Principal Aspects Regarding the Maintenance of Mammalian Mitochondrial Genome Integrity


Abstract
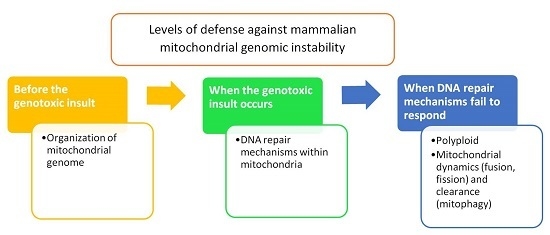
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Organization of Mitochondrial Genome
3. Mitochondrial DNA Repair Machinery
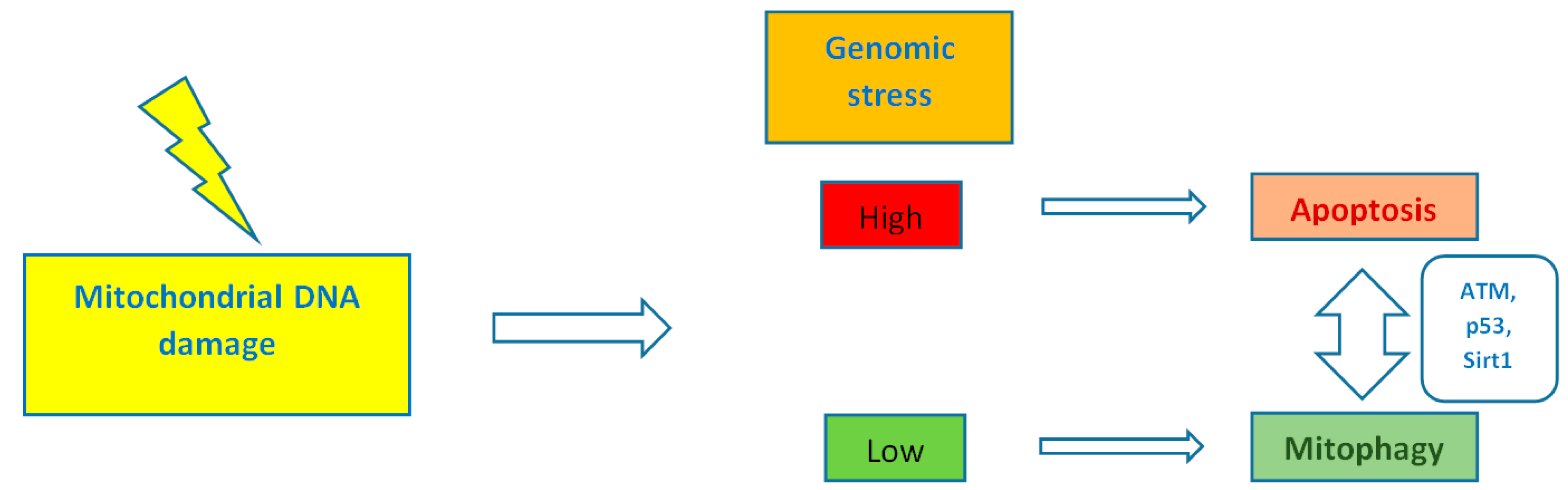
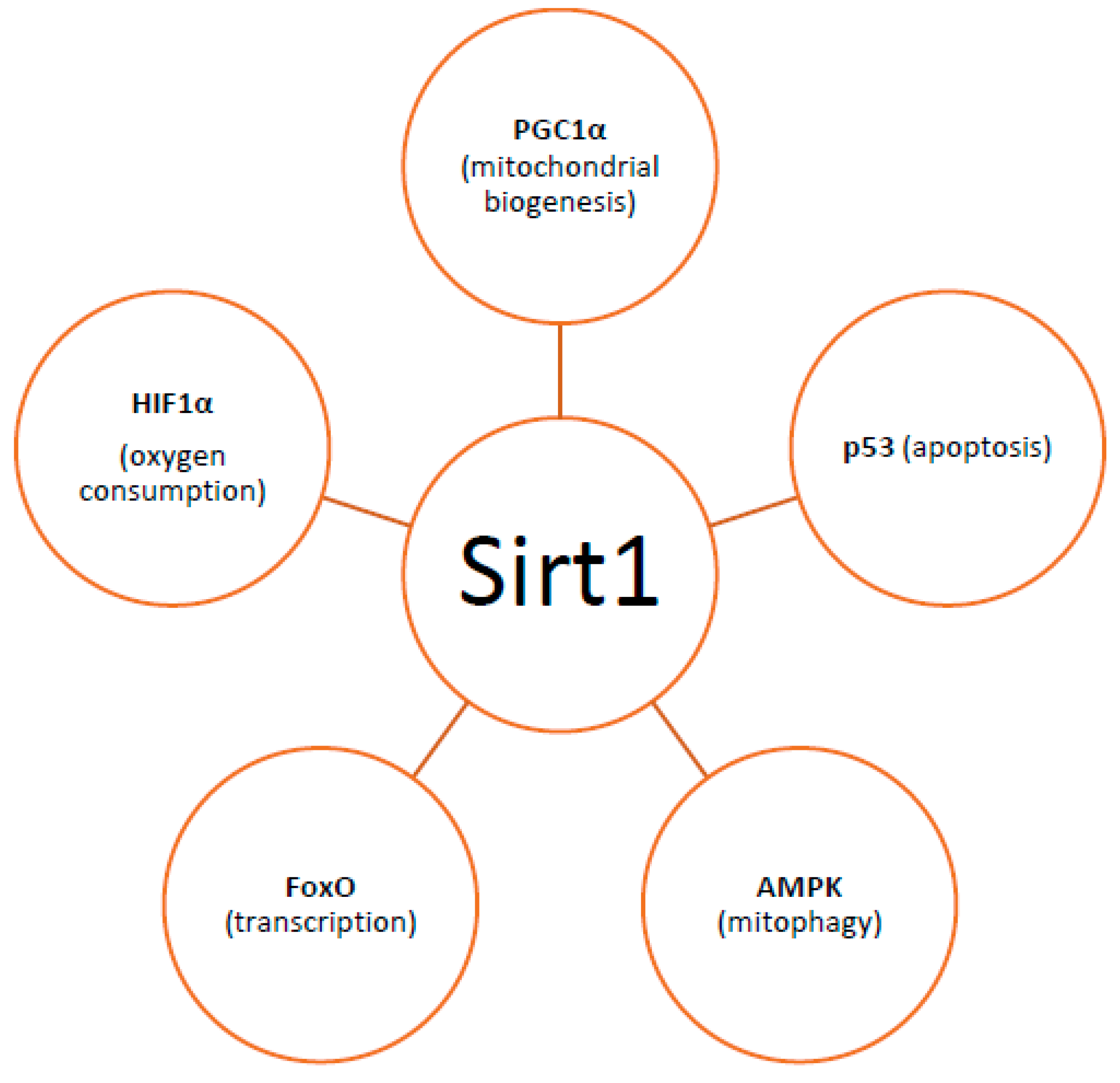
4. Heteroplasmy, Mitochondrial Dynamics, and Mitophagy
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Gonzalez-Freire, M.; de Cabo, R.; Bernier, M.; Sollott, S.J.; Fabbri, E.; Navas, P.; Ferrucci, L. Reconsidering the role of mitochondria in aging. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Garesse, R.; Vallejo, C.G. Animal mitochondrial biogenesis and function: A regulatory cross-talk between two genomes. Gene 2001, 263, 1–16. [Google Scholar] [CrossRef]
- Bogenhagen, D.F. Mitochondrial DNA nucleoid structure. Biochim. Biophys. Acta 2012, 1819, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Why do we still have maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu. Rev. Biochem. 2007, 76, 781–821. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.C.; Gomes, K.M.; Ferreira, J.C. Impact of exercise training on redox signaling in cardiovascular diseases. Food Chem. Toxicol. 2013, 62, 107–119. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Georgakilas, A.G.; Bonner, W.M. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat. Res. 2010, 704, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Cline, S.D. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta 2012, 1819, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Gayarre, J.; Duran-Trío, L.; Criado Garcia, O.; Aguado, C.; Juana-López, L.; Crespo, I.; Knecht, E.; Bovolenta, P.; Rodríguez de Córdoba, S. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell. Biol. 2016, 18, 777–789. [Google Scholar] [Green Version]
- Nass, M.M.; Nass, S. Intramitochondrial fibers with DNA characteristics. I. Fixation and electron staining reactions. J. Cell. Biol. 1963, 19, 593–611. [Google Scholar] [CrossRef] [PubMed]
- Schatz, G.; Haslbrunner, E.; Tuppy, H. Deoxyribonucleic acid associated with yeast mitochondria. Biochem. Biophys. Res. Commun. 1964, 15, 127–132. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A. Sanger Fsequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Bendich, A.J. The size and form of chromosomes are constant in the nucleus, but highly variable in bacteria, mitochondria and chloroplasts. Bioessays 2007, 29, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Bebenek, K.; Kunkel, T.A. Functions of DNA polymerases. Adv. Protein Chem. 2004, 69, 137–165. [Google Scholar] [PubMed]
- Ropp, P.A.; Copeland, W.C. Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase gamma. Genomics 1996, 36, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C.; Longley, M.J. Mitochondrial genome maintenance in health and disease. DNA Repair (Amst.) 2014, 19, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Longley, M.J.; Nguyen, D.; Kunkel, T.A.; Copeland, W.C. The fidelity of human DNA polymerase γ with and without exonucleolytic proofreading and the p55 accessory subunit. J. Biol. Chem. 2001, 276, 38555–38562. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Pham, X.H.; Pellegrini, M.; Falkenberg, M. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004, 23, 2423–2429. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C. Inherited mitochondrial diseases of DNA replication. Annu. Rev. Med. 2008, 59, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Longley, M.J.; Graziewicz, M.A.; Bienstock, R.J.; Copeland, W.C. Consequences of mutations in human DNA polymerase γ. Gene 2005, 354, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, J.D.; Copeland, W.C. Mitochondrial DNA replication and disease: Insights from DNA polymerase γ mutations. Cell. Mol. Life Sci. 2011, 68, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Mott, J.L.; Chang, S.W.; Denniger, G.; Feng, Z.; Zassenhaus, H.P. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics 2000, 69, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Tyynismaa, H.; Sembongi, H.; Bokori-Brown, M.; Granycome, C.; Ashley, N.; Poulton, J.; Jalanko, A.; Spelbrink, J.N.; Holt, I.J.; Suomalainen, A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum. Mol. Genet. 2004, 13, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- Spelbrink, J.N.; Li, F.Y.; Tiranti, V.; Nikali, K.; Yuan, Q.P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Majander, A.; Haltia, M.; Somer, H.; Lönnqvist, J.; Savontaus, M.L.; Peltonen, L. Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J. Clin. Investig. 1992, 90, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Servidei, S.; Gellera, C.; Bertini, E.; DiMauro, S.; DiDonato, S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the d-loop region. Nature 1989, 339, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Nikali, K.; Suomalainen, A.; Saharinen, J.; Kuokkanen, M.; Spelbrink, J.N.; Lönnqvist, T.; Peltonen, L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum. Mol. Genet. 2005, 14, 2981–2990. [Google Scholar] [CrossRef] [PubMed]
- Hakonen, A.H.; Goffart, S.; Marjavaara, S.; Paetau, A.; Cooper, H.; Mattila, K.; Lampinen, M.; Sajantila, A.; Lönnqvist, T.; Spelbrink, J.N.; et al. Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum. Mol. Genet. 2008, 17, 3822–3835. [Google Scholar] [CrossRef] [PubMed]
- Lӧnnqvist, T.; Paetau, A.; Nikali, K.; von Boguslawski, K.; Pihko, H. Infantile onset spinocerebellar ataxia with sensory neuropathy (IOSCA): Neuropathological features. J. Neurol. Sci. 1998, 161, 57–65. [Google Scholar] [CrossRef]
- Nikkanen, J.; Forsstrӧm, S.; Euro, L.; Paetau, I.; Kohnz, R.A.; Wang, L.; Chilov, D.; Viinamäki, J.; Roivainen, A.; Marjamäki, P.; et al. Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell. Metabol. 2016, 23, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1α. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Hansson, A.; Hance, N.; Dufour, E.; Rantanen, A.; Hultenby, K.; Clayton, D.A.; Wibom, R.; Larsson, N.G. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc. Natl. Acad. Sci. USA 2004, 101, 3136–3141. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation 2007, 115, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- François, R.; Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Bayeva, M.; Gheorghiade, M.; Ardehali, H. Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol. 2013, 61, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.; Fortin, D.; Deloménie, C.; Momken, I.; Veksler, V.; Ventura-Clapier, R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. 2003, 551, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W. Mitochondrial DNA nucleoids determine mitochondrial genetics and dysfunction. Int. J. Biochem. Cell. Biol. 2009, 41, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Kukat, C.; Wurm, C.A.; Spåhr, H.; Falkenberg, M.; Larsson, N.G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [PubMed]
- Iborra, F.J.; Kimura, H.; Cook, P.R. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2004, 2, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobs, S.; Wurm, C.A. Super-resolution microscopy of mitochondria. Curr. Opin. Chem. Biol. 2014, 20, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Parisi, M.A.; Clayton, D.A. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science 1991, 252, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Kanki, T.; Ohgaki, K.; Gaspari, M.; Gustafsson, C.M.; Fukuoh, A.; Sasaki, N.; Hamasaki, N.; Kang, D. Architectural role of mitochondrial transcription factor a in maintenance of human mitochondrial DNA. Mol. Cell. Biol. 2004, 24, 9823–9834. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, M.; Gaspari, M.; Rantanen, A.; Trifunovic, A.; Larsson, N.G.; Gustafsson, C.M. Mitochondrial transcription factors B1 and B2activate transcription of human mtDNA. Nat. Genet. 2002, 31, 289–294. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, V.; Seidel-Rogol, B.L.; Shadel, G.C. A human mitochondrial transcription factor is related to RNA adenine methyltransferases and binds S-adenosylmethionine. Mol. Cell. Biol. 2002, 22, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.I.; Kanki, T.; Muta, T.; Ukaji, K.; Abe, Y.; Nakayama, H.; Takio, K.; Hamasaki, N.; Kang, D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003, 31, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wilhelmsson, H.; Graff, C.; Li, H.; Oldfors, A.; Rustin, P.; Brüning, J.C.; Kahn, C.R.; Clayton, D.A.; Barsh, G.S.; et al. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat. Genet. 1999, 21, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Garrido, N.; Griparic, L.; Jokitalo, E.; Wartiovaara, J.; van der Bliek, A.M.; Spelbrink, J.N. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell 2003, 14, 1583–1596. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 2008, 283, 3665–3675. [Google Scholar] [CrossRef] [PubMed]
- Osman, C.; Haag, M.; Potting, C.; Rodenfels, J.; Dip, P.V.; Wieland, F.T.; Brügger, B.; Westermann, B.; Langer, T. The genetic interactome of prohibitins: Coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J. Cell. Biol. 2009, 184, 583–596. [Google Scholar] [CrossRef] [PubMed]
- McClung, J.K.; Jupe, E.R.; Liu, X.T.; Dell’Orco, R.T. Prohibitin: Potential role in senescence, development, and tumor suppression. Exp. Gerontol. 1995, 30, 99–124. [Google Scholar] [CrossRef]
- Nijtmans, L.G.; Artal, S.M.; Grivell, L.A.; Coates, P.J. The mitochondrial PHB complex: Rolesin mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell. Mol. Life Sci. 2002, 59, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, K.; Sumitani, M.; Satoh, M.; Endo, H. Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Exp. Cell Res. 2008, 314, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Dargazanli, S.; Tatsuta, T.; Geimer, S.; Löwer, B.; Wunderlich, F.T.; von Kleist-Retzow, J.C.; Waisman, A.; Westermann, B.; Langer, T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008, 22, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, K.; Ohta, E.; Kagawa, Y.; Endo, H. Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J. Biol. Chem. 2006, 281, 36401–36410. [Google Scholar] [CrossRef] [PubMed]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P. OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain 2008, 131, 338–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, G.; Amati-Bonneau, P.; Blakely, E.L.; Stewart, J.D.; He, L.; Schaefer, A.M.; Griffiths, P.G.; Ahlqvist, K.; Suomalainen, A.; Reynier, P.; et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: A novel disorder of mtDNA maintenance. Brain 2008, 131, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkwirth, C.; Langer, T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta 2009, 793, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, G.; Wang, S.; Chellappan, S. Differential regulation of Rb family proteins and prohibitin during camptothecin-induced apoptosis. Oncogene 2002, 21, 4539–4548. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.M.; Yang, Y.; Ylikalli, E.; Khairullin, R.; Woldegebriel, R.; Lin, K.L.; Euro, L.; Palin, E.; Wolf, A.; Trokovic, R.; et al. ATPase-deficient mitochondrial inner membrane protein ATAD3A disturbs mitochondrial dynamics in dominant hereditary spastic paraplegia. Hum. Mol. Genet. 2017, 26, 1432–1443. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Rousseau, D.J. ATAD3, a vital membrane bound mitochondrial ATPase involved in tumor progression. J. Bioenerg. Biomembr. 2012, 44, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.A. Replication of animal mitochondrial DNA. Cell 1982, 28, 693–705. [Google Scholar] [CrossRef]
- Gilquin, B.; Taillebourg, E.; Cherradi, N. The AAA+ATPase ATAD3A controls mitochondrial dynamics at the interface of the inner and outer membrane. Mol. Cell. Biol. 2010, 30, 1984–1996. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.Y.; Chang, C.L.; Hsu, S.H.; Huang, C.Y.; Chiang, S.F.; Chiou, S.H.; Huang, C.H.; Hsiao, Y.T.; Lin, T.Y.; Chiang, I.P.; et al. ATPase family AAA domain-containing 3A is a novel anti-apoptotic factor in lung adenocarcinoma cells. J. Cell Sci. 2010, 123, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Mao, C.C.; Reyes, A.; Sembongi, H.; Di Re, M.; Granycome, C.; Clippingdale, A.B.; Fearnley, I.M.; Harbour, M.; Robinson, A.J.; et al. The AAA+ protein ATAD3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J. Cell Biol. 2007, 176, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.; Frazier, A.E.; Durigon, R.; Patel, H.; Jones, A.W.; Dalla Rosa, I.; Lake, N.J.; Compton, A.G.; Mountford, H.S.; Tucker, E.J.; et al. ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 2017, 140, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Ahn, J.; Walker, K.K.; Hoffman, W.H.; Evans, R.M.; Levine, A.J.; George, D.L. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev. 1999, 13, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- Achanta, G.; Sasaki, R.; Feng, L.; Carew, J.S.; Lu, W.; Pelicano, H.; Keating, M.J.; Huang, P. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA pol γ. EMBO J. 2005, 24, 3482–3492. [Google Scholar] [CrossRef] [PubMed]
- Lilling, G.; Novitsky, E.; Sidi, Y.; Bakhanashvili, M. p53-associated 3′→5′ exonuclease activity in nuclear and cytoplasmic compartments of the cells. Oncogene 2003, 22, 233–245. [Google Scholar] [PubMed]
- Heyne, K.; Mannebach, S.; Wuertz, E.; Knaup, K.X.; Mahyar-Roemer, M.; Roemer, K. Identification of a putative p53 binding sequence within the human mitochondrial genome. FEBS Lett. 2004, 578, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Bakhanashvili, M.; Grinberg, S.; Bondal, E.; Simon, A.J.; Moshitch-Moshkovitz, S.; Rahav, G. p53 in mitochondria enhances the accuracy of DNA synthesis. Cell Death Differ. 2008, 15, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Marchenko, N.D.; Zaika, A.; Moll, U.M. Death signal-induced localization of p53 protein to mitochondria. J. Biol. Chem. 2000, 275, 16202–16212. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, H.T.; Lehtinen, S.K.; Spelbrink, J.N. No sex please, we’re mitochondria: A hypothesis on the somatic unit of inheritance of mammalian mtDNA. Bioessays 2000, 22, 564–572. [Google Scholar] [CrossRef]
- D’ Aurelio, M.; Gajewski, C.D.; Lin, M.T.; Mauck, W.M.; Shao, L.Z.; Lenaz, G.; Moraes, C.T.; Manfredi, G. Heterologous mitochondrial DNA recombination in human cells. Hum. Mol. Genet. 2004, 13, 3171–3179. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.W.; Schon, E.A.; Hernandez, E.; Davidson, M.M. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J. Cell Biol. 2008, 181, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Doublié, S. Base excision repair in the mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity-critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Tatarenkov, A.; Avise, J.C. Rapid concerted evolution in animal mitochondrial DNA. Proc. Biol. Sci. 2007, 274, 1795–1798. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear-DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Anson, R.M.; Hudson, E.; Bohr, V.A. Mitochondrial endogenous oxidative damage has been overestimated. FASEB J. 2000, 14, 355–360. [Google Scholar] [PubMed]
- Alexeyev, M.F. Is there more to aging than mitochondrial DNA and reactive oxygen species? FEBS J. 2009, 276, 5768–5787. [Google Scholar] [CrossRef] [PubMed]
- Burney, S.; Caulfield, J.L.; Niles, J.C.; Wishnok, J.S.; Tannenbaum, S.R. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 1999, 424, 37–49. [Google Scholar] [CrossRef]
- Burney, S.; Niles, J.C.; Dedon, P.C.; Tannenbaum, S.R. DNA damage in deoxynucleo-sides and oligonucleotides treated with peroxynitrite. Chem. Res. Toxicol. 1999, 12, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Goszcz, K.; Deakin, S.J.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Antioxidants in cardiovascular therapy: Panacea or false hope? Front. Cardiovasc. Med. 2015, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hyperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [PubMed]
- Singer, T.P.; Ramsay, R.R. Mechanism of the neurotoxicity of MPTP: An update. FEBS Lett. 1990, 274, 1–8. [Google Scholar] [PubMed]
- Maybury, B.D. Mitochondrial DNA damage is uncommon in cancer but can promote aggressive behavior. Anticancer Res. 2013, 33, 3543–3552. [Google Scholar] [PubMed]
- Boesch, P.; Weber-Lotfi, F.; Ibrahim, N. DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim. Biophys. Acta 2013, 1813, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.A.; Mayard, S.; Hashiguchi, K.; Souza-Pinto, N.C.; Bohr, V.A. Localization of mitochondrial DNA base excision repair to an inner membrane-associated particulate fraction. Nucl. Acids Res. 2005, 33, 3722–3732. [Google Scholar] [CrossRef] [PubMed]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-Hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; Sunesen, M.; Bohr, V.A.; Stevnsner, T. Repair of 8-oxoG is slower in endogenous nuclear genes than in mitochondrial DNA and is without strand bias. DNA Repair (Amst.) 2002, 1, 261–273. [Google Scholar] [CrossRef]
- Akbari, M.; Visnes, T.; Krokan, H.E.; Otterlei, M. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair 2008, 7, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Boesch, P.; Ibrahim, N.; Dietrich, A.; Lightowlers, R.N. Membrane association of mitochondrial DNA facilitates base excision repair in mammalian mitochondria. Nucleic. Acids Res. 2010, 38, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Qian, L.; Sung, J.S.; de Souza-Pinto, N.C.; Zheng, L.; Bogenhagen, D.F.; Bohr, V.A.; Wilson, D.M.; Shen, B.; Demple, B. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol. Cell. Biol. 2008, 28, 4975–4987. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Tann, A.W.; Longley, M.J.; Copeland, W.C.; Mitra, S. Long patch base excision repair in mammalian mitochondrial genomes. J. Biol. Chem. 2008, 283, 26349–26356. [Google Scholar] [CrossRef] [PubMed]
- Stierum, R.H.; Dianov, G.L.; Bohr, V.A. Single-nucleotide patch base excision repair of uracil in DNA by mitochondrial protein extracts. Nucleic. Acids Res. 1999, 27, 3712–3719. [Google Scholar] [CrossRef] [PubMed]
- Hanssen-Bauer, A.; Solvang-Garten, K.; Sundheim, O.; Peña-Diaz, J.; Andersen, S.; Slupphaug, G.; Krokan, H.E.; Wilson, D.M.; Akbari, M.; Otterlei, M. XRCC1 coordinates disparate responses and multiprotein repair complexes depending on the nature and context of the DNA damage. Environ. Mol. Mutagen. 2011, 52, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Maynard, S.; Bayne, A.C.; Sykora, P.; Tian, J.; de Souza-Pinto, N.C.; Croteau, D.L.; Bohr, V.A. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair (Amst.) 2010, 9, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Kanno, S.; Akbari, M.; Kulikowicz, T.; Baptiste, B.A.; Leandro, G.S.; Lu, H.; Tian, J.; May, A.; Becker, K.A. DNA polymerase beta participates in mitochondrial DNA repair. Mol. Cell. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kamenisch, Y.; Fousteri, M.; Knoch, J.; von Thaler, A.K.; Fehrenbacher, B.; Kato, H.; Becker, T.; Dollé, M.E.; Kuiper, R.; Majora, M.; et al. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J. Exp. Med. 2010, 207, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Wilson, D.M.; Bohr, V.A. Repair of persistent strand breaks in the mitochondrial genome. Mech. Ageing Dev. 2012, 133, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.A.; Matheson, E.C.; Hall, A.G.; Lightowlers, R.N. Mismatch repair activity in mammalian mitochondria. Nucleic. Acids Res. 2003, 31, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- De Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair (Amst.) 2009, 8, 704–719. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C.; Kasiviswanathan, R.; Longley, M.J. Analysis of translesion DNA synthesis by the mitochondrial DNA polymerase γ. Methods Mol. Biol. 2016, 1351, 19–26. [Google Scholar] [PubMed]
- Graziewicz, M.A.; Longley, M.J.; Copeland, W.C. DNA polymerase γ in mitochondrial DNA replication and repair. Chem. Rev. 2006, 106, 383–405. [Google Scholar] [CrossRef] [PubMed]
- Bailey, L.J.; Doherty, A.J. Mitochondrial DNA replication: A PrimPol perspective. Biochem. Soc. Trans. 2017, 45, 513–529. [Google Scholar] [CrossRef] [PubMed]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Futami, K.; Shimamoto, A.; Furuichi, Y. Mitochondrial and nuclear localization of human PIF1 helicase. Biol. Pharm. Bull. 2007, 30, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Dunaway, S.; Ivessa, A.S. The role of Pif1p, a DNA helicase in Saccharomyces cerevisiae, in maintaining mitochondrial DNA. Mitochondrion 2007, 7, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Foury, F.; Kolodynski, J. PIF mutation blocks recombination between mitochondrial ρ+ and ρ− genomes having tandemly arrayed repeat units in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1983, 80, 5345–5349. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; Zhou, J.; Kaplan, D.L.; Spelbrink, J.N.; Mergny, J.L.; Brosh, R.M., Jr. DNA sequences proximal to human mitochondrial DNA deletion breakpoints prevalent in human disease form G-quadruplexes, a class of DNA structures inefficiently unwound by the mitochondrial replicative Twinkle helicase. J. Biol. Chem. 2014, 289, 29975–29993. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Berg-Alonso, L.; Augé, G.; Fragaki, K.; Kolesar, J.E.; Lespinasse, F.; Lacas-Gervais, S.; Burel-Vandenbos, F.; Villa, E.; Belmonte, F.; et al. Inactivation of Pif1 helicase causes a mitochondrial myopathy in mice. Mitochondrion 2016, 30, 126–137. [Google Scholar] [CrossRef] [PubMed]
- LeDoux, S.P.; Wilson, G.L.; Beecham, E.J.; Stevnsner, T.; Wassermann, K.; Bohr, V.A. Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis 1992, 13, 1967–1973. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.A.; Saffhill, R.; O’Connor, P.J. Repair of alkylated purines in the hepatic DNA of mitochondria and nuclei in the rat. Carcinogenesis 1988, 9, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.S.; Huh, N.; Rajewsky, M.F.; Kuroki, T. Enzymatic removal of O6-ethylguanine from mitochondrial DNA in rat tissues exposed to N-ethyl-N-nitrosoureain vivo. J. Biol. Chem. 1988, 263, 6854–6856. [Google Scholar] [PubMed]
- Baccarelli, A.A.; Byun, H.M. Platelet mitochondrial DNA methylation: A potential new marker of cardiovascular disease. Clin. Epigenetics 2015, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.M.; Panni, T.; Motta, V.; Hou, L.; Nordio, F.; Apostoli, P.; Bertazzi, P.A.; Baccarelli, A.A. Effects of airborne pollutants on mitochondrial DNA methylation. Part. Fibre Toxicol. 2013, 10, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, H.M.; Baccarelli, A.A. Environmental exposure and mitochondrial epigenetics: Study design and analytical challenges. Hum. Genet. 2014, 133, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Castegna, A.; Iacobazzi, F.; Spera, I.; Scala, I.; Andria, G.; Iacobazzi, V. Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down’s syndrome. Mol. Genet. Metab. 2011, 102, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Xiong, L.; Ji, Z.; Cheng, W.; Yang, H. Correlation between increased ND2 expression and demethylated displacement loop of mtDNA in colorectal cancer. Mol. Med. Rep. 2012, 6, 125–130. [Google Scholar] [PubMed]
- Lakshmipathy, U.; Campbell, C. Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res. 1999, 27, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, B.; Padua, R.A.; Campbell, C. Mammalian mitochondria possess homologous DNA recombination activity. J. Biol. Chem. 1996, 271, 27536–27543. [Google Scholar]
- Coffey, G.; Lakshmipathy, U.; Campbell, C. Mammalian mitochondrial extracts possess DNA end-binding activity. Nucleic Acids Res. 1999, 27, 3348–3354. [Google Scholar] [CrossRef] [PubMed]
- Dynan, W.S.; Yoo, S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998, 26, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Lakshmipathy, U.; Campbell, C. The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol. Cell. Biol. 1999, 19, 3869–3876. [Google Scholar] [CrossRef] [PubMed]
- Tadi, K.S.; Sebastian, R.; Dahal, S.; Babu, R.K.; Choudhary, B.; Raghavan, S.C. Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol. Biol. Cell. 2016, 27, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Meissner, C.; Bruse, P.; Mohamed, S.A.; Schulz, A.; Warnk, H.; Storm, T.; Oehmichen, M. The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: A useful biomarker or more? Exp. Gerontol. 2008, 43, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Cortopassi, G.A.; Arnheim, N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990, 18, 6927–6933. [Google Scholar] [CrossRef] [PubMed]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J. Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial DNA. Microbiol. Mol. Biol. Rev. 2013, 77, 476–496. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Palacin, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Kalkhoran, S.B.; Hernández-Reséndiz, S.; Samangouei, P.; Ong, S.G.; Hausenloy, D.J. Mitochondrial-shaping proteins in cardiac health and disease-the long and the short of it! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W. Mitochondrial dynamism and heart disease: Changing shape and shaping change. EMBO Mol. Med. 2015, 7, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell. Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM proteinkinase: Regulating the cellularresponse to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Turenne, G.A.; Paul, P.; Laflair, L.; Price, B.D. Activation of p53 transcriptional activity requires ATM’s kinase domain and multiple N-terminal serine residues of p53. Oncogene 2001, 20, 5100–5110. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Sirt1 and the mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [PubMed]
- Yoshii, S.R.; Mizushima, N. Autophagy machinery in the context of mammalian mitophagy. Biochim. Biophys. Acta 2015, 1853, 2797–2801. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negativecontrol of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively down regulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Clay Montier, L.L.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genom. 2009, 36, 125–131. [Google Scholar] [CrossRef]
- Rotig, A.; Poulton, J. Genetic causes of mitochondrial DNA depletion in humans. Biochim. Biophys. Acta 2009, 1792, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, D.T.; McAllister, K.; Worth, L. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ. Health Perspect. 2014, 122, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacman, S.R.; Williams, S.L.; Moraes, C.T. Intra- and inter-molecular recombination of mitochondrial DNA after in vivo induction of multiple double-strand breaks. Nucleic Acids Res. 2009, 37, 4218–4226. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C. Defects in mitochondrial DNA replication and human disease. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.P.; Sarzi, E.; Aubert, S.; Chrétien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, B.; Area, E.; Flanigan, K.M.; Ganesh, J.; Jayakar, P.; Swoboda, K.J.; Coku, J.; Naini, A.; Shanske, S.; Tanji, K.; et al. Mitochondrial DNA depletion syndrome due to mutations in the RRM2B gene. Neuromuscul. Disord. 2008, 18, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Menzies, K.J.; Robinson, B.H.; Hood, D.A. Effect of thyroid hormone on mitochondrial properties and oxidative stress in cells from patients with mtDNA defects. Am. J. Physiol. Cell Physiol. 2009, 296, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Nicolini, G.; Iervasi, G. Mitochondria as key targets of cardioprotection in cardiac ischemic disease: Role of thyroid hormone triiodothyronine. Int. J. Mol. Sci. 2015, 16, 6312–6336. [Google Scholar] [CrossRef] [PubMed]
- Pantos, C.; Mourouzis, I. Translating thyroid hormone effects into clinical practice: The relevance of thyroid hormone receptor α1 in cardiac repair. Heart Fail. Rev. 2015, 20, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Pantos, C.; Mourouzis, I.; Cokkinos, D.V. Thyroid hormone and cardiac repair/regeneration: From Prometheus myth to reality? Can. J. Physiol. Pharmacol. 2012, 90, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.C.; Chen, S.L.; Lin, S.L.; Tsai, C.Y.; Chuang, W.Y.; Lin, Y.H.; Huang, Y.H.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Thyroid hormone protects hepatocytes from HBx-induced carcinogenesis by enhancing mitochondrial turnoveral. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasileiou, P.V.S.; Mourouzis, I.; Pantos, C. Principal Aspects Regarding the Maintenance of Mammalian Mitochondrial Genome Integrity. Int. J. Mol. Sci. 2017, 18, 1821. https://doi.org/10.3390/ijms18081821
Vasileiou PVS, Mourouzis I, Pantos C. Principal Aspects Regarding the Maintenance of Mammalian Mitochondrial Genome Integrity. International Journal of Molecular Sciences. 2017; 18(8):1821. https://doi.org/10.3390/ijms18081821
Chicago/Turabian StyleVasileiou, Panagiotis V. S., Iordanis Mourouzis, and Constantinos Pantos. 2017. "Principal Aspects Regarding the Maintenance of Mammalian Mitochondrial Genome Integrity" International Journal of Molecular Sciences 18, no. 8: 1821. https://doi.org/10.3390/ijms18081821
APA StyleVasileiou, P. V. S., Mourouzis, I., & Pantos, C. (2017). Principal Aspects Regarding the Maintenance of Mammalian Mitochondrial Genome Integrity. International Journal of Molecular Sciences, 18(8), 1821. https://doi.org/10.3390/ijms18081821