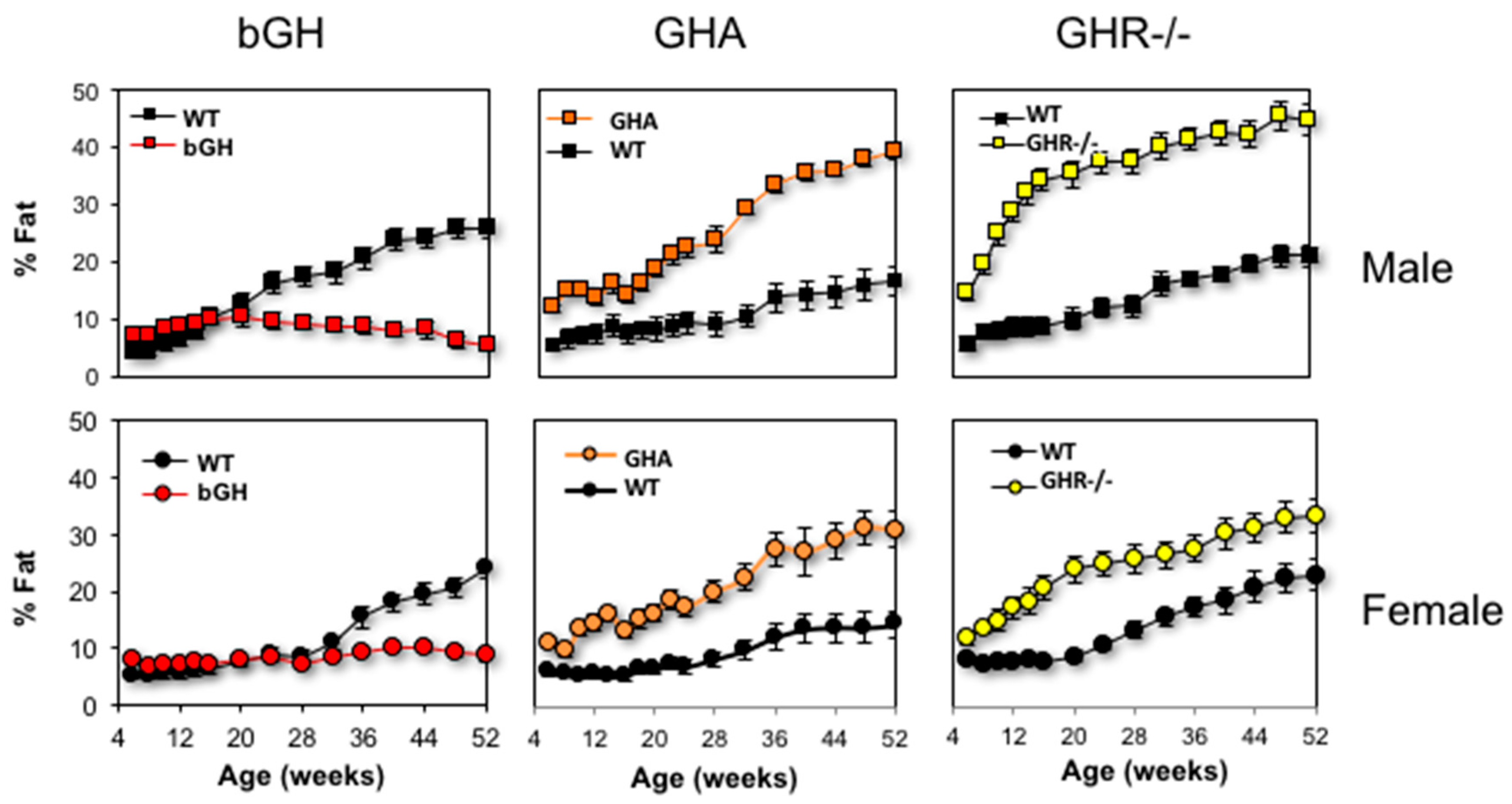
4.1. Body Composition
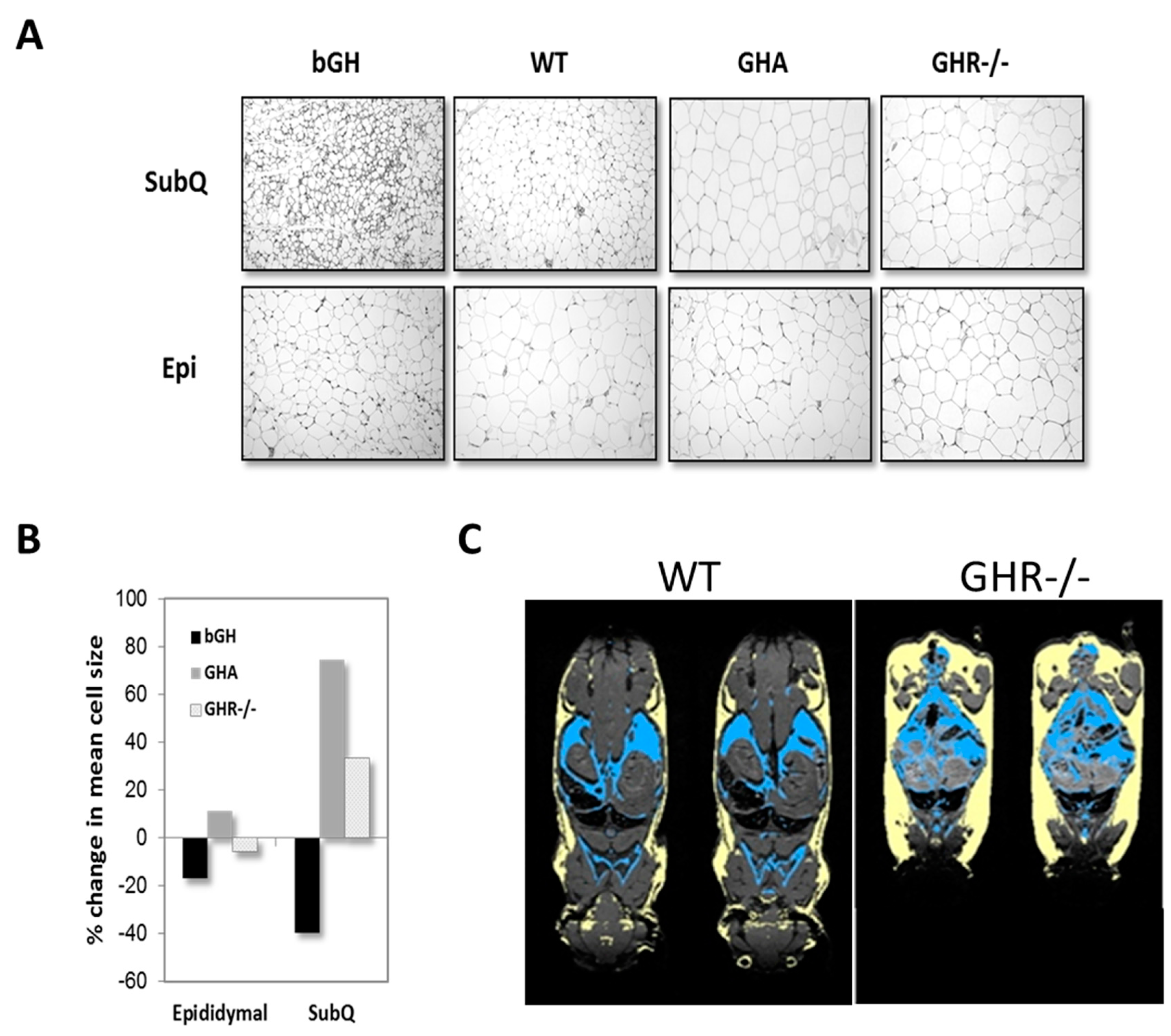
4.2. Depot Specific Differences
4.3. Cellular and Non-Cellular Components Altered by GH
4.4. Brown Adipose Tissue and Beige Adipose Tissue
4.5. Adipokines
5. Does GH or GH Antagonist Have Potential for Treatment of Obesity/Lipodystrophy?
5.1. Obesity
5.2. Lipodystrophy
6. Gaps in Knowledge and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, M.J.; Wu, Y.; Fried, S.K. Adipose tissue heterogeneity: Implication of depot differences in adipose tissue for obesity complications. Mol. Aspects Med. 2013, 34, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef] [PubMed]
- Cohade, C.; Mourtzikos, K.A.; Wahl, R.L. “USA-Fat”: Prevalence is related to ambient outdoor temperature-evaluation with 18F-FDG PET/CT. J. Nucl. Med. 2003, 44, 1267–1270. [Google Scholar] [PubMed]
- Au-Yong, I.T.; Thorn, N.; Ganatra, R.; Perkins, A.C.; Symonds, M.E. Brown adipose tissue and seasonal variation in humans. Diabetes 2009, 58, 2583–2587. [Google Scholar] [CrossRef] [PubMed]
- Giralt, M.; Villarroya, F. White, brown, beige/brite: Different adipose cells for different functions? Endocrinology 2013, 154, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Vitali, A.; Murano, I.; Zingaretti, M.C.; Frontini, A.; Ricquier, D.; Cinti, S. The adipose organ of obesity-prone C57BL/6J mice is composed of mixed white and brown adipocytes. J. Lipid Res. 2012, 53, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.A.; Tao, C.; Gupta, R.K.; Scherer, P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013, 19, 1338–1344. [Google Scholar] [CrossRef] [PubMed]
- Boss, O.; Farmer, S.R. Recruitment of brown adipose tissue as a therapy for obesity-associated diseases. Front. Endocrinol. 2012, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, L.; Lin, J.Z.; Aprahamian, T.R.; Farmer, S.R. Browning of white adipose tissue with roscovitine induces a distinct population of UCP1+ adipocytes. Cell. Metab. 2016, 24, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, K.; Luijten, I.H.; Hasegawa, Y.; Hong, H.; Sonne, S.B.; Kim, M.; Xue, R.; Chondronikola, M.; Cypess, A.M.; Tseng, Y.H.; et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat. Med. 2015, 21, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M.; Patti, M.E.; Gesta, S.; Kahn, B.B.; Kahn, C.R. Intrinsic heterogeneity in adipose tissue of fat-specific insulin receptor knock-out mice is associated with differences in patterns of gene expression. J. Biol. Chem. 2004, 279, 31891–31901. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Laurin, N.; Himms-Hagen, J.; Rudnicki, M.A.; Levy, E.; Robert, M.F.; Pan, L.; Oligny, L.; Mitchell, G.A. The adipose tissue phenotype of hormone-sensitive lipase deficiency in mice. Obes. Res. 2001, 9, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.Y.; Bandiera, R.; Serrels, A.; Martinez-Estrada, O.M.; Qing, W.; Lee, M.; Slight, J.; Thornburn, A.; Berry, R.; McHaffie, S.; et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat. Cell. Biol. 2014, 16, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Lynes, M.D.; Dreyfuss, J.M.; Shamsi, F.; Schulz, T.J.; Zhang, H.; Huang, T.L.; Townsend, K.L.; Li, Y.; Takahashi, H.; et al. Clonal analyses and gene profiling identify genetic biomarkers of the thermogenic potential of human brown and white preadipocytes. Nat. Med. 2015, 21, 760–768. [Google Scholar] [CrossRef] [PubMed]
- McGuire, M.J.; Ishii, M. Leptin dysfunction and Alzheimer's disease: Evidence from cellular, animal, and human studies. Cell. Mol. Neurobiol. 2016, 36, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Abella, V.; Scotece, M.; Conde, J.; Pino, J.; Gonzalez-Gay, M.A.; Gomez-Reino, J.J.; Mera, A.; Lago, F.; Gomez, R.; Gualillo, O. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 2017, 13, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B. Direct and indirect effects of leptin on adipocyte metabolism. Biochim. Biophys. Acta 2014, 1842, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Horikoshi, M.; Yamauchi, T.; Yago, H.; Miyazaki, O.; Ebinuma, H.; Imai, Y.; Nagai, R.; Kadowaki, T. Measurement of the high-molecular weight form of adiponectin in plasma is useful for the prediction of insulin resistance and metabolic syndrome. Diabetes Care 2006, 29, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Hirose, H.; Yamamoto, Y.; Seino-Yoshihara, Y.; Kawabe, H.; Saito, I. Serum high-molecular-weight adiponectin as a marker for the evaluation and care of subjects with metabolic syndrome and related disorders. J Atheroscler. Thromb. 2010, 17, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Pessin, J.E. Adipokines mediate inflammation and insulin resistance. Front. Endocrinol. 2013, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, M.; Maruoka, S.; Katayose, S. Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur. J. Endocrinol. 2002, 147, 173–180. [Google Scholar] [CrossRef] [PubMed]
- De Souza Batista, C.M.; Yang, R. Z.; Lee, M.J.; Glynn, N.M.; Yu, D.Z.; Pray, J.; Ndubuizu, K.; Patil, S.; Schwartz, A.; Kligman, M.; et al. Omentin plasma levels and gene expression are decreased in obesity. Diabetes 2007, 56, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Hana, V.; Silha, J.V.; Justova, V.; Lacinova, Z.; Stepan, J.J.; Murphy, L.J. The effects of GH replacement in adult GH-deficient patients: Changes in body composition without concomitant changes in the adipokines and insulin resistance. Clin. Endocrinol. 2004, 60, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Curat, C.A.; Wegner, V.; Sengenes, C.; Miranville, A.; Tonus, C.; Busse, R.; Bouloumie, A. Macrophages in human visceral adipose tissue: Increased accumulation in obesity and a source of resistin and visfatin. Diabetologia 2006, 49, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, J.; Cereijo, R.; Villarroya, F. An endocrine role for brown adipose tissue? Am. J. Physiol. Endocrinol. Metab. 2013, 305, E567–E572. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.X.; Zhao, X.Y.; Meng, Z.X.; Kern, M.; Dietrich, A.; Chen, Z.; Cozacov, Z.; Zhou, D.; Okunade, A.L.; Su, X.; et al. The brown fat-enriched secreted factor Nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nat. Med. 2014, 20, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Arner, P. Regional adipocity in man. J. Endocrinol. 1997, 155, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. The adipose organ. Prostaglandins Leukot. Essent. Fatty Acids 2005, 73, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Lidell, M.E.; Betz, M.J.; Dahlqvist Leinhard, O.; Heglind, M.; Elander, L.; Slawik, M.; Mussack, T.; Nilsson, D.; Romu, T.; Nuutila, P.; et al. Evidence for two types of brown adipose tissue in humans. Nat. Med. 2013, 19, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Three years with adult human brown adipose tissue. Ann. N. Y. Acad. Sci. 2010, 1212, E20–E36. [Google Scholar] [CrossRef] [PubMed]
- Sackmann-Sala, L.; Berryman, D.E.; Munn, R.D.; Lubbers, E.R.; Kopchick, J.J. Heterogeneity among white adipose tissue depots in male C57BL/6J mice. Obesity 2012, 20, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Berryman, D.E.; List, E.O.; Sackmann-Sala, L.; Lubbers, E.; Munn, R.; Kopchick, J.J. Growth hormone and adipose tissue: Beyond the adipocyte. Growth Horm. IGF Res. 2011, 21, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Troike, K.M.; Henry, B.E.; Jensen, E.A.; Young, J.A.; List, E.O.; Kopchick, J.J.; Berryman, D.E. Impact of Growth Hormone on Regulation of Adipose Tissue. Compr. Physiol. 2017, 7, 819–840. [Google Scholar] [PubMed]
- Kuk, J.L.; Katzmarzyk, P.T.; Nichaman, M.Z.; Church, T.S.; Blair, S.N.; Ross, R. Visceral fat is an independent predictor of all-cause mortality in men. Obesity 2006, 14, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; She, L.; Liu, B.Q.; Vuguin, P.; Cohen, P.; Wang, J.; Rossetti, L. Surgical removal of visceral fat reverses hepatic insulin resistance. Diabetes 1999, 48, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Muzumdar, R.; Allison, D.B.; Huffman, D.M.; Ma, X.; Atzmon, G.; Einstein, F.H.; Fishman, S.; Poduval, A.D.; McVei, T.; Keith, S.W.; et al. Visceral adipose tissue modulates mammalian longevity. Aging Cell 2008, 7, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Kahn, C.R. Transplantation of adipose tissue and stem cells: Role in metabolism and disease. Nat. Rev. Endocrinol. 2010, 6, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Morbeck, D.E.; von Zglinicki, T.; van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raguso, C.A.; Kyle, U.; Kossovsky, M.P.; Roynette, C.; Paoloni-Giacobino, A.; Hans, D.; Genton, L.; Pichard, C. A 3-year longitudinal study on body composition changes in the elderly: Role of physical exercise. Clin. Nutr. 2006, 25, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. The sexual dimorphism of obesity. Mol. Cell. Endocrinol. 2015, 402, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.F.; Chiapa, A.L.; Rodriquez, M.; Phelps, D.R.; Cardarelli, K.M.; Vishwanatha, J.K.; Bae, S.; Cardarelli, R. Visceral fat, waist circumference, and BMI: Impact of race/ethnicity. Obesity 2008, 16, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Katzmarzyk, P.T.; Bray, G.A.; Greenway, F.L.; Johnson, W.D.; Newton, R.L., Jr.; Ravussin, E.; Ryan, D.H.; Smith, S.R.; Bouchard, C. Racial differences in abdominal depot-specific adiposity in white and African American adults. Am. J. Clin. Nutr. 2010, 91, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Barreau, C.; Labit, E.; Guissard, C.; Rouquette, J.; Boizeau, M.L.; Gani Koumassi, S.; Carriere, A.; Jeanson, Y.; Berger-Muller, S.; Dromard, C.; et al. Regionalization of browning revealed by whole subcutaneous adipose tissue imaging. Obesity 2016, 24, 1081–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman-Boehm, I.; Bluher, M.; Redel, H.; Sion-Vardy, N.; Ovadia, S.; Avinoach, E.; Shai, I.; Kloting, N.; Stumvoll, M.; Bashan, N.; et al. Macrophage infiltration into omental versus subcutaneous fat across different populations: Effect of regional adiposity and the comorbidities of obesity. J. Clin. Endocrinol. Metab. 2007, 92, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Kwok, K.H.; Lam, K.S.; Xu, A. Heterogeneity of white adipose tissue: Molecular basis and clinical implications. Exp. Mol. Med. 2016, 48, e215. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Bhatia-Dey, N.; Kanherkar, R.R.; Stair, S.E.; Makarev, E.O.; Csoka, A.B. Cellular Senescence as the Causal Nexus of Aging. Front. Genet. 2016, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Tchkonia, T.; LeBrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 2015, 64, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J. Clin. Investig. 2017, 127, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Stout, M.B.; Justice, J.N.; Nicklas, B.J.; Kirkland, J.L. Physiological aging: Links among adipose tissue dysfunction, diabetes, and frailty. Physiology 2017, 32, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Maquoi, E.; Munaut, C.; Colige, A.; Collen, D.; Lijnen, H.R. Modulation of adipose tissue expression of murine matrix metalloproteinases and their tissue inhibitors with obesity. Diabetes 2002, 51, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.; Unal, R.; Zhu, B.; Rasouli, N.; McGehee, R.E., Jr.; Peterson, C.A.; Kern, P.A. Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. J. Clin. Endocrinol. Metab. 2011, 96, E1990–E1998. [Google Scholar] [CrossRef] [PubMed]
- Pasarica, M.; Gowronska-Kozak, B.; Burk, D.; Remedios, I.; Hymel, D.; Gimble, J.; Ravussin, E.; Bray, G.A.; Smith, S.R. Adipose tissue collagen VI in obesity. J. Clin. Endocrinol. Metab. 2009, 94, 5155–5162. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Muise, E.S.; Iyengar, P.; Wang, Z.V.; Chandalia, M.; Abate, N.; Zhang, B.B.; Bonaldo, P.; Chua, S.; Scherer, P.E. Metabolic dysregulation and adipose tissue fibrosis: Role of collagen VI. Mol. Cell. Biol. 2009, 29, 1575–1591. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z.; Ginsberg, S.; Lilos, P.; Arbiv, M.; Vaisman, N. Body composition in untreated adult patients with Laron syndrome (primary GH insensitivity). Clin. Endocrinol. 2006, 65, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S. Acromegaly pathogenesis and treatment. J. Clin. Invest. 2009, 119, 3189–3202. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S. Pathogenesis of pituitary tumors. Nat. Rev. Endocrinol. 2011, 7, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Katznelson, L.; Laws, E.R., Jr.; Melmed, S.; Molitch, M.E.; Murad, M.H.; Utz, A.; Wass, J.A. Acromegaly: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 3933–3951. [Google Scholar] [CrossRef] [PubMed]
- Abreu, A.; Tovar, A.P.; Castellanos, R.; Valenzuela, A.; Giraldo, C.M.; Pinedo, A.C.; Guerrero, D.P.; Barrera, C.A.; Franco, H.I.; Ribeiro-Oliveira, A., Jr.; et al. Challenges in the diagnosis and management of acromegaly: A focus on comorbidities. Pituitary 2016, 19, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Colao, A.; Ferone, D.; Marzullo, P.; Lombardi, G. Systemic complications of acromegaly: Epidemiology, pathogenesis, and management. Endocr. Rev. 2004, 25, 102–152. [Google Scholar] [CrossRef] [PubMed]
- Katznelson, L.; Atkinson, J.L.; Cook, D.M.; Ezzat, S.Z.; Hamrahian, A.H.; Miller, K.K. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the Diagnosis and Treatment of Acromegaly—2011 update: Executive summary. Endocr. Pract. 2011, 17, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Rokkas, T.; Pistiolas, D.; Sechopoulos, P.; Margantinis, G.; Koukoulis, G. Risk of colorectal neoplasm in patients with acromegaly: A meta-analysis. World J. Gastroenterol. 2008, 14, 3484–3489. [Google Scholar] [CrossRef] [PubMed]
- Rogozinski, A.; Furioso, A.; Glikman, P.; Junco, M.; Laudi, R.; Reyes, A.; Lowenstein, A. Thyroid nodules in acromegaly. Arq. Bras. Endocrinol. Metabol. 2012, 56, 300–304. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, M.C.; Nascimento, G.C.; Nascimento, A.G.; Carvalho, V.C.; Lopes, M.H.; Montenegro, R.; Montenegro, R., Jr.; Vilar, L.; Albano, M.F.; Alves, A.R.; et al. Thyroid cancer in patients with acromegaly: A case-control study. Pituitary 2013, 16, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, P.J. Cancers associated with acromegaly. Neuroendocrinology 2006, 83, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Kopchick, J.J.; Laron, Z. Is the Laron Mouse an Accurate Model of Laron Syndrome? Mol. Genet. Metab. 1999, 68, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Berryman, D.E.; List, E.O.; Coschigano, K.T.; Behar, K.; Kim, J.K.; Kopchick, J.J. Comparing adiposity profiles in three mouse models with altered GH signaling. Growth Horm. IGF Res. 2004, 14, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.J.; Chung, M.Y.; List, E.O.; Walker, J.; Okada, S.; Kopchick, J.J.; Berryman, D.E. Age-related changes in body composition of bovine growth hormone transgenic mice. Endocrinology 2009, 150, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Berryman, D.E.; List, E.O.; Kohn, D.T.; Coschigano, K.T.; Seeley, R.J.; Kopchick, J.J. Effect of growth hormone on susceptibility to diet-induced obesity. Endocrinology 2006, 147, 2801–2808. [Google Scholar] [CrossRef] [PubMed]
- Kopchick, J.J.; List, E.O.; Kelder, B.; Gosney, E.S.; Berryman, D.E. Evaluation of growth hormone (GH) action in mice: Discovery of GH receptor antagonists and clinical indications. Mol. Cell. Endocrinol. 2014, 386, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Miquet, J.G.; Freund, T.; Martinez, C.S.; Gonzalez, L.; Diaz, M.E.; Micucci, G.P.; Zotta, E.; Boparai, R.K.; Bartke, A.; Turyn, D.; et al. Hepatocellular alterations and dysregulation of oncogenic pathways in the liver of transgenic mice overexpressing growth hormone. Cell Cycle 2013, 12, 1042–1057. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A. Can growth hormone (GH) accelerate aging? Evidence from GH-transgenic mice. Neuroendocrinology 2003, 78, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Mullis, P.E. Genetics of growth hormone deficiency. Endocrinol. Metab. Clin. N. Am. 2007, 36, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Alatzoglou, K.S.; Webb, E.A.; le Tissier, P.; Dattani, M.T. Isolated growth hormone deficiency (GHD) in childhood and adolescence: Recent advances. Endocr. Rev. 2014, 35, 376–432. [Google Scholar] [CrossRef] [PubMed]
- Smuel, K.; Kauli, R.; Lilos, P.; Laron, Z. Growth, development, puberty and adult height before and during treatment in children with congenital isolated growth hormone deficiency. Growth Horm. IGF Res. 2015, 25, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Molitch, M.E.; Clemmons, D.R.; Malozowski, S.; Merriam, G.R.; Vance, M.L. Evaluation and treatment of adult growth hormone deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1587–1609. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.L.; Merriam, G.R.; Kargi, A.Y. Adult growth hormone deficiency—Benefits, side effects, and risks of growth hormone replacement. Front Endocrinol 2013, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Wight, D.C.; Mehta, B.V.; Wagner, T.E.; Kopchick, J.J. Glycine 119 of bovine growth hormone is critical for growth-promoting activity. Mol. Endocrinol. 1991, 5, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Berryman, D.E.; Lubbers, E.R.; Magon, V.; List, E.O.; Kopchick, J.J. A dwarf mouse model with decreased GH/IGF-1 activity that does not experience life-span extension: Potential impact of increased adiposity, leptin, and insulin with advancing age. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Coschigano, K.T.; Holland, A.N.; Riders, M.E.; List, E.O.; Flyvbjerg, A.; Kopchick, J.J. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin and IGF-1 levels and increased lifespan. Endocrinology 2003, 144, 3799–3810. [Google Scholar] [CrossRef] [PubMed]
- Sornson, M.W.; Wu, W.; Dasen, J.S.; Flynn, S.E.; Norman, D.J.; O’Connell, S.M.; Gukovsky, I.; Carriere, C.; Ryan, A.K.; Miller, A.P.; et al. Pituitary lineage determination by the Prophet of Pit-1 homeodomain factor defective in Ames dwarfism. Nature 1996, 384, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A.; Wright, J.C.; Mattison, J.A.; Ingram, D.K.; Miller, R.A.; Roth, G.S. Extending the lifespan of long-lived mice. Nature 2001, 414, 412. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, M.S.; Rocha, J.S.; Masternak, M.M.; al Regaiey, K.A.; Bartke, A. Targeted disruption of growth hormone receptor interferes with the beneficial actions of calorie restriction. Proc. Natl. Acad. Sci. USA 2006, 103, 7901–7905. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Crenshaw, E.B., 3rd; Rawson, E.J.; Simmons, D.M.; Swanson, L.W.; Rosenfeld, M.G. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU-domain gene pit-1. Nature 1990, 347, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Flurkey, K.; Papaconstantinou, J.; Miller, R.A.; Harrison, D.E. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc. Natl. Acad. Sci. USA 2001, 98, 6736–6741. [Google Scholar] [CrossRef] [PubMed]
- Luque, R.M.; Lin, Q.; Cordoba-Chacon, J.; Subbaiah, P.V.; Buch, T.; Waisman, A.; Vankelecom, H.; Kineman, R.D. Metabolic Impact of Adult-Onset, Isolated, Growth Hormone Deficiency (AOiGHD) Due to Destruction of Pituitary Somatotropes. PLoS ONE 2011, 6, e15767. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z.; Pertzelan, A.; Karp, M. Pituitary dwarfism with high serum levels of growth hormone. Isr. J. Med. Sci. 1968, 4, 883–894. [Google Scholar] [PubMed]
- Laron, Z.; Kauli, R. Fifty seven years of follow-up of the Israeli cohort of Laron Syndrome patients-From discovery to treatment. Growth Horm. IGF Res. 2016, 28, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Agladioglu, S.Y.; Cetinkaya, S.; Savas Erdeve, S.; Onder, A.; Kendirci, H.N.; Bas, V.N.; Aycan, Z. Diabetes mellitus with Laron syndrome: Case report. J. Pediatr. Endocrinol. Metab. 2013, 26, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Rosenbloom, A.L.; Balasubramanian, P.; Teran, E.; Guevara-Aguirre, M.; Guevara, C.; Procel, P.; Alfaras, I.; de Cabo, R.; Di Biase, S.; et al. GH receptor deficiency in Ecuadorian adults is associated with obesity and enhanced insulin sensitivity. J. Clin. Endocrinol. Metab. 2015, 100, 2589–2596. [Google Scholar] [CrossRef] [PubMed]
- Shevah, O.; Laron, Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: A preliminary report. Growth Horm. IGF Res. 2007, 17, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Steuerman, R.; Shevah, O.; Laron, Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur. J. Endocrinol. 2011, 164, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef] [PubMed]
- Kopchick, J.J.; Laron, Z. Laron Syndrome—From Man to Mouse; Laron, Z., Kopchick, J., Eds.; Springer: Berlin, Germany, 2011. [Google Scholar]
- Zhou, Y.; Xu, B.C.; Maheshwari, H.G.; He, L.; Reed, M.; Lozykowski, M.; Okada, S.; Cataldo, L.; Coschigamo, K.; Wagner, T.E.; et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc. Natl. Acad. Sci. USA 1997, 94, 13215–13220. [Google Scholar] [CrossRef] [PubMed]
- Berryman, D.E.; List, E.O.; Palmer, A.J.; Chung, M.Y.; Wright-Piekarski, J.; Lubbers, E.; O’Connor, P.; Okada, S.; Kopchick, J.J. Two-year body composition analyses of long-lived GHR null mice. J. Gerontol. A. Biol. Sci. Med. Sci. 2010, 65, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Lubbers, E.R.; List, E.O.; Jara, A.; Sackman-Sala, L.; Cordoba-Chacon, J.; Gahete, M.D.; Kineman, R.D.; Boparai, R.; Bartke, A.; Kopchick, J.J.; et al. Adiponectin in mice with altered GH action: Links to insulin sensitivity and longevity? J. Endocrinol. 2013, 216, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Ikeno, Y.; Hubbard, G.B.; Lee, S.; Cortez, L.A.; Lew, C.M.; Webb, C.R.; Berryman, D.E.; List, E.O.; Kopchick, J.J.; Bartke, A. Reduced incidence and delayed occurrence of fatal neoplastic diseases in growth hormone receptor/binding protein knockout mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Brown-Borg, H.M.; Rakoczy, S.G.; Sharma, S.; Bartke, A. Long-living growth hormone receptor knockout mice: Potential mechanisms of altered stress resistance. Exp. Gerontol. 2009, 44, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Junnila, R.K.; Duran-Ortiz, S.; Suer, O.; Sustarsic, E.G.; Berryman, D.E.; List, E.O.; Kopchick, J.J. Disruption of the GH Receptor Gene in Adult Mice Increases Maximal Lifespan in Females. Endocrinology 2016, 157, 4502–4513. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, B.A.; Brummer, R.J.; Eden, S.; Bosaeus, I.; Lindstedt, G. Body composition in acromegaly: The effect of treatment. Clin. Endocrinol. 1989, 31, 481–490. [Google Scholar] [CrossRef]
- Moller, N.; Jorgensen, J.O. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr. Rev. 2009, 30, 152–177. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, B.A.; Brummer, R.J.; Eden, S.; Bosaeus, I. Body composition in acromegaly. Clin. Endocrinol. 1989, 30, 121–130. [Google Scholar] [CrossRef]
- Gibney, J.; Wolthers, T.; Burt, M.G.; Leung, K.C.; Umpleby, A.M.; Ho, K.K. Protein metabolism in acromegaly: Differential effects of short- and long-term treatment. J. Clin. Endocrinol. Metab. 2007, 92, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Kaps, M.; Moura, A.S.; Safranski, T.J.; Lamberson, W.R. Components of growth in mice hemizygous for a MT/bGH transgene. J. Anim. Sci. 1999, 77, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, F.; Lochmuller, E.M.; Koller, B.; Wehr, U.; Weusten, A.; Rambeck, W.; Hoeflich, A.; Wolf, E. Body composition, bone mass and microstructural analysis in GH-transgenic mice reveals that skeletal changes are specific to bone compartment and gender. Growth Horm. IGF Res. 2002, 12, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Bohlooly, Y.M.; Fitzgerald, S.M.; Frick, F.; Ljungberg, A.; Ahren, B.; Tornell, J.; Bergstrom, G.; Oscarsson, J. Bovine growth hormone transgenic mice are resistant to diet-induced obesity but develop hyperphagia, dyslipidemia, and diabetes on a high-fat diet. Endocrinology 2005, 146, 920–930. [Google Scholar] [CrossRef] [PubMed]
- List, E.O.; Palmer, A.J.; Berryman, D.E.; Bower, B.; Kelder, B.; Kopchick, J.J. Growth hormone improves body composition, fasting blood glucose, glucose tolerance and liver triacylglycerol in a mouse model of diet-induced obesity and type 2 diabetes. Diabetologia 2009, 52, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Boot, A.M.; Engels, M.A.; Boerma, G.J.; Krenning, E.P.; de Muinck Keizer-Schrama, S.M. Changes in bone mineral density, body composition, and lipid metabolism during growth hormone (GH) treatment in children with GH deficiency. J. Clin. Endocrinol. Metab. 1997, 82, 2423–2428. [Google Scholar] [CrossRef] [PubMed]
- de Boer, H.; Blok, G.J.; Voerman, H.J.; de Vries, P.M.; van der Veen, E.A. Body composition in adult growth hormone-deficient men, assessed by anthropometry and bioimpedance analysis. J. Clin. Endocrinol. Metab. 1992, 75, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Arnao, J.; Jabbar, A.; Fulcher, K.; Besser, G.M.; Ross, R.J. Effects of growth hormone replacement on physical performance and body composition in GH deficient adults. Clin. Endocrinol. 1999, 51, 53–60. [Google Scholar] [CrossRef]
- Bartke, A.; Westbrook, R. Metabolic characteristics of long-lived mice. Front. Genet. 2012, 3, 288. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.M.; Fang, Y.; Miquet, J.G.; Sun, L.Y.; Masternak, M.M.; Bartke, A. Long-lived hypopituitary Ames dwarf mice are resistant to the detrimental effects of high-fat diet on metabolic function and energy expenditure. Aging Cell 2016, 15, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Heiman, M.L.; Tinsley, F.C.; Mattison, J.A.; Hauck, S.; Bartke, A. Body composition of prolactin-, growth hormone-, and thyrotropin-deficient Ames dwarf mice. Endocrine 2003, 20, 149–154. [Google Scholar] [CrossRef]
- Robertson, K.; Kopchick, J.J.; Liu, J.L. Growth hormone receptor gene deficiency causes delayed insulin responsiveness in skeletal muscles without affecting compensatory islet cell overgrowth in obese mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E491–E498. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Householder, L.A.; Lubbers, E.R.; List, E.O.; Troike, K.; Vesel, C.; Duran-Ortiz, S.; Kopchick, J.J.; Berryman, D.E. Growth hormone receptor antagonist transgenic mice are protected from hyperinsulinemia and glucose intolerance despite obesity when placed on a HF diet. Endocrinology 2015, 156, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Freda, P.U.; Shen, W.; Heymsfield, S.B.; Reyes-Vidal, C.M.; Geer, E.B.; Bruce, J.N.; Gallagher, D. Lower visceral and subcutaneous but higher intermuscular adipose tissue depots in patients with growth hormone and insulin-like growth factor I excess due to acromegaly. J. Clin. Endocrinol. Metab. 2008, 93, 2334–2343. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, B.A.; Eden, S.; Lonn, L.; Kvist, H.; Stokland, A.; Lindstedt, G.; Bosaeus, I.; Tolli, J.; Sjostrom, L.; Isaksson, O.G. Treatment of adults with growth hormone (GH) deficiency with recombinant human GH. J. Clin. Endocrinol. Metab. 1993, 76, 309–317. [Google Scholar] [PubMed]
- Benencia, F.; Harshman, S.; Duran-Ortiz, S.; Lubbers, E.R.; List, E.O.; Householder, L.; Alnaeeli, M.; Liang, X.; Welch, L.; Kopchick, J.J.; Berryman, D.E. Male bovine GH transgenic mice have decreased adiposity with an adipose depot-specific increase in immune cell populations. Endocrinology 2015, 156, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Masternak, M.M.; Bartke, A.; Wang, F.; Spong, A.; Gesing, A.; Fang, Y.; Salmon, A.B.; Hughes, L.F.; Liberati, T.; Boparai, R.; et al. Metabolic effects of intra-abdominal fat in GHRKO mice. Aging Cell 2012, 11, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.; Zhi, X.; Hossain, T.; Bartke, A.; Spong, A.; Gesing, A.; Masternak, M.M. The contribution of visceral fat to improved insulin signaling in Ames dwarf mice. Aging Cell 2014, 13, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Bennis, M.T.; Schneider, A.; Victoria, B.; Do, A.; Wiesenborn, D.S.; Spinel, L.; Gesing, A.; Kopchick, J.J.; Siddiqi, S.A.; Masternak, M.M. The role of transplanted visceral fat from the long-lived growth hormone receptor knockout mice on insulin signaling. Geroscience 2017, 39, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Kelder, B.; Berryman, D.E.; Clark, R.; Li, A.; List, E.O.; Kopchick, J.J. CIDE-A gene expression is decreased in white adipose tissue of growth hormone receptor/binding protein gene disrupted mice and with high-fat feeding of normal mice. Growth Horm. IGF Res. 2007, 17, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Sackmann-Sala, L.; Berryman, D.E.; Lubbers, E.R.; Zhang, H.; Vesel, C.B.; Troike, K.M.; Gosney, E.S.; List, E.O.; Kopchick, J.J. Age-related and depot-specific changes in white adipose tissue of growth hormone receptor-null mice. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 69, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Stout, M.B.; Tchkonia, T.; Pirtskhalava, T.; Palmer, A.K.; List, E.O.; Berryman, D.E.; Lubbers, E.R.; Escande, C.; Spong, A.; Masternak, M.M.; et al. Growth hormone action predicts age-related white adipose tissue dysfunction and senescent cell burden in mice. Aging 2014, 6, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Stout, M.B.; Swindell, W.R.; Zhi, X.; Rohde, K.; List, E.O.; Berryman, D.E.; Kopchick, J.J.; Gesing, A.; Fang, Y.; Masternak, M.M. Transcriptome profiling reveals divergent expression shifts in brown and white adipose tissue from long-lived GHRKO mice. Oncotarget 2015, 6, 26702–26715. [Google Scholar] [CrossRef] [PubMed]
- Olarescu, N.C.; Berryman, D.E.; Householder, L.A.; Lubbers, E.R.; List, E.O.; Benencia, F.; Kopchick, J.J.; Bollerslev, J. GH action influences adipogenesis of mouse adipose tissue-derived mesenchymal stem cells. J. Endocrinol. 2015, 226, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.Y.; Lobie, P.E. The mechanism of effect of growth hormone on preadipocyte and adipocyte function. Obes. Rev. 2000, 1, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Flint, D.J.; Binart, N.; Boumard, S.; Kopchick, J.J.; Kelly, P. Developmental aspects of adipose tissue in GH receptor and prolactin receptor gene disrupted mice: Site-specific effects upon proliferation, differentiation and hormone sensitivity. J. Endocrinol. 2006, 191, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Comisford, R.; Lubbers, E.R.; Householder, L.A.; Suer, O.; Tchkonia, T.; Kirkland, J.L.; List, E.O.; Kopchick, J.J.; Berryman, D.E. Growth hormone receptor antagonist transgenic mice have increased subcutaneous adipose tissue mass, altered glucose homeostasis and no change in white adipose tissue cellular senescence. Gerontology 2016, 62, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, O.; Goldberg, E.L.; Camell, C.D.; Youm, Y.H.; Kopchick, J.J.; Nguyen, K.Y.; Bartke, A.; Sun, L.Y.; Dixit, V.D. Growth hormone receptor deficiency protects against age-related NLRP3 inflammasome activation and immune senescence. Cell Rep. 2016, 14, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Longobardi, S.; Keay, N.; Ehrnborg, C.; Cittadini, A.; Rosen, T.; Dall, R.; Boroujerdi, M.A.; Bassett, E.E.; Healy, M.L.; Pentecost, C.; et al. Growth hormone (GH) effects on bone and collagen turnover in healthy adults and its potential as a marker of GH abuse in sports: A double blind, placebo-controlled study. J. Clin. Endocrinol. Metab. 2000, 85, 1505–1512. [Google Scholar] [PubMed]
- Doessing, S.; Holm, L.; Heinemeier, K.M.; Feldt-Rasmussen, U.; Schjerling, P.; Qvortrup, K.; Larsen, J.O.; Nielsen, R.H.; Flyvbjerg, A.; Kjaer, M. GH and IGF1 levels are positively associated with musculotendinous collagen expression: Experiments in acromegalic and GH deficiency patients. Eur. J. Endocrinol. 2010, 163, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.T.; Cowley, M.J.; Lee, P.; Birzniece, V.; Kaplan, W.; Ho, K.K. Identification of novel GH-regulated pathway of lipid metabolism in adipose tissue: A gene expression study in hypopituitary men. J. Clin. Endocrinol. Metab. 2011, 96, E1188–E1196. [Google Scholar] [CrossRef] [PubMed]
- Berryman, D.E.; Householder, L.A.; Comisford, R.; Troike, K.; Wilson, C.; Henry, B.E.; List, E.O.; Kopchick, J.J. Increasing fibrosis: A novel means by which growth hormone may limit white adipose tissue (WAT) expansion. Growth Horm. IGF Res. 2016, 30 (Suppl. S1), 47. [Google Scholar]
- Darcy, J.; McFadden, S.; Fang, Y.; Huber, J.A.; Zhang, C.; Sun, L.Y.; Bartke, A. Brown adipose tissue function is enhanced in long-lived, male Ames dwarf mice. Endocrinology 2016, 157, 4744–4753. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Knapp, J.R.; Kopchick, J.J. Enlargement of interscapular brown adipose tissue in growth hormone antagonist transgenic and in growth hormone receptor gene-disrupted dwarf mice. Exp. Biol. Med. 2003, 228, 207–215. [Google Scholar] [CrossRef]
- Hioki, C.; Yoshida, T.; Kogure, A.; Takakura, Y.; Umekawa, T.; Yoshioka, K.; Shimatsu, A.; Yoshikawa, T. Effects of growth hormone (GH) on mRNA levels of uncoupling proteins 1, 2, and 3 in brown and white adipose tissues and skeletal muscle in obese mice. Horm. Metab. Res. 2004, 36, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.S.; Xu, A.; Tan, K.C.; Wong, L.C.; Tiu, S.C.; Tam, S. Serum adiponectin is reduced in acromegaly and normalized after correction of growth hormone excess. J. Clin. Endocrinol. Metab. 2004, 89, 5448–5453. [Google Scholar] [CrossRef] [PubMed]
- Silha, J.V.; Krsek, M.; Hana, V.; Marek, J.; Jezkova, J.; Weiss, V.; Murphy, L.J. Perturbations in adiponectin, leptin and resistin levels in acromegaly: Lack of correlation with insulin resistance. Clin. Endocrinol. 2003, 58, 736–742. [Google Scholar] [CrossRef]
- Sucunza, N.; Barahona, M.J.; Resmini, E.; Fernandez-Real, J.M.; Ricart, W.; Farrerons, J.; Rodriguez Espinosa, J.; Marin, A.M.; Puig, T.; Webb, S.M. A link between bone mineral density and serum adiponectin and visfatin levels in acromegaly. J. Clin. Endocrinol. Metab. 2009, 94, 3889–3896. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Vidal, C.; Fernandez, J.C.; Bruce, J.N.; Crisman, C.; Conwell, I.M.; Kostadinov, J.; Geer, E.B.; Post, K.D.; Freda, P.U. Prospective study of surgical treatment of acromegaly: Effects on ghrelin, weight, adiposity, and markers of CV risk. J. Clin. Endocrinol. Metab. 2014, 99, 4124–4132. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Al-Regaiey, K.A.; Masternak, M.M.; Bartke, A. Adipocytokines and lipid levels in Ames dwarf and calorie-restricted mice. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Kanety, H.; Hemi, R.; Ginsberg, S.; Pariente, C.; Yissachar, E.; Barhod, E.; Funahashi, T.; Laron, Z. Total and high molecular weight adiponectin are elevated in patients with Laron syndrome despite marked obesity. Eur. J. Endocrinol. 2009, 161, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Stenholm, S.; Metter, E.J.; Roth, G.S.; Ingram, D.K.; Mattison, J.A.; Taub, D.D.; Ferrucci, L. Relationship between plasma ghrelin, insulin, leptin, interleukin 6, adiponectin, testosterone and longevity in the Baltimore longitudinal study of aging. Aging Clin. Exp. Res. 2011, 23, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Takayama, M.; Abe, Y.; Hirose, N. Adipokines and aging. J Atheroscler. Thromb. 2011, 18, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Ciresi, A.; Amato, M.C.; Pizzolanti, G.; Giordano, C. Serum visfatin levels in acromegaly: Correlation with disease activity and metabolic alterations. Growth Horm. IGF Res. 2015, 25, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciresi, A.; Pizzolanti, G.; Leotta, M.; Guarnotta, V.; Teresi, G.; Giordano, C. Resistin, visfatin, leptin and omentin are differently related to hormonal and metabolic parameters in growth hormone-deficient children. J. Endocrinol. Investig. 2016, 39, 1023–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meazza, C.; Elsedfy, H.H.; Pagani, S.; Bozzola, E.; el Kholy, M.; Bozzola, M. Metabolic parameters and adipokine profile in growth hormone deficient (GHD) children before and after 12-month GH treatment. Horm. Metab. Res. 2014, 46, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Brooks, N.E.; Hjortebjerg, R.; Henry, B.E.; List, E.O.; Kopchick, J.J.; Berryman, D.E. Fibroblast growth factor 21, fibroblast growth factor receptor 1, and beta-Klotho expression in bovine growth hormone transgenic and growth hormone receptor knockout mice. Growth Horm. IGF Res. 2016, 30–31, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.H. Obesity, growth hormone and weight loss. Mol. Cell. Endocrinol. 2010, 316, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.H.; Hvidberg, A.; Juul, A.; Main, K.M.; Gotfredsen, A.; Skakkebaek, N.E.; Hilsted, J.; Skakkebae, N.E. Massive weight loss restores 24-hour growth hormone release profiles and serum insulin-like growth factor-I levels in obese subjects. J. Clin. Endocrinol. Metab. 1995, 80, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Berryman, D.E.; Glad, C.A.; List, E.O.; Johannsson, G. The GH/IGF-1 axis in obesity: Pathophysiology and therapeutic considerations. Nat. Rev. Endocrinol. 2013, 9, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Lewitt, M.S. The Role of The Growth Hormone/Insulin-Like Growth Factor System in Visceral Adiposity. Biochem. Insights 2017, 10, 1178626417703995. [Google Scholar] [CrossRef] [PubMed]
- Johannsson, G.; Marin, P.; Lonn, L.; Ottosson, M.; Stenlof, K.; Bjorntorp, P.; Sjostrom, L.; Bengtsson, B. A. Growth hormone treatment of abdominally obese men reduces abdominal fat mass, improves glucose and lipoprotein metabolism, and reduces diastolic blood pressure. J. Clin. Endocrinol. Metab. 1997, 82, 727–734. [Google Scholar] [PubMed]
- Skaggs, S.R.; Crist, D.M. Exogenous human growth hormone reduces body fat in obese women. Horm. Res. 1991, 35, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Richelsen, B.; Pedersen, S.B.; Borglum, J.D.; Moller-Pedersen, T.; Jorgensen, J.; Jorgensen, J.O. Growth hormone treatment of obese women for 5 wk: Effect on body composition and adipose tissue LPL activity. Am. J. Physiol. 1994, 266, E211–E216. [Google Scholar] [PubMed]
- Kim, K.R.; Nam, S.Y.; Song, Y.D.; Lim, S.K.; Lee, H.C.; Huh, K.B. Low-dose growth hormone treatment with diet restriction accelerates body fat loss, exerts anabolic effect and improves growth hormone secretory dysfunction in obese adults. Horm. Res. 1999, 51, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Franco, C.; Brandberg, J.; Lonn, L.; Andersson, B.; Bengtsson, B.A.; Johannsson, G. Growth hormone treatment reduces abdominal visceral fat in postmenopausal women with abdominal obesity: A 12-month placebo-controlled trial. J. Clin. Endocrinol. Metab. 2005, 90, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Bredella, M.A.; Lin, E.; Brick, D.J.; Gerweck, A.V.; Harrington, L.M.; Torriani, M.; Thomas, B.J.; Schoenfeld, D.A.; Breggia, A.; Rosen, C.J.; et al. Effects of GH in women with abdominal adiposity: A 6-month randomized, double-blind, placebo-controlled trial. Eur. J. Endocrinol. 2012, 166, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, D.R.; Snyder, D.K.; Williams, R.; Underwood, L.E. Growth hormone administration conserves lean body mass during dietary restriction in obese subjects. J. Clin. Endocrinol. Metab. 1987, 64, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Snyder, D.K.; Clemmons, D.R.; Underwood, L.E. Treatment of obese, diet-restricted subjects with growth hormone for 11 weeks: Effects on anabolism, lipolysis, and body composition. J. Clin. Endocrinol. Metab. 1988, 67, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Snyder, D.K.; Underwood, L.E.; Clemmons, D.R. Anabolic effects of growth hormone in obese diet-restricted subjects are dose dependent. Am. J. Clin. Nutr. 1990, 52, 431–437. [Google Scholar] [PubMed]
- Tagliaferri, M.; Scacchi, M.; Pincelli, A.I.; Berselli, M.E.; Silvestri, P.; Montesano, A.; Ortolani, S.; Dubini, A.; Cavagnini, F. Metabolic effects of biosynthetic growth hormone treatment in severely energy-restricted obese women. Int. J. Obes. Relat. Metab. Disord. 1998, 22, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Mekala, K.C.; Tritos, N.A. Effects of recombinant human growth hormone therapy in obesity in adults—A metaanalysis. J. Clin. Endocrinol. Metab. 2009, 94, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Shadid, S.; Jensen, M.D. Effects of growth hormone administration in human obesity. Obes. Res. 2003, 11, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Snyder, D.K.; Underwood, L.E.; Clemmons, D.R. Persistent lipolytic effect of exogenous growth hormone during caloric restriction. Am. J. Med. 1995, 98, 129–134. [Google Scholar] [CrossRef]
- Nam, S.Y.; Kim, K.R.; Cha, B.S.; Song, Y.D.; Lim, S.K.; Lee, H.C.; Huh, K.B. Low-dose growth hormone treatment combined with diet restriction decreases insulin resistance by reducing visceral fat and increasing muscle mass in obese type 2 diabetic patients. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.W.; Kim, C.S.; Nam, J.H.; Kim, H.J.; Nam, J.S.; Park, J.S.; Kang, E.S.; Cha, B.S.; Lim, S.K.; Kim, K.R.; et al. Effects of growth hormone on insulin resistance and atherosclerotic risk factors in obese type 2 diabetic patients with poor glycaemic control. Clin. Endocrinol. 2006, 64, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Masternak, M.M.; al-Regaiey, K.A.; Bartke, A. Adipocytokines and the regulation of lipid metabolism in growth hormone transgenic and calorie-restricted mice. Endocrinology 2007, 148, 2845–2853. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Setser, J.; Zhao, H.; Stannard, B.; Haluzik, M.; Glatt, V.; Bouxsein, M.L.; Kopchick, J.J.; leRoith, D. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. J. Clin. Investig. 2004, 113, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A. Impact of reduced insulin-like growth factor-1/insulin signaling on aging in mammals: Novel findings. Aging Cell 2008, 7, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Alderman, J.M.; Flurkey, K.; Brooks, N.L.; Naik, S.B.; Gutierrez, J.M.; Srinivas, U.; Ziara, K.B.; Jing, L.; Boysen, G.; Bronson, R.; et al. Neuroendocrine inhibition of glucose production and resistance to cancer in dwarf mice. Exp. Gerontol. 2009, 44, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Egecioglu, E.; Bjursell, M.; Ljungberg, A.; Dickson, S.L.; Kopchick, J.J.; Bergstrom, G.; Svensson, L.; Oscarsson, J.; Tornell, J.; Bohlooly, Y.M. Growth hormone receptor deficiency results in blunted ghrelin feeding response, obesity, and hypolipidemia in mice. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E317–E325. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, C.G.; Chou, S.H.; Mantzoros, C.S. Lipodystrophy: Pathophysiology and advances in treatment. Nat. Rev. Endocrinol. 2011, 7, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Stanley, T.L.; Grinspoon, S.K. GH/GHRH axis in HIV lipodystrophy. Pituitary 2009, 12, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Wanke, C.; Gerrior, J.; Kantaros, J.; Coakley, E.; Albrecht, M. Recombinant human growth hormone improves the fat redistribution syndrome (lipodystrophy) in patients with HIV. AIDS 1999, 13, 2099–2103. [Google Scholar] [CrossRef] [PubMed]
- Bickel, M.; Zangos, S.; Jacobi, V.; Lutz, T.; Knecht, G.; Goebel, F.; Staszewski, S.; Klauke, S. A randomized, open-label study to compare the effects of two different doses of recombinant human growth hormone on fat reduction and fasting metabolic parameters in HIV-1-infected patients with lipodystrophy. HIV Med. 2006, 7, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Yogi, A.; Ishizuka, N.; Genka, I.; Gatanaga, H.; Teruya, K.; Tachikawa, N.; Kikuchi, Y.; Oka, S. Effectiveness of subcutaneous growth hormone in HIV-1 patients with moderate to severe facial lipoatrophy. Intern. Med. 2007, 46, 359–362. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elevated GH | GH deficiency (GHD) | GH insensitivity | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Clinical | Mouse | Clinical | Mouse | Mouse | Mouse | Clinical | Mouse | Mouse | |
| Acromegaly Gigantism | bGH | GHD * | GHA ** | AOiGHD | Ames | Laron | aGHRKO **** | GHR-/- | |
| GH defect | Hypersecretion of GH commonly due to pituitary adenoma | Transgenic for bovine GH | Many variations depending on age and etiology | Transgenic for GHR antagonist gene | Ablation of somatotrophs with an inducible system | Mutation in Prop1 | Hereditary conditions usually caused by GHR receptor defects | Knockdown of Ghr gene via an inducible system | Disruption of Ghr gene |
| GH action | ↑↑ with onset of adenoma | ↑↑ from birth | ↓ onset varies based on etiology | ↓ throughout life due to GH antagonism | ↓ beginning at time of induction (10–12 weeks) | GH deficiency (as well as prolactin and TSH) | Absent from birth | ↓ beginning at time of induction (6 weeks) | Absent GHR from birth |
| GH | ↑↑ | ↑↑ | ↓ | ↑ | ↓ | ↑ | ↑ | ↑ | |
| IGF-1 | ↑↑ | ↑↑ | ↓ | ↓ | ↓↓ | ↓↓ | ↓ | ↓↓ | |
| Growth and body weight | ↑↑ * | ↑↑ | ↓ ↔ * | ↓ | ↔ | ↓↓ | ↓↓ | ↓ | ↓↓ |
| Insulin sensitivity | ↓ | ↓ | ↑ | ↑ | ↓↔ | ↑ | ↑↓ *** | ↓ | ↑ |
| Lifespan | ↓ | ↓ | ↔ | No data | ↔ | ↑↑ | ↔ | ↔ male; ↑ female | ↑ |
| Model System | Research Focus | Findings | Citation |
|---|---|---|---|
| GHR-/- mice | Proliferation and differentiation of preadipocytes | SubQ derived preadipocytes proliferate, differentiate and respond to hormones in a similar manner to controls
Perigonadal preadipocytes from GHR-/- mice fail to differentiate and proliferate normally | [131] |
| GHR-/- mice | CideA RNA expression | ↓ cell-death-inducing DFF45-like effector-A (CideA) levels in subQ AT
No difference in CideA expression retroperitoneal or epididymal | [132] |
| GHR-/- | Proteomic analysis of depot differences with age | Lower levels of Glut4 protein in subQ AT of GHR-/- mice, no difference in epididymal AT
Retroperitoneal depot particularly affected by GHR deletion and age | [133] |
| bGH, GHA, GHR-/-, AoiGHD, Ames | Adiponectin expression | Circulating adiponectin levels correlated strongly with subQ fat mass
Higher adiponectin levels in subQ AT of GHR-/- mice | [105] |
| bGH mice | Immune cell infiltration in AT; RNA-seq analyses of depots | ↑ immune cell infiltration (macrophage, T cells) mainly in subQ and mesenteric depots with little change in epididymal AT
↑ in gene expression pathways related to T cell infiltration/activation in subQ, but not epididymal AT | [127] |
| bGH mice, Snell, Ames, GHR-/- and GH injected mice | Cellular senescence in AT | bGH females: ↑ cellular senescence in all depots except periovarian
GH injected WT females: ↑ cellular senescence in subscapular and mesenteric depots GHR-/- females: ↓ cellular senescence all depots except mesenteric Ames: ↓ cellular senescence in paraovarian, mesenteric and subQ | [134] |
| GHR-/- | Depot whole-genome microarrays | Gene expression differences in gene expression related to metabolic function and inflammation among epididymal, subQ, retroperitoneal AT | [135] |
| GHR-/-, bGH | AT-derived mesenchymal stem cells | Increased differentiation in cells isolated from subcutaneous AT vs. epididymal | [136] |
| bGH | GHA | AOiGHD | Ames | aGHRKO | GHR-/- | |
|---|---|---|---|---|---|---|
| GH Defect | Transgenic for bovine GH | GHR antagonist gene | Adult GH deficiency | Homozygous recessive mutation in Prop1 (Ames) | Adult induction of GHR deletion | Disruption of GHR gene |
| WAT | ||||||
| Mass | ↑ young ↓ old | ↑↑ | ↑ (after induction) | ↑ | ↑ (after induction) | ↑↑ |
| Depot mass differences | All depots | ↑ subQ | ↑ subQ/Retro | ↑ subQ | ↑ subQ; ↑ Epi for males only | ↑ subQ |
| Diet-induced obesity | resistant | Increased susceptibility; impairment in glucose homeostasis with advancing age | Increased susceptibility; preservation of improved glucose homoeostasis | Increased susceptibility; preservation of improved glucose homoeostasis | ND | Increased susceptibility; preservation of improved glucose homoeostasis |
| Adipokines | ||||||
| Leptin | ↓ | ↑↑ | ↑ | ↑/↔ | ↑ | ↑/↔ |
| Adiponectin | ↓ | ↑ | ↔ | ↑ | ↑ | ↑↑ |
| Resistin | ↓ | ↑ | ND | ↔ | ↑ in females | ↑ |
| Senescence | ↑ | ↔ | ND | ND | ND | ↓ |
| Immune Cells ** | ↑ macrophage, T cells | ND | ND | ND | ND | ↓ macrophage inflammation |
| BAT | ||||||
| Mass | ↑ ↔ *** | ↑ | ND | ↑ | ↑ in females | ↑ |
| UCP1 content | ↑ | ↑ | ND | ↑ | ND | ↑↓ *** |
| References | [75,76,77,78,79,178] | [75,87,105,124,179] | [94,105] | [105,121,180,181] | [108] | [75,77,104,105,145,182] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berryman, D.E.; List, E.O. Growth Hormone’s Effect on Adipose Tissue: Quality versus Quantity. Int. J. Mol. Sci. 2017, 18, 1621. https://doi.org/10.3390/ijms18081621
Berryman DE, List EO. Growth Hormone’s Effect on Adipose Tissue: Quality versus Quantity. International Journal of Molecular Sciences. 2017; 18(8):1621. https://doi.org/10.3390/ijms18081621
Chicago/Turabian StyleBerryman, Darlene E., and Edward O. List. 2017. "Growth Hormone’s Effect on Adipose Tissue: Quality versus Quantity" International Journal of Molecular Sciences 18, no. 8: 1621. https://doi.org/10.3390/ijms18081621
APA StyleBerryman, D. E., & List, E. O. (2017). Growth Hormone’s Effect on Adipose Tissue: Quality versus Quantity. International Journal of Molecular Sciences, 18(8), 1621. https://doi.org/10.3390/ijms18081621