Abstract
Normal pregnancy is a state of hypercoagulability with diminishing fibrinolytic activity, which is mainly caused by an increase of plasminogen activator inhibitor type 1 (PAI-1). PAI-1 is the main inhibitor of plasminogen activators, including tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). In human placentas, PAI-1 is expressed in extravillous interstitial trophoblasts and vascular trophoblasts. During implantation and placentation, PAI-1 is responsible for inhibiting extra cellular matrix (ECM) degradation, thereby causing an inhibition of trophoblasts invasion. In the present study, we have reviewed the literature of various reproductive diseases where PAI-1 plays a role. PAI-1 levels are increased in patients with recurrent pregnancy losses (RPL), preeclampsia, intrauterine growth restriction (IUGR), gestational diabetes mellitus (GDM) in the previous pregnancy, endometriosis and polycystic ovary syndrome (PCOS). In general, an increased expression of PAI-1 in the blood is associated with an increased risk for infertility and a worse pregnancy outcome. GDM and PCOS are related to the genetic role of the 4G/5G polymorphism of PAI-1. This review provides an overview of the current knowledge of the role of PAI-1 in reproductive diseases. PAI-1 represents a promising monitoring biomarker for reproductive diseases and may be a treatment target in the near future.
1. Introduction
The fibrinolytic system plays a role in several physiological and pathophysiological processes, such as hemostatic balance, tissue remodeling, tumor invasion, angiogenesis and reproduction [1]. Normal pregnancy is a state of hypercoagulability with remarkable changes in all aspects of hemostasis—an increase of clotting factors and coagulability and a decrease of anticoagulants and fibrinolytic activity, thereby influencing placental function during pregnancy and meeting delivery’s hemostatic challenge [2]. Fibrinolytic system is depressed during pregnancy, and this change partly explains the higher incidence of thromboembolic complications such as recurrent pregnancy losses, preeclampsia and intrauterine growth restriction [2]. The diminishing fibrinolytic activity is mainly caused by a continuous increase of the major inhibitor of the fibrinolytic system: plasminogen activator inhibitor type 1 (PAI-1) [2]. PAI-1 is responsible for approximately 60% of the PA-inhibitory activity in the plasma [3] and is the key inhibitor of fibrinolysis compared with PAI-2 and PAI-3 during pregnancy [4].
PAI-1 gene deficiency shows a transient impaired placentation in mice [5], while, in humans, PAI-1 gene deficiency is associated with abnormal bleeding after a trauma or surgery [6,7]. Transgenic mice that overexpress PAI-1 exhibit thrombotic occlusion [8]. Former studies in humans suggest that increased PAI-1 levels are found to be crucial mediators of vascular disease, fibrosis, tumor metastasis, diabetes, and reproductive diseases [9,10,11,12]. PAI-1 acts as a major inhibitor of fibrinolysis, its overexpression leads to fibrin accumulation and insufficient placentation. In this review, we focus on the complex roles of PAI-1 in normal placentation and reproductive diseases, including recurrent pregnancy losses, preeclampsia, intrauterine growth restriction, endometriosis and polycystic ovary syndrome.
2. Fibrinolytic System and PAI-1 (Plasminogen Activator Inhibitor Type 1)
The prime fibrinolytic protease of the fibrinolytic system is plasminogen, which can be activated by urokinase-type plasminogen activator (uPA) and the tissue-type plasminogen activator (tPA) [1,8]. Plasminogen can then be converted into plasmin, and eventually cleaves fibrin into cross-linked fibrin degradation products (Figure 1) [8]. Plasminogen activator inhibitors include PAI-1, PAI-2, PAI-3, C1-esterase inhibitor and protease nexin (Figure 1) [8]. Plasmin inhibitors are α2-plasmin inhibitor (α2-PI), α2-macroglobulin (α2-MG) and protease nexin (Figure 1) [8]. Both uPA and tPA are serine proteases that cleave a single Arg-Val peptide bond to transfer plasminogen to plasmin; uPA functions mainly in pericellular proteolysis while tPA is involved in the circulation [13]. uPA plays an important role in a variety of physiological and pathological processes including tissue destruction, inflammatory reactions and invasion of trophoblasts [14] and cancer cells [15]. Both uPA and tPA consist of a single-chain form and a two-chain form [4,16]. During normal pregnancy, the levels of uPA, PAI-1, PAI-2 and α2-antiplasmin are increased and tPA levels are decreased [17].
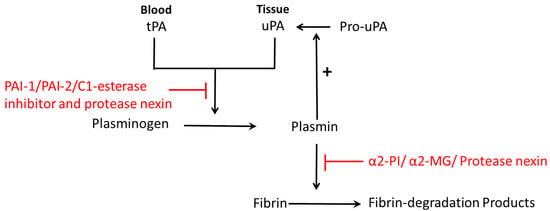
Figure 1.
Schematic diagram of fibrinolysis: plasminogen is activated by plasminogen activator (tPA in blood or uPA in tissue), and then converted to plasmin. Then plasmin cleaves fibrin into fibrin-degradation products. Plasminogen activators inhibitors are PAI-1, PAI-2, C1-esterase inhibitor and protease nexin. Plasmin inhibitors are α2-plasmin inhibitor (α2-PI), α2-macroglobulin (α2-MG) and protease nexin. Pro-uPA can be converted to uPA, which is catalyzed by plasmin, the product of plasminogen.
PAI-1 is the primary inhibitor of tPA in the plasma during pregnancy [4]. It is a single-chain glycoprotein consisting of 379 or 381 amino acids (N-terminal heterogeneity) and belongs to the serine family of protease inhibitor, with a molecular weight of about 45 kDa. There are three different forms of PAI-1: active, inactive and substrate form. The active form can inhibit tPA or uPA by forming a 1:1 stoichiometric complex with each enzyme and the inactive form does not react with the target proteinase [18]. The conformational conversion from the active into the inactive form is completed by the P1-P1’ in a reactive center loop (RCL) of the serpin cleave, followed by the insertion of the RCL into the β-sheet A of the serpin [19]. PAI-1 gene in humans is located on chromosome 7 (q21.3-q22), extends approximately 12.200 base pairs and consists of nine exons and eight introns [20]. PAI-1 gene has several polymorphisms and the 4G allele of the 4G/5G polymorphism is related to high PAI-1 levels [21]. 4G polymorphism is located in the PAI-1 promotor, which is 675 bp upstream from the start site of transcription in the promoter region [21]. Circulating PAI-1 is mainly found in platelets, whilst a large range of cells can further express PAI-1, such as fibroblasts, smooth muscle cells, endothelial cells, hepatocytes, inflammatory cells and placental cells [22].
Both forms of tPA are inhibited by PAI-1, whereas PAI-2 inhibits mainly the two-chain form [23]. PAI-2 consists of two molecular forms: the low molecular weight (LMW) form with 43–48 kDa is intracellular and non-glycosylated, while the high molecular weight (HMW) form with 60 kDa is secreted and glycosylated [24]. PAI-2 is mainly expressed by placental trophoblasts and macrophages [23]. PAI-3, also known as protein C inhibitor (PCI), is prominently expressed in male reproductive organs and its low levels in seminal plasma are associated with infertility [25].
3. Role of PAI-1 in the Female Reproduction System
During a healthy pregnancy, PAI-1 levels in the plasma gradually elevate during the second trimester of pregnancy and reach a maximum at 32–40 weeks of pregnancy. Within 5–8 weeks after delivery, PAI-1 levels fall again to the levels before the occurrence of pregnancy [2]. The concentration of PAI-1 in the plasma of healthy non-pregnant women varies (55 ± 17 ng/mL) [26]. The maximum of PAI-1 concentration during the last trimester of pregnancy is approximately 3–5 times higher than that of non-pregnant women [26]. These temporary changes of PAI-1 during pregnancy are accounted for hormonal influences [27]. Increases in PAI-2 levels are observed during pregnancy and during delivery [23]. Both PAI-1 and PAI-2 decrease quickly following the placenta separation from the uterus, but PAI-2 can still be found in the circulation up to eight weeks postpartum [2].
Trophoblasts can express PAI-1 and PAI-2 in vivo and in vitro [28]. Both PAI-1 and PAI-2 are localized in the cytoplasm of cytotrophoblasts and in the cytoplasm and plasma membrane of intermediate and syncytiotrophoblasts [28]. PAI-1 is localized in invading trophoblasts in the human placenta by immunostaining [29], especially in extravillous interstitial trophoblasts and vascular trophoblasts [30]. In the human placenta, PAI-1 protein and mRNA expression exist in most extravillous cytotrophoblast cells of the decidual layer, especially the chorionic villous tree and in cytotrophoblast cells of the chorionic plate, basal plate and intercotyledonary septae [31]. No expression of PAI-1 has been observed in the basal plate of endometrial stromal cells, chorionic plate connective tissue cells, septal endometrial stromal cells or villous core mesenchyme [31]. PAI-2 is the predominant PAI accumulated in villous syncytiotrophoblasts [29]. Both PAI-1 and PAI-2 mRNAs expressions are detected in cultured cytotrophoblasts isolated from both the first trimester and term placenta [29]. PAI-1 and PAI-2 are also expressed by uterine natural killer (uNK) cells [32].
3.1. PAI-1 Inhibits Trophoblast Invasion
Trophoblast invasion at the maternal-fetal interface is a key process during implantation and placentation, and during this process extravillous cytotrophoblasts (EVT) acquire invasive properties, which are able to invade and remodel maternal tissues (interstitial EVT) and uterine spiral artery (endovascular EVT) [33]. EVT can degrade extracellular matrix (ECM) to promote cell migration to the maternal side [33]. This process is precisely controlled by many factors expressed by maternal cells and trophoblasts (Figure 2) [33].
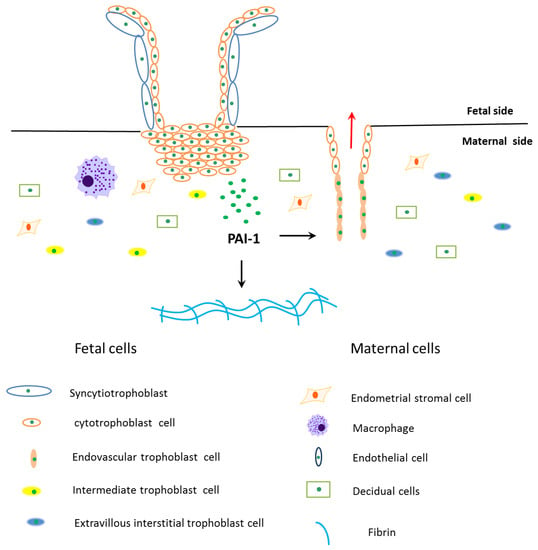
Figure 2.
Schematic diagram of PAI-1’s role during trophoblasts invasion. The fetal side of the human placenta mainly includes cytotrophoblasts and syncytiotrophoblasts, and sycytiotrophoblasts are differentiated and fused by cytotrophoblasts. The cytotrophoblasts invade into the maternal side and differentiate into extravillous interstitial trophoblasts, intermediate trophoblasts and endovascular trophoblasts. Among them, extravillous interstitial trophoblasts and endovascular trophoblasts express plasminogen activator inhibitor type 1 (PAI-1). Furthermore, cells from the maternal side take part in trophoblast invasion, such as endometrial stromal cells, decidual cells, macrophages and endothelial cells. Extravillous trophoblast invasion in early pregnancy is precisely controlled by many factors expressed by trophoblasts and maternal cells, where PAI-1 is the main anti-invasive factor. PAI-1 prevents trophoblast invasion by inhibiting extracellular matrix degradation, which leads to fibrin accumulation in the maternal side. PAI-1 may also play a role in remodeling maternal uterine spiral arteries.
Trophoblasts and malignant tumors use the same biochemical mediators to facilitate invasion, including extracellular matrix degradation and immunosuppression of environmental conditions. PAI-1 can inhibit trophoblasts invasion while promoting tumor cell immigration [34,35]. PAI-1 is a biomarker for malignancies with poor prognosis because it facilitates tumor cell migration and invasion [35]. PAI-1/uPA/uPA receptor (uPAR)/low density lipoprotein receptor-related protein (LRP)/integrin complexes are initiating an “adhesion–detachment–re-adhesion” cycle to promote tumor cell migration [35,36]. Hyperinvasiveness in premalignant and malignant extravillous trophoblasts (JAR and JEG-3 choriocarcinoma cell lines) results from a downregulation of tissue inhibitors of metalloprotease (TIMP)-1 and PAI-1 genes [34].
EVT invasion in early pregnancy occurs in a relatively low-oxygen (3%) environment, which is mediated by a general inhibition of the plasminogen activator system [37], as well as many adhesion molecules, growth factors, cytokines, interleukins, ECM components, and various placental hormones [14,33]. The anti-invasive action of EVT is caused by an upregulation of the tissue inhibitor of TIMP-1 and PAI-1 and a downregulation of uPA [14]. PAI-1 and PAI-2 are expressed in invading human extravillous trophoblast cells and they limit the depth of invasion [37,38]. PAI-1 is found to be absent in the placental bed of ectopic and molar pregnancies, suggesting that no expression of PAI-1 can contribute to an uncontrolled placental invasion [30]. Expression of PAI-1 is elevated in the process of EVT invasion treated by tumor necrosis factor α (TNF-α), and adding PAI-1-inactivating antibodies restores migration [39].
The limitation of EVT invasion is due to reduced ECM degradation (Figure 2), which requires the balance of promoting and restraining factors, such as metalloproteinases (MMPs) and tissue inhibitors of MMPs (TIMPs), uPA and PAI-1 [40]. PAI-1 restrains ECM degradation in three different ways. Firstly, PAI-1 can directly block uPA activity to constrain the proteolytic activity of plasmin [18]. Secondly, PAI-1 binds to uPA and causes its degradation via uPAR and low-density lipoprotein receptor-related protein (LRP) internalization [41]. The direct binding of occupied uPAR to LRP is essential for internalization and clearance of uPA-PAI-1-occupied uPAR [42]. The activity of PAI-1 depends on its interactions with LRP, which leads to the activation of the Janus kinase 2/signal transducer and activator of transcription protein (Jak/-Stat) signaling pathway [43]. Lastly, an increase in the PAI-1 transcript and translation leads to the formation of keloids and fibrosis [44]. Renaud et al. (2005) suggested that macrophages participate in decreasing the depth of trophoblast invasion by secreting TNF-α, which couples TNFR and promotes EVT to release PAI-1 during placentation [45]. Huber et al. (2006) further reported that TNF-α stimulates PAI-1 level of HTR-8/SVneo cells by activation of the nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) pathway [46].
PAI-1 may also play a role in remodeling maternal uterine spiral arteries (Figure 2) [47]. PAI-1 mRNA positive cells in the maternal arteriole co-express cytokeratin, implying that PAI-1 may participate in the process of endovascular cytotrophoblast cells replacing cells of the arteriole wall [47]. Hypoinvasion and failed placental vascular remodeling are associated with reproductive diseases, such as recurrent pregnancy losses (RPL), preeclampsia and intrauterine growth restriction (IUGR) and even maternal as well as fetal death [48,49]. In terms of tumor cells, PAI-1 can both promote and inhibit tumor growth and angiogenesis. Low concentrations of PAI-1 can stimulate tumor angiogenesis while treatment of animals with high doses of PAI-1 suppresses angiogenesis and tumor growth [50].
3.2. Recurrent Pregnancy Losses
Recurrent pregnancy losses (RPL) affects approximately 1% of all couples worldwide [51], and its latest definition is two or more consecutive failed pregnancies as documented by ultrasonography or histopathologic examination before the 20th pregnancy week according to the Practice Committee of the American Society for Reproductive Medicine [52]. The identifiable reasons for RPL include genetic abnormalities, structural abnormalities, infection, endocrine abnormalities, immune dysfunction, and thrombophilic disorders [53]. However, there are still up to 40–50% of pregnancy losses that have unidentifiable causes [54]. PAI-1 plasma levels in RPL patients are increased in comparison to women with healthy pregnancies [55]. Among all the thrombophilic genes, functional PAI-1-675 4G/5G polymorphism is one of the most frequently analyzed PAI-1 genetic variants. However, the contribution of PAI-1-675 4G/5G to unexplained RPL has remained controversial.
Gris et al. (1997) analyzed a study with 500 women with unexplained RPL and demonstrated that increased PAI-1 levels are the most frequent hemostasis-related abnormality connected to unexplained RPL [56]. A recent meta-analysis by Li et al. (2015) including 22 studies with 4306 cases and 3076 controls showed that PAI-1 4G/5G polymorphism is associated with an increased RPL risk (p = 0.0003), especially in the Caucasian subgroup (p < 0.001) [57]. Khosravi et al. (2014) further found that a high prevalence of PAI-1 -675 4G/4G existed in RPL patients as well as in implantation failure (IF) patients (p < 0.001) [58].
Another group suggests that PAI-1-675 4G/5G alone is not responsible for RPL [59,60], and more thrombophilic gene mutations together can help predispose RPL risk. More than three gene mutations among the 10 thrombophilic gene mutations (factor V G1691A, factor V H1299R (R2), factor V Y1702C, factor II pro-thrombin G20210A, factor XIII V34L, b-fibrinogen −455G > A, PAI-1 4G/5G, HPA1 a/b (L33P), MTHFR C677T, and MTHFR A1298C) are more prevalent in RPL patients [61,62]. Endothelial PAI-1 synthesis is induced by angiotensin II, which is generated by angiotensin I-converting enzyme (ACE) and this might be one of the main reasons of elevated PAI-1 concentrations in RPL [63].
In general, associations between PAI-1 4G/5G and unexplained RPL may have been masked by sample-size effects, ethnicity, enrollment criteria or combinations. In order to improve our understanding of the pathobiology of RPL, we need to identify not only novel genetic variants and the interaction, but also how genes, proteins and the environment contribute to RPL. Thus far, PAI-1 inhibiting fibrinolysis and fibrin accumulation are believed to be the principle reasons for RPL, but the understanding of the mechanism of PAI-1 in RPL still has to be further analyzed.
3.3. Preeclampsia
Preeclampsia affects about 2.5% to 3.0% of pregnant women [64] and it is a leading cause of perinatal morbidity and mortality both for the fetus and the mother [65]. Preeclampsia is a pregnancy disorder characterized by hypertension, proteinuria and placental abnormalities [66], which are generally manifested in the second to third trimester of pregnancy [67]. Although the mechanisms responsible for the pathogenesis of preeclampsia are poorly understood, there is an agreement that it is associated with hypoinvasion and failed conversion of maternal endometrial spiral arteries in the placenta [64], both of which are related to PAI-1 and PAI-2.
Maternal PAI-1 levels in the plasma are higher in patients with preeclampsia during the second trimester of pregnancy [67], and its mRNA levels are positively correlated with the severity of preeclampsia during 35–41 weeks of gestation [68]. The ratio of PAI-1 to PAI-2 is increased in women with early-onset preeclampsia (24–32 gestational weeks) in comparison to the control group, but not in late-onset preeclampsia (35–42 gestational weeks) [69]. Estelles et al. (1989) found that both antigenic and functional PAI-1 levels are increased while antigenic and functional levels of PAI-2 are decreased in the third trimester (≥28 weeks) compared with healthy controls [70]. In contrast, in the first trimester (11–13 weeks), PAI-2 in the plasma is observed to be not differently expressed in preeclampsia [71]. PAI-1 antigen levels are positively correlated to proteinuria in women with preeclampsia [72]. Therefore, PAI-1 has been considered as a potentially useful predictor of preeclampsia. Although it is known that smoking may reduce the risk for preeclampsia [73], specifically chronic smoking upregulates PAI-1 levels [74].
It remains uncertain whether increased PAI levels are the primary mechanism leading to preeclampsia or a consequence of the associated endothelial and placental damage [75]. Cytotrophoblasts, which do not express Raf kinase inhibitor protein (RKIP), could be one of the reasons for impaired migration of cytotrophoblasts in preeclampsia, because locostatin (the inhibitor of RKIP) induces PAI-1 expression with the support of activation of NF-κB pathway and finally contributes to an inadequate trophoblast invasion [76].
PAI-1 expression in the plasma is increased following exposure to inflammatory cytokines, including interleukin 1β (IL-1β) [77], vascular endothelial growth factor (VEGF), epidermal growth factor (EGF) and fibroblast growth factor (FGF) or hypoxic conditions [78]. Hypoxia can directly stimulate PAI-1 mRNA and protein expression [35], and can also stimulate hypoxia-inducible transcription factors (HIF-1α and HIF-2α) to induce PAI-1 [79], both of which may also be the mechanisms of preeclampsia. When preeclampsia occurs in combination with increased levels of syncytial PAI-1, intervillous fibrin deposition and infarction may reduce the flow of nutrients from mother to fetus leading to IUGR [80].
Still, there is a dispute of the correlation between PAI-1 polymorphism and preeclampsia. Gerhardt et al. (2005) discovered that women with PAI-1 5G/5G genotype are at risk for early onset of severe preeclampsia (17–35 gestational weeks) [81]. Morgan JA et al. (2013) found that PAI-1-675 4G/5G polymorphism is not associated with preeclampsia with a total of 5003 women involved in the meta-analysis [82].
3.4. Intrauterine Growth Restriction
Intrauterine growth restriction (IUGR) is defined as fetuses with pathological smallness caused by an underlying functional problem [83], occurring in 5–10% of all pregnancies [84]. IUGR is associated with increased risks of neonatal death or disability in the perinatal period [84,85] and predisposes the child to a lifelong increased risk for hypertension, cardiovascular disorders and renal disease [86]. Histopathological studies of IUGR placentas indicate abnormalities of the maternal spiral arterioles, dysregulated villous vasculogenesis, and abundant fibrin deposition, as well as oxidative stress and apoptosis in villous trophoblast [87].
PAI-1 is a potential marker of placental insufficiency and it is associated with fetal hypoxia and angiogenesis in IUGR [84]. PAI-1 levels in the umbilical cord blood are increased in patients with IUGR and it is associated with the plasma’s angiogenic potency measured in vitro [84]. Cytotrophoblast cells isolated from the placenta of IUGR pregnancies express significantly higher levels of PAI-1, with a significant decrease in plasminogen activator activity, compared with trophoblast cells from normal pregnancy cultured in vitro [88]. This localized increased production of PAI-1 may play an important part in restricting endovascular trophoblast invasion in early pregnancy, increasing fibrin deposition and reducing uteroplacental blood flow in IUGR pregnancies [88].
PAI-1 placental levels are increased in pregnancies with IUGR and preeclampsia, but not in patients with isolated IUGR [89]. In contrast, PAI-2 expression is reduced in placentas of both IUGR women with and without preeclampsia compared with normal placentas [89]. In the first trimester, low PAI-2 is associated with a higher risk for the development of IUGR [90]. A significant decrease in PAI-2 in the plasma and amniotic fluid is observed in IUGR groups in comparison with normal pregnancies [91]. PAI-2 levels are correlated with fetal weight of IUGR pregnancies [92,93], indicating that PAI-2 is not only the marker of the quantity and quality of the placenta tissues but also a marker for fetal growth and development [17]. PAI-1 polymorphism (4G/5G) has not been associated with an increased risk of IUGR in the case-control analysis [94,95].
3.5. Gestational Diabetes Mellitus
Gestational diabetes mellitus (GDM) is characterized by impaired glucose tolerance during pregnancy, with the prevalence of 0.6–20% of pregnancies [96]. Women with GDM can have complications including polyhydramnios, fetal macrosomia, preeclampsia, shoulder dystocia and increased risk for operative delivery. Postpartum, patients with GDM have a more than seven-fold increased risk of developing postpartum diabetes compared to women without GDM [97]. Insulin resistance and chronic subclinical inflammation are the two main pathways leading to GDM and possibly previous GDM [98]. Insulin resistance syndrome is associated with increased levels of leptin, TNF-α, tPA, PAI-1 and testosterone [99].
PAI-1 is partly secreted by adipose tissue, and can lead to an impairment of the fibrinolytic system [100]. Salmi et al. (2012) reported that PAI-1 levels are higher in the serum of women with GDM compared to healthy women during the early third trimester pregnancy [101]. Bugatto et al. (2017) stated that PAI-1 levels in maternal uterine blood do not change in women with GDM at the third trimester pregnancy compared to controls [102]. McManus et al. (2014) suggested that GDM women have lower concentrations of PAI-1 in comparison to the age- and weight-matched controls in maternal plasma, as well as in the umbilical artery and umbilical vein [103]. This study further demonstrated that GDM offspring also have decreased PAI-1 concentrations compared to controls [103].
PAI-1 levels in women with GDM are not consistent, but PAI-1 levels have been shown to be increased in women who had GDM during a previous pregnancy [98]. Women with previous GDM have elevated PAI-1 levels 0.25–4 years after delivery [98]. PAI-1 levels are correlated with the insulin sensitivity index (SI), and the PAI-1/SI ratio is increased in women with a previous GDM and impaired insulin [104].
PAI-1 expression is significantly correlated with a variety of adiposity features, including body mass index (BMI), total fat mass, waist circumference, visceral adipose tissue and subcutaneous adipose tissue, total cholesterol triglycerides, fasting plasma glucose and 2 h plasma glucose in the glucose tolerance test, insulin sensitivity as well as pancreatic beta-cell function [100,105,106]. Hyperglycemia inhibits the expression of uPA and PAI-1 by the induction of p38 mitogen-activated protein kinases (p38 MAPK) and peroxisome proliferator-activated receptor γ (PPAR-γ) stress signaling pathways, which are different from the PAI-1 levels induced during preeclampsia [107].
Leipold et al. (2006) reported that the 5G allele of the PAI-1 gene is associated with normal glucose tolerance in pregnant women independent of maternal age or BMI and it is also related to low fasting glucose values in the oral glucose tolerance test [108]. Physical activities can lower cardiometabolic risk in women with previous GDM and PAI-1 levels, as well as the level of C-reactive protein, leptin and triglycerides [109].
Furthermore, Glueck et al. (2000) found that the polymorphism of PAI-1 gene is not associated with eclampsia, abruptio placentae and intrauterine fetal death by stepwise logistic regression [110].
3.6. Endometriosis
Endometriosis is defined as the presence of endometrial-like tissue outside the uterus, causing dysmenorrhea, chronic pelvic pain, dyspareunia, and infertility [111]. Pathogenesis of endometriosis includes inflammation, angiogenesis, cytokine/chemokine expression and endocrine alterations such as estrogen receptors (ESRs) and progesterone receptors (PGRs) expression [111]. PAI-1 plays an important role in tumor cell migration and invasion [11], which is also the mechanism of endometriotic cells invasion [112].
Ovarian endometriotic tissues have higher antigenic levels of PAI-1 than normal endometrium [113]. Increased PAI-1 antigen levels in peritoneal fluid from patients with endometriosis contribute to an increase of peritoneal adhesions [114]. In vitro, isolated endometriotic stromal cells release a higher level of PAI-1 compared to the endometrial stromal and epithelial cells of women with endometriosis and controls [112].
There exists a dispute of the association between PAI-1 gene polymorphism 4G/5G and endometriosis [115,116]. PAI-1 4G/5G polymorphism is associated with an increased risk for endometriosis-associated infertility [116]. However, Goodarzi et al. found that PAI-1 genotype distribution is similar in patients with endometriosis and controls [115].
3.7. Polycystic Ovary Syndrome
Polycystic ovary syndrome (PCOS) is the most common endocrinopathy of women in the reproductive age, with a prevalence of up to 10% and symptoms include hyperandrogenism and/or hyperandrogenemia, oligo-ovulation and polycystic ovarian morphology [117]. PCOS is associated with infertility, RPL, type 2 diabetes mellitus and cardiovascular diseases [117]. Many studies in women with PCOS have reported that PAI-1 levels in the plasma are increased in both normal weight and overweight/obese women with PCOS compared with controls with matched body mass index (BMI) [100,118,119]. Serum PAI-1 activity is related to the BMI and homeostasis model assessment (HOMA) score [120], insulin levels and insulin sensitivity indices [121]. PAI-1 levels decline after treatment with sibutramine and metformin in normal weight and overweight women with PCOS [118,122].
Oligo-ovulation (defined as delayed menses >35 days or <8 spontaneous hemorrhagic episodes per year) is not completely understood yet [123]. uPA plays an essential role in the early growing follicles during cell proliferation and migration, and in the early corpus luteum (CL) formation related to ECM degradation and angiogenesis [124]. PAI-1 is localized in the granulosa and theca cells, indicating it possibly plays a role in human ovulation, but its role in PCOS needs to be further explained [125]. Increased PAI-1 expression in CL of rat and monkey at a later stage is correlated with a sharp decrease in progesterone production of CL [124]. The role of PAI-1 in ovulation needs to be studied further in humans. PAI-1 is a predominant independent risk factor for miscarriages in women with PCOS [126]. Elevated plasma PAI-1 levels are associated with an increased risk for both type 2 diabetes mellitus and cardiovascular diseases (i.e., coronary disease/ischemic stroke) [127,128,129].
PAI-1 4G/5G polymorphism is associated with susceptibility to PCOS in European, Turkish, and Asian populations [130,131].
4. Conclusions
PAI-1 is expressed in the placenta and maternal plasma of pregnant women. In the human placenta, PAI-1 is localized in invading trophoblasts, especially in extravillous trophoblasts. By inhibiting ECM degradation, PAI-1 plays a vital role in the prevention of trophoblast invasion in RPL, preeclampsia and IUGR. Increased expression of PAI-1 in the plasma is found in RPL, preeclampsia, IUGR, GDM in previous pregnancies, endometriosis and PCOS. Similar to tumor cells, PAI-1 promotes endometriotic cells invasion during endometriosis. In PCOS, PAI-1 may regulate ovulation but this hypothesis still requires further research. PAI-1 4G/5G polymorphism is associated with GDM and PCOS; however, contradictory results are found in patients with RPL, preeclampsia and endometriosis. PAI-1 expression can be reduced through physical exercise and pharmacological interventions, such as the oral intake of thiazolidinedines (troglitazone or pioglitazone) [100], statins [100] and metformin [118]. Altered PAI-1 levels have further been found in other pathologies. Studies on cardiovascular diseases have displayed a strong positive correlation between PAI-1 levels in the serum and cardiovascular risks for myocardial infarction (MI), recurrent MI, angina pectoris and atherosclerosis [9]. In addition, PAI-1 is a pivotal mediator of vascular diseases, cancer, asthma, insulin resistance and diabetes [100]. In the future, PAI-1 may function as a biological monitoring parameter and possibly represents a therapeutic approach in reproductive pathologies.
Acknowledgments
China Scholarship Council supported scholarships for Yao Ye, Xi Zhang and Junyan Zhu. The sponsors did not participate in the manuscript writing.
Author Contributions
Yao Ye and Aurelia Vattai performed the literature search, and wrote and revised the manuscript; Xi Zhang, Junyan Zhu, Christian J. Thaler, Sven Mahner, Udo Jeschke and Viktoria von Schönfeldt revised the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| PAI | plasminogen activator inhibitor |
| tPA | tissue-type plasminogen activator |
| uPA | urokinase-type plasminogen activator |
| ECM | extra cellular matrix |
| α2-PI | α2-plasmin inhibitor |
| α2-MG | α2-macroglobulin |
| RCL | reactive center loop |
| LMW | low molecular weight |
| HMW | high molecular weight |
| PCI | protein C inhibitor |
| uNK | uterine natural killer |
| EVT | extravillous cytotrophoblast |
| uPAR | uPA receptor |
| LRP | low density lipoprotein receptor-related protein |
| TIMP | tissue inhibitors of metalloprotease |
| TNF-α | tumor necrosis factor α |
| MMP | metalloproteinase |
| Jak/-Stat | Janus kinase 2/signal transducer and activator of transcription protein |
| NF-κB | nuclear factor κ-light-chain-enhancer of activated B cells |
| ACE | angiotensin I-converting enzyme |
| IL-1β | interleukin 1β |
| VEGF | vascular endothelial growth factor |
| EGF | epidermal growth factor |
| FGF | fibroblast growth factor |
| HIF | hypoxia-inducible transcription factors |
| p38 MAPK | p38 mitogen-activated protein kinases |
| PPAR-γ | peroxisome proliferator-activated receptor γ |
| RKIP | raf kinase inhibitor protein |
| ESR | estrogen receptor |
| PGR | progesterone receptor |
| SI | sensitivity index |
| BMI | body mass index |
| HOMA | homeostasis model assessment |
| CL | corpus luteum |
| RPL | recurrent pregnancy losses |
| IF | implantation failure |
| IUGR | intrauterine growth restriction |
| GDM | gestational diabetes mellitus |
| PCOS | polycystic ovary syndrome |
| MI | myocardial infarction |
References
- Zorio, E.; Gilabert-Estelles, J.; Espana, F.; Ramon, L.A.; Cosin, R.; Estelles, A. Fibrinolysis: The key to new pathogenetic mechanisms. Curr. Med. Chem. 2008, 15, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Hellgren, M. Hemostasis during normal pregnancy and puerperium. Semin. Thromb. Hemost. 2003, 29, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Kluft, C.; Jie, A.F.; Sprengers, E.D.; Verheijen, J.H. Identification of a reversible inhibitor of plasminogen activators in blood plasma. FEBS Lett. 1985, 190, 315–318. [Google Scholar] [CrossRef]
- Jorgensen, M.; Philips, M.; Thorsen, S.; Selmer, J.; Zeuthen, J. Plasminogen activator inhibitor-1 is the primary inhibitor of tissue-type plasminogen activator in pregnancy plasma. Thromb. Haemost. 1987, 58, 872–878. [Google Scholar] [PubMed]
- Labied, S.; Blacher, S.; Carmeliet, P.; Noel, A.; Frankenne, F.; Foidart, J.M.; Munaut, C. Transient reduction of placental angiogenesis in PAI-1-deficient mice. Physiol. Genom. 2011, 43, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Fay, W.P.; Parker, A.C.; Condrey, L.R.; Shapiro, A.D. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: Characterization of a large kindred with a null mutation in the PAI-1 gene. Blood 1997, 90, 204–208. [Google Scholar] [PubMed]
- Heiman, M.; Gupta, S.; Shapiro, A.D. The obstetric, gynaecological and fertility implications of homozygous PAI-1 deficiency: Single-centre experience. Haemophilia 2014, 20, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Cesarman-Maus, G.; Hajjar, K.A. Molecular mechanisms of fibrinolysis. Br. J. Haematol. 2005, 129, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Yasar Yildiz, S.; Kuru, P.; Toksoy Oner, E.; Agirbasli, M. Functional stability of plasminogen activator inhibitor-1. Sci. World J. 2014, 2014, 858293. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; McGowan, P.M.; Harbeck, N.; Thomssen, C.; Schmitt, M. uPA and PAI-1 as biomarkers in breast cancer: Validated for clinical use in level-of-evidence-1 studies. Breast Cancer Res. 2014, 16, 428. [Google Scholar] [CrossRef] [PubMed]
- Lyon, C.J.; Hsueh, W.A. Effect of plasminogen activator inhibitor-1 in diabetes mellitus and cardiovascular disease. Am. J. Med. 2003, 115, 62–68. [Google Scholar] [CrossRef]
- Lijnen, H.R.; Collen, D. Mechanisms of physiological fibrinolysis. Baillieres Clin. Haematol. 1995, 8, 277–290. [Google Scholar] [CrossRef]
- Chakraborty, C.; Gleeson, L.M.; McKinnon, T.; Lala, P.K. Regulation of human trophoblast migration and invasiveness. Can. J. Physiol. Pharmacol. 2002, 80, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Vassalli, J.D.; Dano, K. Urokinase-type plasminogen activator: Proenzyme, receptor, and inhibitors. J. Cell Biol. 1987, 104, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.C.; Lund, L.R.; Nielsen, L.S.; Dano, K.; Skriver, L. One-chain urokinase-type plasminogen activator from human sarcoma cells is a proenzyme with little or no intrinsic activity. J. Biol. Chem. 1988, 263, 11189–11195. [Google Scholar] [PubMed]
- Brenner, B. Haemostatic changes in pregnancy. Thromb. Res. 2004, 114, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Gils, A.; Declerck, P.J. The structural basis for the pathophysiological relevance of PAI-1 in cardiovascular diseases and the development of potential PAI-1 inhibitors. Thromb. Haemost. 2004, 91, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, P.A.; Egelund, R.; Petersen, H.H. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell. Mol. Life Sci. CMLS 2000, 57, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Gils, A. The pathophysiological relevance of PAI-1 in cardiovascular diseases and the development of monoclonal antibodies as PAI-1 inhibitors. Verh K Acad Geneeskd Belg 2006, 68, 179–198. [Google Scholar] [PubMed]
- Eriksson, P.; Nilsson, L.; Karpe, F.; Hamsten, A. Very-low-density lipoprotein response element in the promoter region of the human plasminogen activator inhibitor-1 gene implicated in the impaired fibrinolysis of hypertriglyceridemia. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Saksela, O.; Rifkin, D.B. Cell-associated plasminogen activation: Regulation and physiological functions. Annu. Rev. Cell Biol. 1988, 4, 93–126. [Google Scholar] [CrossRef] [PubMed]
- Astedt, B.; Lindoff, C.; Lecander, I. Significance of the plasminogen activator inhibitor of placental type (PAI-2) in pregnancy. Semin. Thromb. Hemost. 1998, 24, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, E.K.; Baker, M.S.; Bunn, C.L. Biological and clinical aspects of plasminogen activator inhibitor type 2. Blood 1995, 86, 4007–4024. [Google Scholar] [PubMed]
- Suzuki, K. The multi-functional serpin, protein C inhibitor: Beyond thrombosis and hemostasis. J. Thromb. Haemost. 2008, 6, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, E.K.; Tran-Thang, C.; Gudinchet, A.; Hauert, J.; Nicoloso, G.; Genton, C.; Welti, H.; Bachmann, F. Fibrinolysis in pregnancy: A study of plasminogen activator inhibitors. Blood 1987, 69, 460–466. [Google Scholar] [CrossRef]
- Stirling, Y.; Woolf, L.; North, W.R.; Seghatchian, M.J.; Meade, T.W. Haemostasis in normal pregnancy. Thromb. Haemost. 1984, 52, 176–182. [Google Scholar] [PubMed]
- Hofmann, G.E.; Glatstein, I.; Schatz, F.; Heller, D.; Deligdisch, L. Immunohistochemical localization of urokinase-type plasminogen activator and the plasminogen activator inhibitors 1 and 2 in early human implantation sites. Am. J. Obstet. Gynecol. 1994, 170, 671–676. [Google Scholar] [CrossRef]
- Feinberg, R.F.; Kao, L.C.; Haimowitz, J.E.; Queenan, J.T., Jr.; Wun, T.C.; Strauss, J.F., 3rd; Kliman, H.J. Plasminogen activator inhibitor types 1 and 2 in human trophoblasts. PAI-1 is an immunocytochemical marker of invading trophoblasts. Lab. Investig. J. Tech. Methods Pathol. 1989, 61, 20–26. [Google Scholar]
- Floridon, C.; Nielsen, O.; Holund, B.; Sweep, F.; Sunde, L.; Thomsen, S.G.; Teisner, B. Does plasminogen activator inhibitor-1 (PAI-1) control trophoblast invasion? A study of fetal and maternal tissue in intrauterine, tubal and molar pregnancies. Placenta 2000, 21, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.Y.; Liu, Y.X.; Liu, K.; Byrne, S.; Ny, T.; Feng, Q.; Ockleford, C.D. Expression of tissue type and urokinase type plasminogen activators as well as plasminogen activator inhibitor type-1 and type-2 in human and rhesus monkey placenta. J. Anat. 1999, 194, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Naruse, K.; Lash, G.E.; Bulmer, J.N.; Innes, B.A.; Otun, H.A.; Searle, R.F.; Robson, S.C. The urokinase plasminogen activator (uPA) system in uterine natural killer cells in the placental bed during early pregnancy. Placenta 2009, 30, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.F.; Serakides, R. Intrauterine trophoblast migration: A comparative view of humans and rodents. Cell Adh. Migr. 2016, 10, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Lala, P.K.; Lee, B.P.; Xu, G.; Chakraborty, C. Human placental trophoblast as an in vitro model for tumor progression. Can. J. Physiol. Pharmacol. 2002, 80, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, T.E.; Graham, C.H. Stimulation of plasminogen activator inhibitor-1 expression in immortalized human trophoblast cells cultured under low levels of oxygen. Exp. Cell Res. 1998, 245, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Crippa, M.P. Urokinase-type plasminogen activator. Int. J. Biochem. Cell Biol. 2007, 39, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Lash, G.E.; Otun, H.A.; Innes, B.A.; Bulmer, J.N.; Searle, R.F.; Robson, S.C. Low oxygen concentrations inhibit trophoblast cell invasion from early gestation placental explants via alterations in levels of the urokinase plasminogen activator system. Biol. Reprod. 2006, 74, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wen, H.Y.; Kellems, R.E. Angiotensin II inhibits human trophoblast invasion through AT1 receptor activation. J. Biol. Chem. 2002, 277, 24601–24608. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Pollheimer, J.; Hartmann, J.; Husslein, P.; Aplin, J.D.; Knofler, M. Tumor necrosis factor-α inhibits trophoblast migration through elevation of plasminogen activator inhibitor-1 in first-trimester villous explant cultures. J. Clin. Endocrinol. Metab. 2004, 89, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Estella, C.; Herrer, I.; Atkinson, S.P.; Quinonero, A.; Martinez, S.; Pellicer, A.; Simon, C. Inhibition of histone deacetylase activity in human endometrial stromal cells promotes extracellular matrix remodelling and limits embryo invasion. PLoS ONE 2012, 7, e30508. [Google Scholar] [CrossRef] [PubMed]
- Degryse, B.; Sier, C.F.; Resnati, M.; Conese, M.; Blasi, F. PAI-1 inhibits urokinase-induced chemotaxis by internalizing the urokinase receptor. FEBS Lett. 2001, 505, 249–254. [Google Scholar] [CrossRef]
- Czekay, R.P.; Kuemmel, T.A.; Orlando, R.A.; Farquhar, M.G. Direct binding of occupied urokinase receptor (uPAR) to LDL receptor-related protein is required for endocytosis of uPAR and regulation of cell surface urokinase activity. Mol. Biol. Cell 2001, 12, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Degryse, B.; Neels, J.G.; Czekay, R.P.; Aertgeerts, K.; Kamikubo, Y.; Loskutoff, D.J. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J. Biol. Chem. 2004, 279, 22595–22604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, Y.; Ann, D.K.; Messadi, D.V.; Tuan, T.L.; Kelly, A.P.; Bertolami, C.N.; Le, A.D. Mechanisms of hypoxic regulation of plasminogen activator inhibitor-1 gene expression in keloid fibroblasts. J. Investig. Dermatol. 2003, 121, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.J.; Postovit, L.M.; Macdonald-Goodfellow, S.K.; McDonald, G.T.; Caldwell, J.D.; Graham, C.H. Activated macrophages inhibit human cytotrophoblast invasiveness in vitro. Biol. Reprod. 2005, 73, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.V.; Saleh, L.; Bauer, S.; Husslein, P.; Knofler, M. TNFα-mediated induction of PAI-1 restricts invasion of HTR-8/SVneo trophoblast cells. Placenta 2006, 27, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liu, Y.; Liu, K.; Byrne, S.; Liu, G.; Wang, X.; Li, Z.; Ockleford, C.D. Expression of urokinase, plasminogen activator inhibitors and urokinase receptor in pregnant rhesus monkey uterus during early placentation. Placenta 2000, 21, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.J.; Chakraborty, D.; Kubota, K.; Renaud, S.J.; Rumi, M.A. Adaptive mechanisms controlling uterine spiral artery remodeling during the establishment of pregnancy. Int. J. Dev. Biol. 2014, 58, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular trophoblast invasion: Implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef] [PubMed]
- McMahon, G.A.; Petitclerc, E.; Stefansson, S.; Smith, E.; Wong, M.K.; Westrick, R.J.; Ginsburg, D.; Brooks, P.C.; Lawrence, D.A. Plasminogen activator inhibitor-1 regulates tumor growth and angiogenesis. J. Biol. Chem. 2001, 276, 33964–33968. [Google Scholar] [CrossRef] [PubMed]
- Stirrat, G.M. Recurrent miscarriage. Lancet 1990, 336, 673–675. [Google Scholar] [CrossRef]
- Practice Committee of the American Society for Reproductive Medicine. Definitions of infertility and recurrent pregnancy loss: A committee opinion. Fertil. Steril. 2013. [Google Scholar] [CrossRef]
- Rai, R.; Regan, L. Recurrent miscarriage. Lancet 2006, 368, 601–611. [Google Scholar] [CrossRef]
- Jaslow, C.R.; Carney, J.L.; Kutteh, W.H. Diagnostic factors identified in 1020 women with two versus three or more recurrent pregnancy losses. Fertil. Steril. 2010, 93, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Gris, J.C.; Neveu, S.; Mares, P.; Biron, C.; Hedon, B.; Schved, J.F. Plasma fibrinolytic activators and their inhibitors in women suffering from early recurrent abortion of unknown etiology. J. Lab. Clin. Med. 1993, 122, 606–615. [Google Scholar] [PubMed]
- Gris, J.C.; Ripart-Neveu, S.; Maugard, C.; Tailland, M.L.; Brun, S.; Courtieu, C.; Biron, C.; Hoffet, M.; Hedon, B.; Mares, P. Respective evaluation of the prevalence of haemostasis abnormalities in unexplained primary early recurrent miscarriages. The Nimes Obstetricians and Haematologists (NOHA) Study. Thromb. Haemost. 1997, 77, 1096–1103. [Google Scholar] [PubMed]
- Li, X.; Liu, Y.; Zhang, R.; Tan, J.; Chen, L.; Liu, Y. Meta-analysis of the association between plasminogen activator inhibitor-1 4G/5G polymorphism and recurrent pregnancy loss. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2015, 21, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, F.; Zarei, S.; Ahmadvand, N.; Akbarzadeh-Pasha, Z.; Savadi, E.; Zarnani, A.H.; Sadeghi, M.R.; Jeddi-Tehrani, M. Association between plasminogen activator inhibitor 1 gene mutation and different subgroups of recurrent miscarriage and implantation failure. J. Assist. Reprod. Genet. 2014, 31, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.; Hur, J.; Goodman, C.S.; Jeyendran, R.S.; Coulam, C. Are polymorphisms in the ACE and PAI-1 genes associated with recurrent spontaneous miscarriages? Am. J. Reprod. Immunol. 2009, 62, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Su, M.T.; Lin, S.H.; Chen, Y.C.; Kuo, P.L. Genetic association studies of ACE and PAI-1 genes in women with recurrent pregnancy loss: A systematic review and meta-analysis. Thromb. Haemost. 2013, 109, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Coulam, C.B.; Jeyendran, R.S.; Fishel, L.A.; Roussev, R. Multiple thrombophilic gene mutations rather than specific gene mutations are risk factors for recurrent miscarriage. Am. J. Reprod. Immunol. 2006, 55, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Poursadegh Zonouzi, A.; Chaparzadeh, N.; Ghorbian, S.; Sadaghiani, M.M.; Farzadi, L.; Ghasemzadeh, A.; Kafshdooz, T.; Sakhinia, M.; Sakhinia, E. The association between thrombophilic gene mutations and recurrent pregnancy loss. J. Assist. Reprod. Genet. 2013, 30, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, T.; Lohse, P.; Rogenhofer, N.; Kosian, E.; Pihusch, R.; Thaler, C.J. Polymorphisms in the ACE and PAI-1 genes are associated with recurrent spontaneous miscarriages. Hum. Reprod. 2003, 18, 2473–2477. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.; Sargent, I.L. Latest advances in understanding preeclampsia. Science 2005, 308, 1592–1594. [Google Scholar] [CrossRef] [PubMed]
- Sibai, B.M.; Stella, C.L. Diagnosis and management of atypical preeclampsia-eclampsia. Am. J. Obstet. Gynecol. 2009, 200, 481. [Google Scholar] [CrossRef] [PubMed]
- Irani, R.A.; Xia, Y. The functional role of the renin-angiotensin system in pregnancy and preeclampsia. Placenta 2008, 29, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Bodova, K.B.; Biringer, K.; Dokus, K.; Ivankova, J.; Stasko, J.; Danko, J. Fibronectin, plasminogen activator inhibitor type 1 (PAI-1) and uterine artery Doppler velocimetry as markers of preeclampsia. Dis. Mark. 2011, 30, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Purwosunu, Y.; Sekizawa, A.; Koide, K.; Farina, A.; Wibowo, N.; Wiknjosastro, G.H.; Okazaki, S.; Chiba, H.; Okai, T. Cell-free mRNA concentrations of plasminogen activator inhibitor-1 and tissue-type plasminogen activator are increased in the plasma of pregnant women with preeclampsia. Clin. Chem. 2007, 53, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Wikstrom, A.K.; Nash, P.; Eriksson, U.J.; Olovsson, M.H. Evidence of increased oxidative stress and a change in the plasminogen activator inhibitor (PAI)-1 to PAI-2 ratio in early-onset but not late-onset preeclampsia. Am. J. Obstet. Gynecol. 2009, 201, 597. [Google Scholar] [CrossRef] [PubMed]
- Estelles, A.; Gilabert, J.; Aznar, J.; Loskutoff, D.J.; Schleef, R.R. Changes in the plasma levels of type 1 and type 2 plasminogen activator inhibitors in normal pregnancy and in patients with severe preeclampsia. Blood 1989, 74, 1332–1338. [Google Scholar] [PubMed]
- Akolekar, R.; Cruz Jde, J.; Penco, J.M.; Zhou, Y.; Nicolaides, K.H. Maternal plasma plasminogen activator inhibitor-2 at 11 to 13 weeks of gestation in hypertensive disorders of pregnancy. Hypertens. Pregnancy 2011, 30, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Catarino, C.; Rebelo, I.; Belo, L.; Rocha, S.; Castro, E.B.; Patricio, B.; Quintanilha, A.; Santos-Silva, A. Relationship between maternal and cord blood hemostatic disturbances in preeclamptic pregnancies. Thromb. Res. 2008, 123, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Liu, C.X.; Gong, T.T.; Wu, Q.J.; Wu, L. Cigarette smoking during pregnancy and preeclampsia risk: A systematic review and meta-analysis of prospective studies. Oncotarget 2015, 6, 43667–43678. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.J.; Gray, R.S.; Moore, N.R.; Booth, N.A. The effects of chronic smoking on the fibrinolytic potential of plasma and platelets. Br. J. Haematol. 1997, 97, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Said, J.M.; Tsui, R.; Borg, A.J.; Higgins, J.R.; Moses, E.K.; Walker, S.P.; Monagle, P.T.; Brennecke, S.P. The PAI-1 4G/5G polymorphism is not associated with an increased risk of adverse pregnancy outcome in asymptomatic nulliparous women. J. Thromb. Haemost. 2012, 10, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Ciarmela, P.; Marzioni, D.; Islam, M.S.; Gray, P.C.; Terracciano, L.; Lorenzi, T.; Todros, T.; Petraglia, F.; Castellucci, M. Possible role of RKIP in cytotrophoblast migration: Immunohistochemical and in vitro studies. J. Cell. Physiol. 2012, 227, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Prutsch, N.; Fock, V.; Haslinger, P.; Haider, S.; Fiala, C.; Pollheimer, J.; Knofler, M. The role of interleukin-1β in human trophoblast motility. Placenta 2012, 33, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Anteby, E.Y.; Greenfield, C.; Natanson-Yaron, S.; Goldman-Wohl, D.; Hamani, Y.; Khudyak, V.; Ariel, I.; Yagel, S. Vascular endothelial growth factor, epidermal growth factor and fibroblast growth factor-4 and -10 stimulate trophoblast plasminogen activator system and metalloproteinase-9. Mol. Hum. Reprod. 2004, 10, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Meade, E.S.; Ma, Y.Y.; Guller, S. Role of hypoxia-inducible transcription factors 1α and 2α in the regulation of plasminogen activator inhibitor-1 expression in a human trophoblast cell line. Placenta 2007, 28, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Guller, S. Role of the syncytium in placenta-mediated complications of preeclampsia. Thromb. Res. 2009, 124, 389–392. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gerhardt, A.; Goecke, T.W.; Beckmann, M.W.; Wagner, K.J.; Tutschek, B.; Willers, R.; Bender, H.G.; Scharf, R.E.; Zotz, R.B. The G20210A prothrombin-gene mutation and the plasminogen activator inhibitor (PAI-1) 5G/5G genotype are associated with early onset of severe preeclampsia. J. Thromb. Haemost. 2005, 3, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.A.; Bombell, S.; McGuire, W. Association of plasminogen activator inhibitor-type 1 (-675 4G/5G) polymorphism with pre-eclampsia: Systematic review. PLoS ONE 2013, 8, e56907. [Google Scholar] [CrossRef] [PubMed]
- Dall’Asta, A.; Brunelli, V.; Prefumo, F.; Frusca, T.; Lees, C.C. Early onset fetal growth restriction. Matern. Health Neonatol. Perinat. 2017, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Seferovic, M.D.; Gupta, M.B. Increased Umbilical Cord PAI-1 Levels in Placental Insufficiency Are Associated with Fetal Hypoxia and Angiogenesis. Dis. Markers 2016, 2016, 7124186. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, I.M.; Horbar, J.D.; Badger, G.J.; Ohlsson, A.; Golan, A. Morbidity and mortality among very-low-birth-weight neonates with intrauterine growth restriction. The Vermont Oxford Network. Am. J. Obstet. Gynecol. 2000, 182, 198–206. [Google Scholar] [CrossRef]
- Murphy, V.E.; Smith, R.; Giles, W.B.; Clifton, V.L. Endocrine regulation of human fetal growth: The role of the mother, placenta, and fetus. Endocr. Rev. 2006, 27, 141–169. [Google Scholar] [CrossRef] [PubMed]
- Scifres, C.M.; Nelson, D.M. Intrauterine growth restriction, human placental development and trophoblast cell death. J. Physiol. 2009, 587, 3453–3458. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, B.L.; Bonnar, J. Uteroplacental hemostasis in intrauterine fetal growth retardation. Semin. Thromb. Hemost. 1999, 25, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Estelles, A.; Gilabert, J.; Keeton, M.; Eguchi, Y.; Aznar, J.; Grancha, S.; Espna, F.; Loskutoff, D.J.; Schleef, R.R. Altered expression of plasminogen activator inhibitor type 1 in placentas from pregnant women with preeclampsia and/or intrauterine fetal growth retardation. Blood 1994, 84, 143–150. [Google Scholar] [PubMed]
- Coolman, M.; Timmermans, S.; de Groot, C.J.; Russcher, H.; Lindemans, J.; Hofman, A.; Geurts-Moespot, A.J.; Sweep, F.C.; Jaddoe, V.V.; Steegers, E.A. Angiogenic and fibrinolytic factors in blood during the first half of pregnancy and adverse pregnancy outcomes. Obstet. Gynecol. 2012, 119, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Gilabert, J.; Estelles, A.; Ayuso, M.J.; Espana, F.; Chirivella, M.; Grancha, S.; Mico, J.M.; Aznar, J. Evaluation of plasminogen activators and plasminogen activator inhibitors in plasma and amniotic fluid in pregnancies complicated with intrauterine fetal growth retardation. Gynecol. Obstet. Investig. 1994, 38, 157–162. [Google Scholar] [CrossRef]
- De Boer, K.; ten Cate, J.W.; Sturk, A.; Borm, J.J.; Treffers, P.E. Enhanced thrombin generation in normal and hypertensive pregnancy. Am. J. Obstet. Gynecol. 1989, 160, 95–100. [Google Scholar] [CrossRef]
- Estelles, A.; Gilabert, J.; Espana, F.; Aznar, J.; Galbis, M. Fibrinolytic parameters in normotensive pregnancy with intrauterine fetal growth retardation and in severe preeclampsia. Am. J. Obstet. Gynecol. 1991, 165, 138–142. [Google Scholar] [CrossRef]
- Infante-Rivard, C.; Rivard, G.E.; Guiguet, M.; Gauthier, R. Thrombophilic polymorphisms and intrauterine growth restriction. Epidemiology 2005, 16, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Larciprete, G.; Rossi, F.; Deaibess, T.; Brienza, L.; Barbati, G.; Romanini, E.; Gioia, S.; Cirese, E. Double inherited thrombophilias and adverse pregnancy outcomes: Fashion or science? J. Obstet. Gynaecol. Res. 2010, 36, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Miehle, K.; Stepan, H.; Fasshauer, M. Leptin, adiponectin and other adipokines in gestational diabetes mellitus and pre-eclampsia. Clin. Endocrinol. 2012, 76, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Kwak, S.H.; Jang, H.C. Prevention of type 2 diabetes mellitus in women with previous gestational diabetes mellitus. Korean J. Intern. Med. 2017, 32, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Vrachnis, N.; Belitsos, P.; Sifakis, S.; Dafopoulos, K.; Siristatidis, C.; Pappa, K.I.; Iliodromiti, Z. Role of adipokines and other inflammatory mediators in gestational diabetes mellitus and previous gestational diabetes mellitus. Int. J. Endocrinol. 2012, 2012, 549748. [Google Scholar] [CrossRef] [PubMed]
- Seely, E.W.; Solomon, C.G. Insulin resistance and its potential role in pregnancy-induced hypertension. J. Clin. Endocrinol. Metab. 2003, 88, 2393–2398. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Pahor, M.; Incalzi, R.A. Plasminogen activator inhibitor-1 (PAI-1): A key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc. Ther. 2010, 28, 72–91. [Google Scholar] [CrossRef] [PubMed]
- Salmi, A.A.; Zaki, N.M.; Zakaria, R.; Nor Aliza, A.G.; Rasool, A.H. Arterial stiffness, inflammatory and pro-atherogenic markers in gestational diabetes mellitus. Vasa 2012, 41, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Bugatto, F.; Quintero-Prado, R.; Visiedo, F.M.; Vilar-Sanchez, J.M.; Figueroa-Quinones, A.; Lopez-Tinoco, C.; Torrejon, R.; Bartha, J.L. The influence of lipid and proinflammatory status on maternal uterine blood flow in women with late onset gestational diabetes. Reprod. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- McManus, R.; Summers, K.; de Vrijer, B.; Cohen, N.; Thompson, A.; Giroux, I. Maternal, umbilical arterial and umbilical venous 25-hydroxyvitamin D and adipocytokine concentrations in pregnancies with and without gestational diabetes. Clin. Endocrinol. 2014, 80, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Farhan, S.; Winzer, C.; Tura, A.; Quehenberger, P.; Bieglmaier, C.; Wagner, O.F.; Huber, K.; Waldhausl, W.; Pacini, G.; Kautzky-Willer, A. Fibrinolytic dysfunction in insulin-resistant women with previous gestational diabetes. Eur. J. Clin. Investig. 2006, 36, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Morimitsu, L.K.; Fusaro, A.S.; Sanchez, V.H.; Hagemann, C.C.; Bertini, A.M.; Dib, S.A. Fibrinolytic dysfunction after gestation is associated to components of insulin resistance and early type 2 diabetes in latino women with previous gestational diabetes. Diabetes Res. Clin. Pract. 2007, 78, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Lowe, L.P.; Metzger, B.E.; Lowe, W.L., Jr.; Dyer, A.R.; McDade, T.W.; McIntyre, H.D.; Group, H.S.C.R. Inflammatory mediators and glucose in pregnancy: Results from a subset of the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study. J. Clin. Endocrinol. Metab. 2010, 95, 5427–5434. [Google Scholar] [CrossRef] [PubMed]
- Cawyer, C.R.; Horvat, D.; Leonard, D.; Allen, S.R.; Jones, R.O.; Zawieja, D.C.; Kuehl, T.J.; Uddin, M.N. Hyperglycemia impairs cytotrophoblast function via stress signaling. Am. J. Obstet. Gynecol. 2014, 211, 541. [Google Scholar] [CrossRef] [PubMed]
- Leipold, H.; Knoefler, M.; Gruber, C.; Klein, K.; Haslinger, P.; Worda, C. Plasminogen activator inhibitor 1 gene polymorphism and gestational diabetes mellitus. Obstet. Gynecol. 2006, 107, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Gingras, V.; Vigneault, J.; Weisnagel, S.J.; Tchernof, A.; Robitaille, J. Accelerometry-measured physical activity and inflammation after gestational diabetes. Med. Sci. Sports Exerc. 2013, 45, 1307–1312. [Google Scholar] [CrossRef] [PubMed]
- Glueck, C.J.; Phillips, H.; Cameron, D.; Wang, P.; Fontaine, R.N.; Moore, S.K.; Sieve-Smith, L.; Tracy, T. The 4G/4G polymorphism of the hypofibrinolytic plasminogen activator inhibitor type 1 gene: An independent risk factor for serious pregnancy complications. Metabolism 2000, 49, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Greene, A.D.; Lang, S.A.; Kendziorski, J.A.; Sroga-Rios, J.M.; Herzog, T.J.; Burns, K.A. Endometriosis: Where are we and where are we going? Reproduction 2016, 152, R63–R78. [Google Scholar] [CrossRef] [PubMed]
- Bruse, C.; Guan, Y.; Carlberg, M.; Carlstrom, K.; Bergqvist, A. Basal release of urokinase plasminogen activator, plasminogen activator inhibitor-1, and soluble plasminogen activator receptor from separated and cultured endometriotic and endometrial stromal and epithelial cells. Fertil. Steril. 2005, 83, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Gilabert-Estelles, J.; Estelles, A.; Gilabert, J.; Castello, R.; Espana, F.; Falco, C.; Romeu, A.; Chirivella, M.; Zorio, E.; Aznar, J. Expression of several components of the plasminogen activator and matrix metalloproteinase systems in endometriosis. Hum. Reprod. 2003, 18, 1516–1522. [Google Scholar] [CrossRef] [PubMed]
- Ramon, L.A.; Gilabert-Estelles, J.; Cosin, R.; Gilabert, J.; Espana, F.; Castello, R.; Chirivella, M.; Romeu, A.; Estelles, A. Plasminogen activator inhibitor-1 (PAI-1) 4G/5G polymorphism and endometriosis. Influence of PAI-1 polymorphism on PAI-1 antigen and mRNA expression. Thromb. Res. 2008, 122, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Gentilini, D.; Vigano, P.; Castaldi, D.; Mari, D.; Busacca, M.; Vercellini, P.; Somigliana, E.; di Blasio, A.M. Plasminogen activator inhibitor-1 4G/5G polymorphism and susceptibility to endometriosis in the Italian population. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 146, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Goncalves-Filho, R.P.; Brandes, A.; Christofolini, D.M.; Lerner, T.G.; Bianco, B.; Barbosa, C.P. Plasminogen activator inhibitor-1 4G/5G polymorphism in infertile women with and without endometriosis. Acta Obstet. Gynecol. Scand. 2011, 90, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.O.; Dumesic, D.A.; Chazenbalk, G.; Azziz, R. Polycystic ovary syndrome: Etiology, pathogenesis and diagnosis. Nat. Rev. Endocrinol. 2011, 7, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Koiou, E.; Tziomalos, K.; Katsikis, I.; Delkos, D.; Tsourdi, E.A.; Panidis, D. Disparate effects of pharmacotherapy on plasma plasminogen activator inhibitor-1 levels in women with the polycystic ovary syndrome. Hormones 2013, 12, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Koiou, E.; Tziomalos, K.; Dinas, K.; Katsikis, I.; Kandaraki, E.A.; Tsourdi, E.; Mavridis, S.; Panidis, D. Plasma plasminogen activator inhibitor-1 levels in the different phenotypes of the polycystic ovary syndrome. Endocr. J. 2012, 59, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Orio, F., Jr.; Palomba, S.; Cascella, T.; Tauchmanova, L.; Nardo, L.G.; di Biase, S.; Labella, D.; Russo, T.; Savastano, S.; Tolino, A.; et al. Is plasminogen activator inhibitor-1 a cardiovascular risk factor in young women with polycystic ovary syndrome? Reprod. Biomed. Online 2004, 9, 505–510. [Google Scholar] [CrossRef]
- Tarkun, I.; Canturk, Z.; Arslan, B.C.; Turemen, E.; Tarkun, P. The plasminogen activator system in young and lean women with polycystic ovary syndrome. Endocr. J. 2004, 51, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Palomba, S.; Orio, F., Jr.; Falbo, A.; Russo, T.; Tolino, A.; Zullo, F. Plasminogen activator inhibitor 1 and miscarriage after metformin treatment and laparoscopic ovarian drilling in patients with polycystic ovary syndrome. Fertil. Steril. 2005, 84, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Fauser, B.C.; Tarlatzis, B.C.; Rebar, R.W.; Legro, R.S.; Balen, A.H.; Lobo, R.; Carmina, E.; Chang, J.; Yildiz, B.O.; Laven, J.S.; et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): The Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil. Steril. 2012, 97, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.X. Plasminogen activator/plasminogen activator inhibitors in ovarian physiology. Front. Biosci. 2004, 9, 3356–3373. [Google Scholar] [CrossRef] [PubMed]
- Atiomo, W.U.; Hilton, D.; Fox, R.; Lee, D.; Shaw, S.; Friend, J.; Wilkin, T.J.; Prentice, A.G. Immunohistochemical detection of plasminogen activator inhibitor-1 in polycystic ovaries. Gynecol. Endocrinol. 2000, 14, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Glueck, C.J.; Wang, P.; Fontaine, R.N.; Sieve-Smith, L.; Tracy, T.; Moore, S.K. Plasminogen activator inhibitor activity: An independent risk factor for the high miscarriage rate during pregnancy in women with polycystic ovary syndrome. Metabolism 1999, 48, 1589–1595. [Google Scholar] [CrossRef]
- Meigs, J.B.; O’Donnell, C.J.; Tofler, G.H.; Benjamin, E.J.; Fox, C.S.; Lipinska, I.; Nathan, D.M.; Sullivan, L.M.; D’Agostino, R.B.; Wilson, P.W. Hemostatic markers of endothelial dysfunction and risk of incident type 2 diabetes: The Framingham Offspring Study. Diabetes 2006, 55, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Patterson, C.; Yarnell, J.; Rumley, A.; Ben-Shlomo, Y.; Lowe, G. Which hemostatic markers add to the predictive value of conventional risk factors for coronary heart disease and ischemic stroke? The Caerphilly Study. Circulation 2005, 112, 3080–3087. [Google Scholar] [CrossRef] [PubMed]
- Yarmolinsky, J.; Bordin Barbieri, N.; Weinmann, T.; Ziegelmann, P.K.; Duncan, B.B.; Ines Schmidt, M. Plasminogen activator inhibitor-1 and type 2 diabetes: A systematic review and meta-analysis of observational studies. Sci. Rep. 2016, 6, 17714. [Google Scholar] [CrossRef] [PubMed]
- Sales, M.F.; Soter, M.O.; Candido, A.L.; Fernandes, A.P.; Oliveira, F.R.; Ferreira, A.C.; Sousa, M.O.; Ferreira, C.N.; Gomes, K.B. Correlation between plasminogen activator inhibitor-1 (PAI-1) promoter 4G/5G polymorphism and metabolic/proinflammatory factors in polycystic ovary syndrome. Gynecol. Endocrinol. 2013, 29, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Song, G.G. Plasminogen activator inhibitor-1 4G/5G and the MTHFR 677C/T polymorphisms and susceptibility to polycystic ovary syndrome: A meta-analysis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 175, 8–14. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).