The Effect of Sepsis on the Erythrocyte

{kind=link}
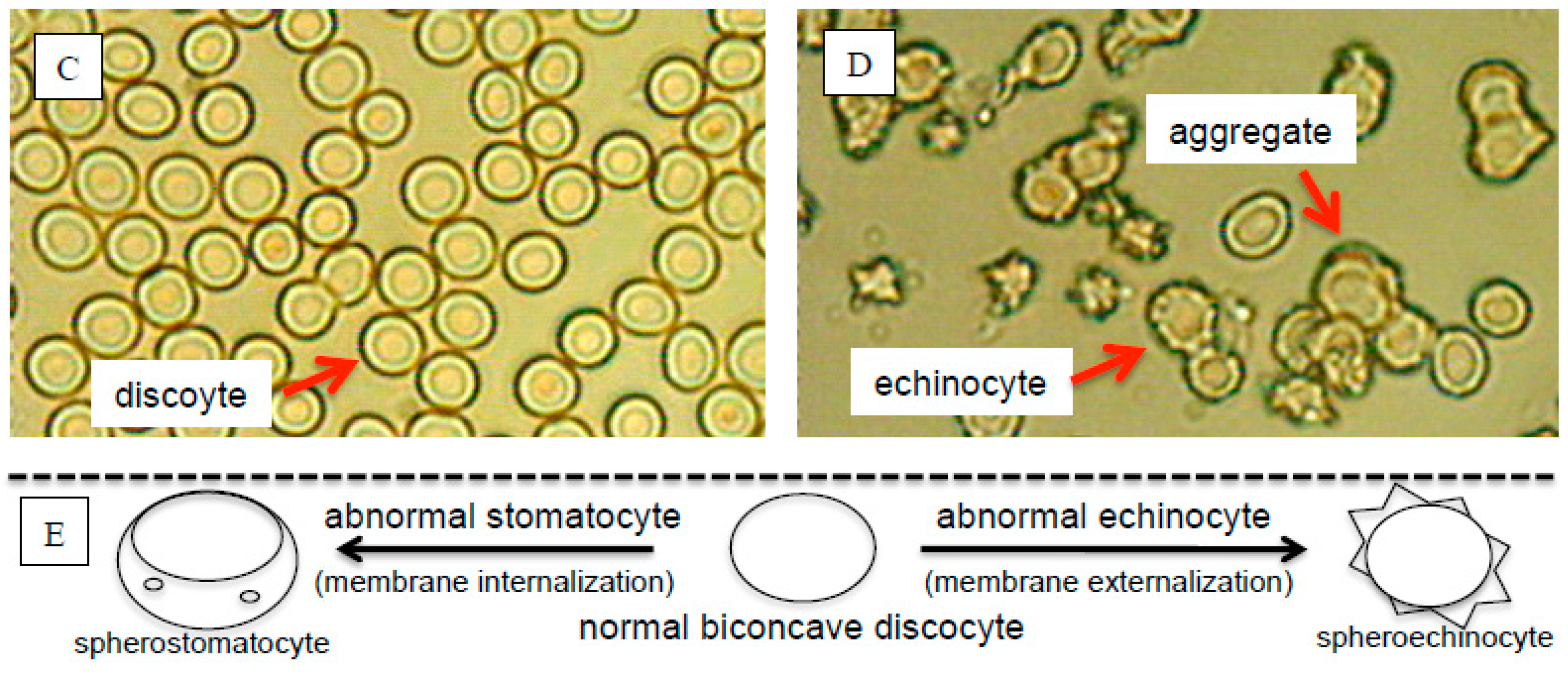{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Erythrocyte Size, Shape and Deformability
2.1. The Effect of Sepsis on Red Blood Cell (Shape) and Distribution Width (RDW)
2.2. Sepsis Induced Changes in RBC Deformability Are Clinically Important
2.3. Septic RBC Subpopulations with Altered Morphology and Decreased Deformability
3. Hemoglobin Oxygen Binding and 2,3-Bisphosphoglycerate (2,3-BPG)
The Effect of Sepsis on RBC 2,3-BPG and the Correlation with Acidemia
4. Sepsis Induced Oxidative Stress, Effects on Erythrocytes and the Importance of Antioxidants
4.1. Sepsis Induced Reduced Antioxidant Status
4.2. Activated Neutrophils and Endothelial Cells as Sources of Reactive Oxygne Species (ROS)
4.3. The Erythrocyte as a Source of Reactive Oxygen Species
4.4. Plasma Xanthine Oxidase as a Source of Reactive Oxygen Species
4.5. Effect of Oxygen Free Radicals and Reactive Oxygen Species on the Erythrocte
4.6. Effect of Sepsis on Erythrocyte Antioxidants
4.6.1. Effect of Sepsis on Erythrocyte Catalase
4.6.2. Effect of Sepsis on Erythrocyte Peroxiredoxin II
5. Sepsis Increases Intracellular Erythrocyte Ca2+
Sepsis and Endotoxemia Alter Intracellular RBC Ca2+ Homeostasis
6. A Hallmark of Sepsis Is Increased Capillary Stopped-Flow
7. The Effect of Sepsis and Bacterial Virulence Factors on RBC Physiology and Survival
7.1. Pyocyanin Decreases RBC Survival
7.2. Bacterial Neuraminidase Is Elevated in Septic Patients and Decreases RBC Survival in Animal Models
8. Alterations in RBC Membrane Proteins in Critically Ill Patients and Trauma Animal Models
Sepsis Induces Band 3 Phosphorylation
9. Sepsis Impairs RBC O2-Dependent ATP Efflux
10. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| 2,3-BPG/2,3-DPG | 2,3-bisphosphoglycerate |
| ATP | Adenosine triphosphate |
| CAT | Catalase |
| H2O2 | Hydrogen peroxide |
| MDA | Malonyldialdehyde |
| NADH | Nicotinamide adenine dinucleotide |
| NADPH | Reduced nicotinamide adenine dinucleotide phosphate |
| PS | Phosphatidylserine |
| PTP | Phosphotyrosine phosphatase |
| PIK | Phosphotyrosine kinase |
| ROS | Reactive oxygen species |
| O2− | Superoxide anion |
| SOD | Superoxide dismutase |
| TBARS | Thiobarbituric acid reactive substances |
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Baskurt, O.K.; Gelmont, D.; Meiselman, H.J. Red blood cell deformability in sepsis. Am. J. Respir. Crit. Care Med. 1998, 157, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.M.; Jagger, J.E.; Sharpe, M.D.; Ellsworth, M.L.; Mehta, S.; Ellis, C.G. Erythrocyte deformability is a nitric oxide-mediated factor in decreased capillary density during sepsis. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2848–H2856. [Google Scholar] [PubMed]
- De Oliveira, Y.P.A.; Pontes-de-Carvalho, L.C.; Couto, R.D.; Noronha-Dutra, A.A. Oxidative stress in sepsis. Possible production of free radicals through an erythrocyte-mediated positive feedback mechanism. BJID 2017, 21, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Piagnerelli, M.; Boudjeltia, K.Z.; Brohee, D.; Piro, P.; Carlier, E.; Vincent, J.L.; Lejeune, P.; Vanhaeverbeek, M. Alterations of red blood cell shape and sialic acid membrane content in septic patients. Crit. Care Med. 2003, 31, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Piagnerelli, M.; Boudjeltia, K.Z.; Brohee, D.; Vincent, J.L.; Vanhaeverbeek, M. Modifications of red blood cell shape and glycoproteins membrane content in septic patients. Adv. Exp. Med. Biol 2003, 510, 109–114. [Google Scholar] [PubMed]
- Piagnerelli, M.; Boudjeltia, K.Z.; Rapotec, A.; Richard, T.; Brohee, D.; Babar, S.; Bouckaert, V.; Simon, A.C.; Toko, J.P.; Walravens, T.; et al. Neuraminidase alters red blood cells in sepsis. Crit. Care Med. 2009, 37, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Piagnerelli, M.; Zouaoui Boudjeltia, K.; Gulbis, B.; Vanhaeverbeek, M.; Vincent, J.L. Anemia in sepsis: The importance of red blood cell membrane changes. TATM 2007, 9, 143–149. [Google Scholar] [CrossRef]
- Simonson, S.G.; Welty-Wolf, K.; Huang, Y.T.; Griebel, J.A.; Caplan, M.S.; Fracica, P.J.; Piantadosi, C.A. Altered mitochondrial redox responses in gram negative septic shock in primates. Circ. Shock 1994, 43, 34–43. [Google Scholar] [PubMed]
- Mofarrahi, M.; Sigala, I.; Guo, Y.; Godin, R.; Davis, E.C.; Petrof, B.; Sandri, M.; Burelle, Y.; Hussain, S.N. Autophagy and skeletal muscles in sepsis. PLoS ONE 2012, 7, e47265. [Google Scholar] [CrossRef] [PubMed]
- Carre, J.E.; Orban, J.C.; Re, L.; Felsmann, K.; Iffert, W.; Bauer, M.; Suliman, H.B.; Piantadosi, C.A.; Mayhew, T.M.; Breen, P.; et al. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am. J. Respir. Crit. Care Med. 2010, 182, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Pinto, B.B.; Dyson, A.; Umbrello, M.; Carre, J.E.; Ritter, C.; Clatworthy, I.; Duchen, M.R.; Singer, M. Improved survival in a long-term rat model of sepsis is associated with reduced mitochondrial calcium uptake despite increased energetic demand. Crit. Care Med. 2017, 45, e840–e848. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.M.; Sharpe, M.D.; Jagger, J.E.; Ellis, C.G. Sepsis impairs microvascular autoregulation and delays capillary response within hypoxic capillaries. Crit. Care 2015, 19, 389. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.G.; Bateman, R.M.; Sharpe, M.D.; Sibbald, W.J.; Gill, R. Effect of a maldistribution of microvascular blood flow on capillary O2 extraction in sepsis. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H156–164. [Google Scholar] [PubMed]
- Lam, C.; Tyml, K.; Martin, C.; Sibbald, W. Microvascular perfusion is impaired in a rat model of normotensive sepsis. J. Clin. Investig. 1994, 94, 2077–2083. [Google Scholar] [CrossRef] [PubMed]
- Sakr, Y.; Dubois, M.J.; De Backer, D.; Creteur, J.; Vincent, J.L. Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit. Care Med. 2004, 32, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Piper, R.D.; Pitt-Hyde, M.L.; Anderson, L.A.; Sibbald, W.J.; Potter, R.F. Leukocyte activation and flow behavior in rat skeletal muscle in sepsis. Am. J. Respir. Crit. Care Med. 1998, 157, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Tyml, K.; Li, F.; Wilson, J.X. Septic impairment of capillary blood flow requires nicotinamide adenine dinucleotide phosphate oxidase but not nitric oxide synthase and is rapidly reversed by ascorbate through an endothelial nitric oxide synthase-dependent mechanism. Crit. Care Med. 2008, 36, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Damiani, E.; Ince, C.; Orlando, F.; Pierpaoli, E.; Cirioni, O.; Giacometti, A.; Mocchegiani, F.; Pelaia, P.; Provinciali, M.; Donati, A. Effects of the infusion of 4% or 20% human serum albumin on the skeletal muscle microcirculation in endotoxemic rats. PLoS ONE 2016, 11, e0151005. [Google Scholar] [CrossRef] [PubMed]
- Boczkowski, J.; Vicaut, E.; Aubier, M. In vivo effects of escherichia coli endotoxemia on diaphragmatic microcirculation in rats. J. Appl. Physiol. 1992, 72, 2219–2224. [Google Scholar]
- Bateman, R.M.; Tokunaga, C.; Kareco, T.; Dorscheid, D.R.; Walley, K.R. Myocardial hypoxia-inducible HIF-1α, VEGF, and GLUT1 gene expression is associated with microvascular and ICAM-1 heterogeneity during endotoxemia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H448–456. [Google Scholar] [CrossRef] [PubMed]
- De Backer, D.; Creteur, J.; Preiser, J.C.; Dubois, M.J.; Vincent, J.L. Microvascular blood flow is altered in patients with sepsis. Am. J. Respir. Crit. Care Med. 2002, 166, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D.; Bateman, R.M.; Ellis, C.G. Effect of sepsis on skeletal muscle oxygen consumption and tissue oxygenation: Interpreting capillary oxygen transport data using a mathematical model. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2535–2544. [Google Scholar] [CrossRef] [PubMed]
- Balagopalakrishna, C.; Manoharan, P.T.; Abugo, O.O.; Rifkind, J.M. Production of superoxide from hemoglobin-bound oxygen under hypoxic conditions. Biochemistry 1996, 35, 6393–6398. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.C.; Said, A.; Corcuera, D.; McLaughlin, D.; Kell, P.; Doctor, A. Hypoxia limits antioxidant capacity in red blood cells by altering glycolytic pathway dominance. FASEB J. 2009, 23, 3159–3170. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, W.H.; Chien, S. Red cell rheology in stomatocyte-echinocyte transformation: Roles of cell geometry and cell shape. Blood 1986, 67, 1110–1118. [Google Scholar] [PubMed]
- Simchon, S.; Jan, K.M.; Chien, S. Influence of reduced red cell deformability on regional blood flow. Am. J. Physiol. 1987, 253, H898–903. [Google Scholar] [PubMed]
- Qadri, S.M.; Donkor, D.A.; Bhakta, V.; Eltringham-Smith, L.J.; Dwivedi, D.J.; Moore, J.C.; Pepler, L.; Ivetic, N.; Nazi, I.; Fox-Robichaud, A.E.; et al. Phosphatidylserine externalization and procoagulant activation of erythrocytes induced by pseudomonas aeruginosa virulence factor pyocyanin. J. Cell. Mol. Med. 2016, 20, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Donadello, K.; Piagnerelli, M.; Reggiori, G.; Gottin, L.; Scolletta, S.; Occhipinti, G.; Zouaoui Boudjeltia, K.; Vincent, J.L. Reduced red blood cell deformability over time is associated with a poor outcome in septic patients. Microvasc Res. 2015, 101, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Langenfeld, J.E.; Livingston, D.H.; Machiedo, G.W. Red cell deformability is an early indicator of infection. Surgery 1991, 110, 398–403. [Google Scholar] [PubMed]
- Moutzouri, A.G.; Skoutelis, A.T.; Gogos, C.A.; Missirlis, Y.F.; Athanassiou, G.M. Red blood cell deformability in patients with sepsis: A marker for prognosis and monitoring of severity. Clin. Hemorheol. Microcirc. 2007, 36, 291–299. [Google Scholar] [PubMed]
- Reggiori, G.; Occhipinti, G.; De Gasperi, A.; Vincent, J.L.; Piagnerelli, M. Early alterations of red blood cell rheology in critically ill patients. Crit. Care Med. 2009, 37, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Totsimon, K.; Biro, K.; Szabo, Z.E.; Toth, K.; Kenyeres, P.; Marton, Z. The relationship between hemorheological parameters and mortality in critically ill patients with and without sepsis. Clin. Hemorheol. Microcirc. 2017, 65, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.M.; Walley, K.R. Microvascular resuscitation as a therapeutic goal in severe sepsis. Crit. Care 2005, 9, S27–32. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.M.; Hodgson, K.C.; Kohli, K.; Knight, D.; Walley, K.R. Endotoxemia increases the clearance of mPEGylated 5000-MW quantum dots as revealed by multiphoton microvascular imaging. J. Biomed. Opt. 2007, 12, 064005. [Google Scholar] [CrossRef] [PubMed]
- Dupire, J.; Socol, M.; Viallat, A. Full dynamics of a red blood cell in shear flow. Proc. Natl. Acad. Sci. USA 2012, 109, 20808–20813. [Google Scholar] [CrossRef] [PubMed]
- Linderkamp, O.; Wu, P.Y.; Meiselman, H.J. Geometry of neonatal and adult red blood cells. Pediatr. Res. 1983, 17, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Deplaine, G.; Safeukui, I.; Jeddi, F.; Lacoste, F.; Brousse, V.; Perrot, S.; Biligui, S.; Guillotte, M.; Guitton, C.; Dokmak, S.; et al. The sensing of poorly deformable red blood cells by the human spleen can be mimicked in vitro. Blood 2011, 117, e88–e95. [Google Scholar] [CrossRef] [PubMed]
- Pivkin, I.V.; Peng, Z.; Karniadakis, G.E.; Buffet, P.A.; Dao, M.; Suresh, S. Biomechanics of red blood cells in human spleen and consequences for physiology and disease. Proc. Natl. Acad. Sci. USA 2016, 113, 7804–7809. [Google Scholar] [CrossRef] [PubMed]
- Condon, M.R.; Kim, J.E.; Deitch, E.A.; Machiedo, G.W.; Spolarics, Z. Appearance of an erythrocyte population with decreased deformability and hemoglobin content following sepsis. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H2177–H2184. [Google Scholar] [CrossRef] [PubMed]
- Linderkamp, O.; Meiselman, H.J. Geometric, osmotic, and membrane mechanical properties of density-separated human red cells. Blood 1982, 59, 1121–1127. [Google Scholar] [PubMed]
- Waugh, R.E.; Narla, M.; Jackson, C.W.; Mueller, T.J.; Suzuki, T.; Dale, G.L. Rheologic properties of senescent erythrocytes: Loss of surface area and volume with red blood cell age. Blood 1992, 79, 1351–1358. [Google Scholar] [PubMed]
- Ghashghaeinia, M.; Cluitmans, J.C.; Akel, A.; Dreischer, P.; Toulany, M.; Koberle, M.; Skabytska, Y.; Saki, M.; Biedermann, T.; Duszenko, M.; et al. The impact of erythrocyte age on eryptosis. Br. J. Haematol. 2012, 157, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Salvagno, G.L.; Sanchis-Gomar, F.; Picanza, A.; Lippi, G. Red blood cell distribution width: A simple parameter with multiple clinical applications. Crit. Rev. Clin. Lab. Sci 2015, 52, 86–105. [Google Scholar] [CrossRef] [PubMed]
- Sadaka, F.; O’Brien, J.; Prakash, S. Red cell distribution width and outcome in patients with septic shock. J. Intensive Care Med. 2013, 28, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Park, J.T.; Kim, E.J.; Han, J.H.; Han, J.S.; Choi, J.Y.; Han, S.H.; Yoo, T.H.; Kim, Y.S.; Kang, S.W.; et al. An increase in red blood cell distribution width from baseline predicts mortality in patients with severe sepsis or septic shock. Crit. Care 2013, 17, R282. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.S.; Kim, H.W.; Oh, H.J.; Kim, Y.C.; Kim, M.H.; Song, J.E.; Oh, D.H.; Ahn, J.Y.; Kim, S.B.; Jeong, S.J.; et al. Red blood cell distribution width is an independent predictor of mortality in patients with gram-negative bacteremia. Shock 2012, 38, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.H.; Kim, K.; Lee, J.H.; Kang, C.; Kim, T.; Park, H.M.; Kang, K.W.; Kim, J.; Rhee, J.E. Red cell distribution width is a prognostic factor in severe sepsis and septic shock. Am. J. Emerg. Med. 2013, 31, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Fontana, V.; Spadaro, S.; Bond, O.; Cavicchi, F.Z.; Annoni, F.; Donadello, K.; Vincent, J.L.; De Backer, D.; Taccone, F.S. No relationship between red blood cell distribution width and microcirculatory alterations in septic patients. Clin. Hemorheol. Microcirc. 2017, 66, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Patel, K.V.; Ferrucci, L.; Sun, K.; Roy, C.N.; Guralnik, J.M.; Fried, L.P. Serum antioxidants and inflammation predict red cell distribution width in older women: The women’s health and aging study I. Clin. Nutr. 2010, 29, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, S. Oxidative stress in the regulation of normal and neoplastic hematopoiesis. Antioxid. Redox Signal. 2008, 10, 1923–1940. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.C.; Hwang, T.L.; Chen, H.M.; Chen, M.F.; Sun, Y.F.; Lau, Y.T. Sepsis correlated with increased erythrocyte Na+ content and Na+-K+ pump activity. J. Biomed. Sci. 2003, 10, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Purtle, S.W.; Horkan, C.M.; Moromizato, T.; Gibbons, F.K.; Christopher, K.B. Nucleated red blood cells, critical illness survivors and postdischarge outcomes: A cohort study. Crit. Care 2017, 21, 154. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.V.; Mohanty, J.G.; Kanapuru, B.; Hesdorffer, C.; Ershler, W.B.; Rifkind, J.M. Association of the red cell distribution width with red blood cell deformability. Adv. Exp. Med. Biol. 2013, 765, 211–216. [Google Scholar] [PubMed]
- Todd, J.C., 3rd; Mollitt, D.L. Sepsis-induced alterations in the erythrocyte membrane. Am. Surg. 1994, 60, 954–957. [Google Scholar] [PubMed]
- Hurd, T.C.; Dasmahapatra, K.S.; Rush, B.F., Jr.; Machiedo, G.W. Red blood cell deformability in human and experimental sepsis. Arch. Surg. 1988, 123, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, N.; Berhes, M.; Kiss, F.; Hajdu, E.; Deak, A.; Molnar, A.; Szabo, J.; Fulesdi, B. Early hemorheological changes in a porcine model of intravenously given E. coli induced fulminant sepsis. Clin. Hemorheol. Microcirc. 2015, 61, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Machiedo, G.W.; Powell, R.J.; Rush, B.F., Jr.; Swislocki, N.I.; Dikdan, G. The incidence of decreased red blood cell deformability in sepsis and the association with oxygen free radical damage and multiple-system organ failure. Arch. Surg. 1989, 124, 1386–1389. [Google Scholar] [CrossRef] [PubMed]
- Langenfeld, J.E.; Machiedo, G.W.; Lyons, M.; Rush, B.F., Jr.; Dikdan, G.; Lysz, T.W. Correlation between red blood cell deformability and changes in hemodynamic function. Surgery 1994, 116, 859–867. [Google Scholar] [PubMed]
- McKenney, J.; Valeri, C.R.; Mohandas, N.; Fortier, N.; Giorgio, A.; Snyder, L.M. Decreased in vivo survival of hydrogen peroxide-damaged baboon red blood cells. Blood 1990, 76, 206–211. [Google Scholar] [PubMed]
- Miller, L.D.; Oski, F.A.; Diaco, J.F.; Sugerman, H.J.; Gottlieb, A.J.; Davidson, D.; Delivoria-Papadopoulos, M. The affinity of hemoglobin for oxygen: Its control and in vivo significance. Surgery 1970, 68, 187–194. [Google Scholar] [PubMed]
- Watkins, G.M.; Rabelo, A.; Pizak, L.F.; Sheldon, G.F. The left shifted oxyhemoglobin curve in sepsis: A preventable defect. Ann. Surg. 1974, 180, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Weisel, R.D.; Vito, L.; Dennis, R.C.; Valeri, C.R.; Hechtman, H.B. Myocardial depression during sepsis. Am. J. Surg. 1977, 133, 512–521. [Google Scholar] [CrossRef]
- Myburgh, J.A.; Webb, R.K.; Worthley, L.I. The P50 is reduced in critically ill patients. Intensive Care Med. 1991, 17, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Duhm, J.; Gerlach, E. On the mechanisms of the hypoxia-induced increase of 2,3-diphosphoglycerate in erythrocytes. Studies on rat erythrocytes in vivo and on human erythrocytes in vitro. Pflug. Arch. 1971, 326, 254–269. [Google Scholar] [CrossRef]
- Spasojevic, I.; Zakrzewska, J.; Bacic, G.G. 31P nmr spectroscopy and polarographic combined study of erythrocytes treated with 5-fluorouracil: Cardiotoxicity-related changes in atp, 2,3-BPG, and O2 metabolism. Ann. N. Y. Acad. Sci. 2005, 1048, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim Eel, D.; McLellan, S.A.; Walsh, T.S. Red blood cell 2,3-diphosphoglycerate concentration and in vivo P50 during early critical illness. Crit. Care Med. 2005, 33, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G., Jr.; McDevitt, N.B.; Proctor, H.J. Erythrocyte 2,3-diphosphoglycerate in endotoxic shock in the subhuman primate: Response to fluid and-or methylprednisolone succinate. Ann. Surg. 1974, 180, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.J.; Koch, D.; Morris, D.; Clague, A.; Purdie, D.M. Reduced red cell 2,3-diphosphoglycerate concentrations in critical illness without decreased in vivo P50. Anaesth. Intensive Care 2001, 29, 479–483. [Google Scholar] [PubMed]
- Morgan, T.J. The oxyhaemoglobin dissociation curve in critical illness. Crit. Care Resusc 1999, 1, 93–100. [Google Scholar] [PubMed]
- Chillar, R.K.; Slawsky, P.; Desforges, J.F. Red cell 2,3-diphosphoglycerate and adenosine triphosphate in patients with shock. Br. J. Haematol. 1971, 21, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.M.; Sharpe, M.D.; Goldman, D.; Lidington, D.; Ellis, C.G. Inhibiting nitric oxide overproduction during hypotensive sepsis increases local oxygen consumption in rat skeletal muscle. Crit. Care Med. 2008, 36, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Marik, P.E.; Khangoora, V.; Rivera, R.; Hooper, M.H.; Catravas, J. Hydrocortisone, vitamin C, and thiamine for the treatment of severe sepsis and septic shock: A retrospective before-after study. Chest 2017, 151, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, H.; Qadri, S.M.; Foller, M.; Lang, F. Inhibition of suicidal erythrocyte death by vitamin C. Nutrition 2010, 26, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Powell, R.J.; Machiedo, G.W.; Rush, B.F., Jr.; Dikdan, G. Effect of α-tocopherol on red cell deformability and survival in sepsis. Curr Surg. 1989, 46, 380–382. [Google Scholar] [PubMed]
- Goode, H.F.; Cowley, H.C.; Walker, B.E.; Howdle, P.D.; Webster, N.R. Decreased antioxidant status and increased lipid peroxidation in patients with septic shock and secondary organ dysfunction. Crit. Care Med. 1995, 23, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Fain, O.; Paries, J.; Jacquart, B.; le Moel, G.; Kettaneh, A.; Stirnemann, J.; Heron, C.; Sitbon, M.; Taleb, C.; Letellier, E.; et al. Hypovitaminosis c in hospitalized patients. Eur J. Intern. Med. 2003, 14, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Cross, C.E.; Forte, T.; Stocker, R.; Louie, S.; Yamamoto, Y.; Ames, B.N.; Frei, B. Oxidative stress and abnormal cholesterol metabolism in patients with adult respiratory distress syndrome. J. Lab. Clin. Med. 1990, 115, 396–404. [Google Scholar] [PubMed]
- Metnitz, P.G.; Bartens, C.; Fischer, M.; Fridrich, P.; Steltzer, H.; Druml, W. Antioxidant status in patients with acute respiratory distress syndrome. Intensive Care Med. 1999, 25, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Cowley, H.C.; Bacon, P.J.; Goode, H.F.; Webster, N.R.; Jones, J.G.; Menon, D.K. Plasma antioxidant potential in severe sepsis: A comparison of survivors and nonsurvivors. Crit. Care Med. 1996, 24, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Richard, C.; Lemonnier, F.; Thibault, M.; Couturier, M.; Auzepy, P. Vitamin E deficiency and lipoperoxidation during adult respiratory distress syndrome. Crit. Care Med. 1990, 18, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Doise, J.M.; Aho, L.S.; Quenot, J.P.; Guilland, J.C.; Zeller, M.; Vergely, C.; Aube, H.; Blettery, B.; Rochette, L. Plasma antioxidant status in septic critically ill patients: A decrease over time. Fundam. Clin. Pharmacol. 2008, 22, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Rattan, V.; Hans, C.P.; Cruz, S.D.; Pathak, R.; Rao, P.; Mehta, S. Assessment of oxidative stress and antioxidant status in patients of acute renal failure with sepsis. WJPR 2015, 5, 1457–1464. [Google Scholar]
- Pascual, C.; Karzai, W.; Meier-Hellmann, A.; Oberhoffer, M.; Horn, A.; Bredle, D.; Reinhart, K. Total plasma antioxidant capacity is not always decreased in sepsis. Crit. Care Med. 1998, 26, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Dewas, C.; Dang, P.M.; Gougerot-Pocidalo, M.A.; El-Benna, J. TNF-α induces phosphorylation of p47phox in human neutrophils: Partial phosphorylation of p47phox is a common event of priming of human neutrophils by TNF-α and granulocyte-macrophage colony-stimulating factor. J. Immunol. 2003, 171, 4392–4398. [Google Scholar] [CrossRef] [PubMed]
- El Benna, J.; Faust, L.P.; Babior, B.M. The phosphorylation of the respiratory burst oxidase component p47phox during neutrophil activation. Phosphorylation of sites recognized by protein kinase C and by proline-directed kinases. J. Biol. Chem. 1994, 269, 23431–23436. [Google Scholar] [PubMed]
- Babior, B.M. Phagocytes and oxidative stress. Am. J. Med. 2000, 109, 33–44. [Google Scholar] [CrossRef]
- Todd, J.C., 3rd; Mollitt, D.L. Leukocyte modulation inhibits endotoxin-induced disruption of intracellular calcium homeostasis. J. Trauma 1995, 39, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.C., 3rd; Poulos, N.D.; Davidson, L.W.; Mollitt, D.L. Role of the leukocyte in endotoxin-induced alterations of the red cell membrane. Second place winner of the conrad jobst award in the gold medal paper competition. Am. Surg. 1993, 59, 9–12. [Google Scholar] [PubMed]
- Weiss, S.J. The role of superoxide in the destruction of erythrocyte targets by human neutrophils. J. Biol. Chem. 1980, 255, 9912–9917. [Google Scholar] [PubMed]
- Weiss, S.J. Neutrophil-mediated methemoglobin formation in the erythrocyte. The role of superoxide and hydrogen peroxide. J. Biol. Chem. 1982, 257, 2947–2953. [Google Scholar] [PubMed]
- Balagopalakrishna, C.; Abugo, O.O.; Horsky, J.; Manoharan, P.T.; Nagababu, E.; Rifkind, J.M. Superoxide produced in the heme pocket of the β-chain of hemoglobin reacts with the β-93 cysteine to produce a thiyl radical. Biochemistry 1998, 37, 13194–13202. [Google Scholar] [CrossRef] [PubMed]
- Luchtemberg, M.N.; Petronilho, F.; Constantino, L.; Gelain, D.P.; Andrades, M.; Ritter, C.; Moreira, J.C.; Streck, E.L.; Dal-Pizzol, F. Xanthine oxidase activity in patients with sepsis. Clin. Biochem. 2008, 41, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Uyesaka, N.; Hasegawa, S.; Ishioka, N.; Ishioka, R.; Shio, H.; Schechter, A.N. Effects of superoxide anions on red cell deformability and membrane proteins. Biorheology 1992, 29, 217–229. [Google Scholar] [PubMed]
- Baskurt, O.K.; Temiz, A.; Meiselman, H.J. Effect of superoxide anions on red blood cell rheologic properties. Free Radic. Biol. Med. 1998, 24, 102–110. [Google Scholar] [CrossRef]
- Snyder, L.M.; Fortier, N.L.; Trainor, J.; Jacobs, J.; Leb, L.; Lubin, B.; Chiu, D.; Shohet, S.; Mohandas, N. Effect of hydrogen peroxide exposure on normal human erythrocyte deformability, morphology, surface characteristics, and spectrin-hemoglobin cross-linking. J. Clin. Investig. 1985, 76, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.A.; Bilto, Y.Y.; Juma, M.; Irhimeh, M.R. Exposure of human erythrocytes to oxygen radicals causes loss of deformability, increased osmotic fragility, lipid peroxidation and protein degradatioin. Clin. Hemorheol. Microcirc. 2000, 23, 13–21. [Google Scholar] [PubMed]
- Jain, S.K.; Mohandas, N.; Clark, M.R.; Shohet, S.B. The effect of malonyldialdehyde, a product of lipid peroxidation, on the deformability, dehydration and 51Cr-survival of erythrocytes. Br. J. Haematol. 1983, 53, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Ross, J.D.; Levy, G.J.; Little, R.L.; Duett, J. The accumulation of malonyldialdehyde, an end product of membrane lipid peroxidation, can cause potassium leak in normal and sickle red blood cells. Biochem. Med. Metab. Biol. 1989, 42, 60–65. [Google Scholar] [CrossRef]
- Agar, N.S.; Sadrzadeh, S.M.; Hallaway, P.E.; Eaton, J.W. Erythrocyte catalase. A somatic oxidant defense? J. Clin. Investig. 1986, 77, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Karapetsa, M.; Pitsika, M.; Goutzourelas, N.; Stagos, D.; Tousia Becker, A.; Zakynthinos, E. Oxidative status in icu patients with septic shock. Food Chem. Toxicol. 2013, 61, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, M.O.; Petronilho, F.; Andrades, M.; Constantino, L.; Mina, F.G.; Moreira, J.C.; Dal-Pizzol, F.; Ritter, C. Plasma superoxide dismutase activity and mortality in septic patients [corrected]. J. Trauma 2010, 69, E102–E106. [Google Scholar] [CrossRef] [PubMed]
- Warner, A.; Bencosme, A.; Healy, D.; Verme, C. Prognostic role of antioxidant enzymes in sepsis: Preliminary assessment. Clin. Chem. 1995, 41, 867–871. [Google Scholar] [PubMed]
- Bayer, S.B.; Maghzal, G.; Stocker, R.; Hampton, M.B.; Winterbourn, C.C. Neutrophil-mediated oxidation of erythrocyte peroxiredoxin 2 as a potential marker of oxidative stress in inflammation. FASEB J. 2013, 27, 3315–3322. [Google Scholar] [CrossRef] [PubMed]
- Bayer, S.B.; Low, F.M.; Hampton, M.B.; Winterbourn, C.C. Interactions between peroxiredoxin 2, hemichrome and the erythrocyte membrane. Free Radic. Res. 2016, 50, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, E.; Mohanty, J.G.; Friedman, J.S.; Rifkind, J.M. Role of peroxiredoxin-2 in protecting RBCs from hydrogen peroxide-induced oxidative stress. Free Radic. Res. 2013, 47, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Low, F.M.; Hampton, M.B.; Peskin, A.V.; Winterbourn, C.C. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood 2007, 109, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, A.; Makhro, A.; Wang, J.; Lipp, P.; Kaestner, L. Calcium in red blood cells-A perilous balance. Int. J. Mol. Sci. 2013, 14, 9848–9872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, T.K.; Carlson, R.W.; Geheb, M.A. Prevalence and clinical implications of hypocalcemia in acutely ill patients in a medical intensive care setting. Am. J. Med. 1988, 84, 209–214. [Google Scholar] [CrossRef]
- Todd, J.C., 3rd; Mollitt, D.L. Effect of sepsis on erythrocyte intracellular calcium homeostasis. Crit. Care Med. 1995, 23, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Ruef, P.; Ehrhard, M.; Frommhold, D.; Koch, L.; Fritzsching, B.; Poeschl, J. Lipid a decreases human erythrocytes deformability by increasing intracellular Ca2+: Effects of verapamil, staurosporine and the rho-kinase inhibitor Y-27632. Clin. Hemorheol. Microcirc. 2011, 49, 315–322. [Google Scholar] [PubMed]
- Lau, Y.T.; Hsieh, C.C.; Liu, M.S.; Hwang, T.L.; Chen, M.F.; Cheng, H.S. Erythrocyte Ca2+ pump is defective during sepsis. Circ. Shock 1994, 44, 121–125. [Google Scholar] [PubMed]
- Smith, B.D.; la Celle, P.L.; Siefring, G.E., Jr.; Lowe-Krentz, L.; Lorand, L. Effects of the calcium-mediated enzymatic cross-linking of membrane proteins on cellular deformability. J. Membr. Biol. 1981, 61, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Piper, R.D.; Pitt-Hyde, M.; Li, F.; Sibbald, W.J.; Potter, R.F. Microcirculatory changes in rat skeletal muscle in sepsis. Am. J. Respir. Crit. Care Med. 1996, 154, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Eichelbronner, O.; Sibbald, W.J.; Chin-Yee, I.H. Intermittent flow increases endotoxin-induced adhesion of human erythrocytes to vascular endothelial cells. Intensive Care Med. 2003, 29, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Foller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol. Cell. Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef] [PubMed]
- Manodori, A.B.; Barabino, G.A.; Lubin, B.H.; Kuypers, F.A. Adherence of phosphatidylserine-exposing erythrocytes to endothelial matrix thrombospondin. Blood 2000, 95, 1293–1300. [Google Scholar] [PubMed]
- Kempe, D.S.; Akel, A.; Lang, P.A.; Hermle, T.; Biswas, R.; Muresanu, J.; Friedrich, B.; Dreischer, P.; Wolz, C.; Schumacher, U.; et al. Suicidal erythrocyte death in sepsis. J. Mol. Med. 2007, 85, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Foller, M.; Biswas, R.; Mahmud, H.; Akel, A.; Shumilina, E.; Wieder, T.; Goetz, F.; Lang, F. Effect of peptidoglycans on erythrocyte survival. Int. J. Med. Microbiol. 2009, 299, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Towhid, S.T.; Pakladok, T.; Alesutan, I.; Gotz, F.; Gulbins, E.; Lang, F. Effect of bacterial peptidoglycan on erythrocyte death and adhesion to endothelial cells. Int. J. Med. Microbiol. 2013, 303, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Mahmud, H.; Foller, M.; Biswas, R.; Lang, K.S.; Bohn, E.; Gotz, F.; Lang, F. Lipopeptides in the triggering of erythrocyte cell membrane scrambling. Cell. Physiol. Biochem. 2008, 22, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun Bhuyan, A.; Nguyen, M.T.; Bissinger, R.; Gotz, F.; Lang, F. Lipopeptide-induced suicidal erythrocyte death correlates with the degree of acylation. Cell. Physiol. Biochem. 2017, 41, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Towhid, S.T.; Mia, S.; Pakladok, T.; Alesutan, I.; Borst, O.; Gawaz, M.; Gulbins, E.; Lang, F. Sphingomyelinase-induced adhesion of eryptotic erythrocytes to endothelial cells. Am. J. Physiol. Cell. Physiol. 2012, 303, C991–C999. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, F.C.; Mate, S.; Bakas, L.; Herlax, V. Induction of eryptosis by low concentrations of E. coli α-hemolysin. BBA Biomembr. 2015, 1848, 2779–2788. [Google Scholar] [CrossRef] [PubMed]
- Foller, M.; Shumilina, E.; Lam, R.; Mohamed, W.; Kasinathan, R.; Huber, S.; Chakraborty, T.; Lang, F. Induction of suicidal erythrocyte death by listeriolysin from listeria monocytogenes. Cell. Physiol. Biochem. 2007, 20, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Lutz, H.U.; Bogdanova, A. Mechanisms tagging senescent red blood cells for clearance in healthy humans. Front. Physiol. 2013, 4, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.; Lang, E.; Foller, M. Physiology and pathophysiology of eryptosis. Transfus. Med. Hemother. 2012, 39, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Grebe, R.; Wolff, H.; Schmid-Schonbein, H. Influence of red cell surface charge on red cell membrane curvature. Pflug. Arch. 1988, 413, 77–82. [Google Scholar] [CrossRef]
- Durocher, J.R.; Payne, R.C.; Conrad, M.E. Role of sialic acid in erythrocyte survival. Blood 1975, 45, 11–20. [Google Scholar] [PubMed]
- Piagnerelli, M.; Cotton, F.; van Nuffelen, M.; Vincent, J.L.; Gulbis, B. Modifications in erythrocyte membrane protein content are not responsible for the alterations in rheology seen in sepsis. Shock 2012, 37, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Caprio, K.; Condon, M.R.; Deitch, E.A.; Xu, D.Z.; Feketova, E.; Machiedo, G.W. Alteration of α-spectrin ubiquitination after hemorrhagic shock. Am. J. Surg. 2008, 196, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Condon, M.R.; Feketova, E.; Machiedo, G.W.; Deitch, E.A.; Spolarics, Z. Augmented erythrocyte band-3 phosphorylation in septic mice. BBA Mol. Basis Dis. 2007, 1772, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.M.; Sharpe, M.D.; Ellis, C.G. Bench-to-bedside review: Microvascular dysfunction in sepsis—Hemodynamics, oxygen transport, and nitric oxide. Crit. Care 2003, 7, 359–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagger, J.E.; Bateman, R.M.; Ellsworth, M.L.; Ellis, C.G. Role of erythrocyte in regulating local O2 delivery mediated by hemoglobin oxygenation. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2833–H2839. [Google Scholar] [PubMed]
- Ellsworth, M.L.; Forrester, T.; Ellis, C.G.; Dietrich, H.H. The erythrocyte as a regulator of vascular tone. Am. J. Physiol. 1995, 269, H2155–H2161. [Google Scholar] [PubMed]
- Crawford, J.H.; Isbell, T.S.; Huang, Z.; Shiva, S.; Chacko, B.K.; Schechter, A.N.; Darley-Usmar, V.M.; Kerby, J.D.; Lang, J.D., Jr.; Kraus, D.; et al. Hypoxia, red blood cells, and nitrite regulate no-dependent hypoxic vasodilation. Blood 2006, 107, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Bell, J.B.; Mohanty, J.G.; Nagababu, E.; Rifkind, J.M. Nitrite enhances RBC hypoxic ATP synthesis and the release of ATP into the vasculature: A new mechanism for nitrite-induced vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1494–H1503. [Google Scholar] [CrossRef] [PubMed]
- Umbrello, M.; Dyson, A.; Pinto, B.B.; Fernandez, B.O.; Simon, V.; Feelisch, M.; Singer, M. Short-term hypoxic vasodilation in vivo is mediated by bioactive nitric oxide metabolites, rather than free nitric oxide derived from haemoglobin-mediated nitrite reduction. J. Physiol. 2014, 592, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yan, Y.; Zeng, M.; Zhang, J.; Hanes, M.A.; Ahearn, G.; McMahon, T.J.; Dickfeld, T.; Marshall, H.E.; Que, L.G.; et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 2004, 116, 617–628. [Google Scholar] [CrossRef]
- Isbell, T.S.; Sun, C.W.; Wu, L.C.; Teng, X.; Vitturi, D.A.; Branch, B.G.; Kevil, C.G.; Peng, N.; Wyss, J.M.; Ambalavanan, N.; et al. Sno-hemoglobin is not essential for red blood cell-dependent hypoxic vasodilation. Nat. Med. 2008, 14, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hess, D.T.; Qian, Z.; Hausladen, A.; Fonseca, F.; Chaube, R.; Reynolds, J.D.; Stamler, J.S. Hemoglobin βCys93 is essential for cardiovascular function and integrated response to hypoxia. Proc. Natl. Acad. Sci. USA 2015, 112, 6425–6430. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, C.W.; Honavar, J.; Townes, T.; Patel, R.P. Role of the b93cys, ATP and adenosine in red cell dependent hypoxic vasorelaxation. Int. J. Physiol. Pathophysiol. Pharmacol. 2013, 5, 21–31. [Google Scholar] [PubMed]
- Hoffman, J.F. ATP compartmentation in human erythrocytes. Curr. Opin. Hematol. 1997, 4, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Locovei, S.; Bao, L.; Dahl, G. Pannexin 1 in erythrocytes: Function without a gap. Proc. Natl. Acad. Sci. USA 2006, 103, 7655–7659. [Google Scholar] [CrossRef] [PubMed]
- Bergfeld, G.R.; Forrester, T. Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnia. Cardiovasc. Res. 1992, 26, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Messana, I.; Orlando, M.; Cassiano, L.; Pennacchietti, L.; Zuppi, C.; Castagnola, M.; Giardina, B. Human erythrocyte metabolism is modulated by the O2-linked transition of hemoglobin. FEBS Lett. 1996, 390, 25–28. [Google Scholar] [CrossRef]
- Lewis, I.A.; Campanella, M.E.; Markley, J.L.; Low, P.S. Role of band 3 in regulating metabolic flux of red blood cells. Proc. Natl. Acad. Sci. USA 2009, 106, 18515–18520. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; McKenna, M.M.; Krump, N.A.; Zheng, S.; Mendelsohn, L.; Thein, S.L.; Garrett, L.J.; Bodine, D.M.; Low, P.S. Reversible binding of hemoglobin to band 3 constitutes the molecular switch that mediates O2 regulation of erythrocyte properties. Blood 2016, 128, 2708–2716. [Google Scholar] [CrossRef] [PubMed]
- Sega, M.F.; Chu, H.; Christian, J.; Low, P.S. Interaction of deoxyhemoglobin with the cytoplasmic domain of murine erythrocyte band 3. Biochemistry 2012, 51, 3264–3272. [Google Scholar] [CrossRef] [PubMed]
- Campanella, M.E.; Chu, H.; Low, P.S. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Rozier, M.D.; Zata, V.J.; Ellsworth, M.L. Lactate interferes with atp release from red blood cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H3038–H3042. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.H.; Murthy, S.N.; Steck, T.L. Effect of red cell membrane binding on the catalytic activity of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 1982, 257, 1438–1442. [Google Scholar] [PubMed]
- Zipser, Y.; Kosower, N.S. Phosphotyrosine phosphatase associated with band 3 protein in the human erythrocyte membrane. Biochem. J. 1996, 314, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.L.; Rathinavelu, P.; Arese, P.; Geahlen, R.L.; Low, P.S. Role of band 3 tyrosine phosphorylation in the regulation of erythrocyte glycolysis. J. Biol. Chem. 1991, 266, 4106–4111. [Google Scholar] [PubMed]
- Zipser, Y.; Piade, A.; Barbul, A.; Korenstein, R.; Kosower, N.S. Ca2+ promotes erythrocyte band 3 tyrosine phosphorylation via dissociation of phosphotyrosine phosphatase from band 3. Biochem. J. 2002, 368, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Barbul, A.; Zipser, Y.; Nachles, A.; Korenstein, R. Deoxygenation and elevation of intracellular magnesium induce tyrosine phosphorylation of band 3 in human erythrocytes. FEBS Lett. 1999, 455, 87–91. [Google Scholar] [CrossRef]
- Murthy, S.N.; Kaul, R.K.; Kohler, H. Hemoglobin binds to the amino-terminal 23-residue fragment of human erythrocyte band 3 protein. Hoppe Seyler’s Z. Physiol. Chem. 1984, 365, 9–17. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bateman, R.M.; Sharpe, M.D.; Singer, M.; Ellis, C.G. The Effect of Sepsis on the Erythrocyte. Int. J. Mol. Sci. 2017, 18, 1932. https://doi.org/10.3390/ijms18091932
Bateman RM, Sharpe MD, Singer M, Ellis CG. The Effect of Sepsis on the Erythrocyte. International Journal of Molecular Sciences. 2017; 18(9):1932. https://doi.org/10.3390/ijms18091932
Chicago/Turabian StyleBateman, Ryon M., Michael D. Sharpe, Mervyn Singer, and Christopher G. Ellis. 2017. "The Effect of Sepsis on the Erythrocyte" International Journal of Molecular Sciences 18, no. 9: 1932. https://doi.org/10.3390/ijms18091932
APA StyleBateman, R. M., Sharpe, M. D., Singer, M., & Ellis, C. G. (2017). The Effect of Sepsis on the Erythrocyte. International Journal of Molecular Sciences, 18(9), 1932. https://doi.org/10.3390/ijms18091932