Complementary RNA-Sequencing Based Transcriptomics and iTRAQ Proteomics Reveal the Mechanism of the Alleviation of Quinclorac Stress by Salicylic Acid in Oryza sativa ssp. japonica

 and
and 
Abstract
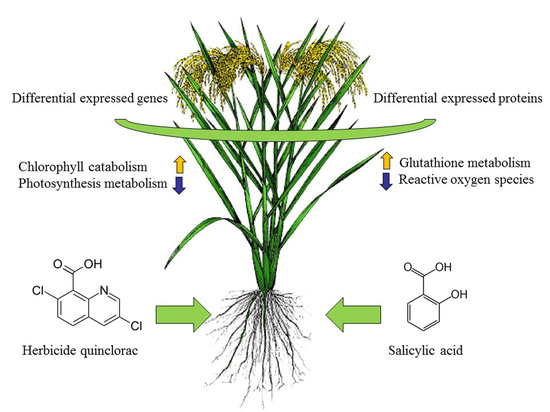
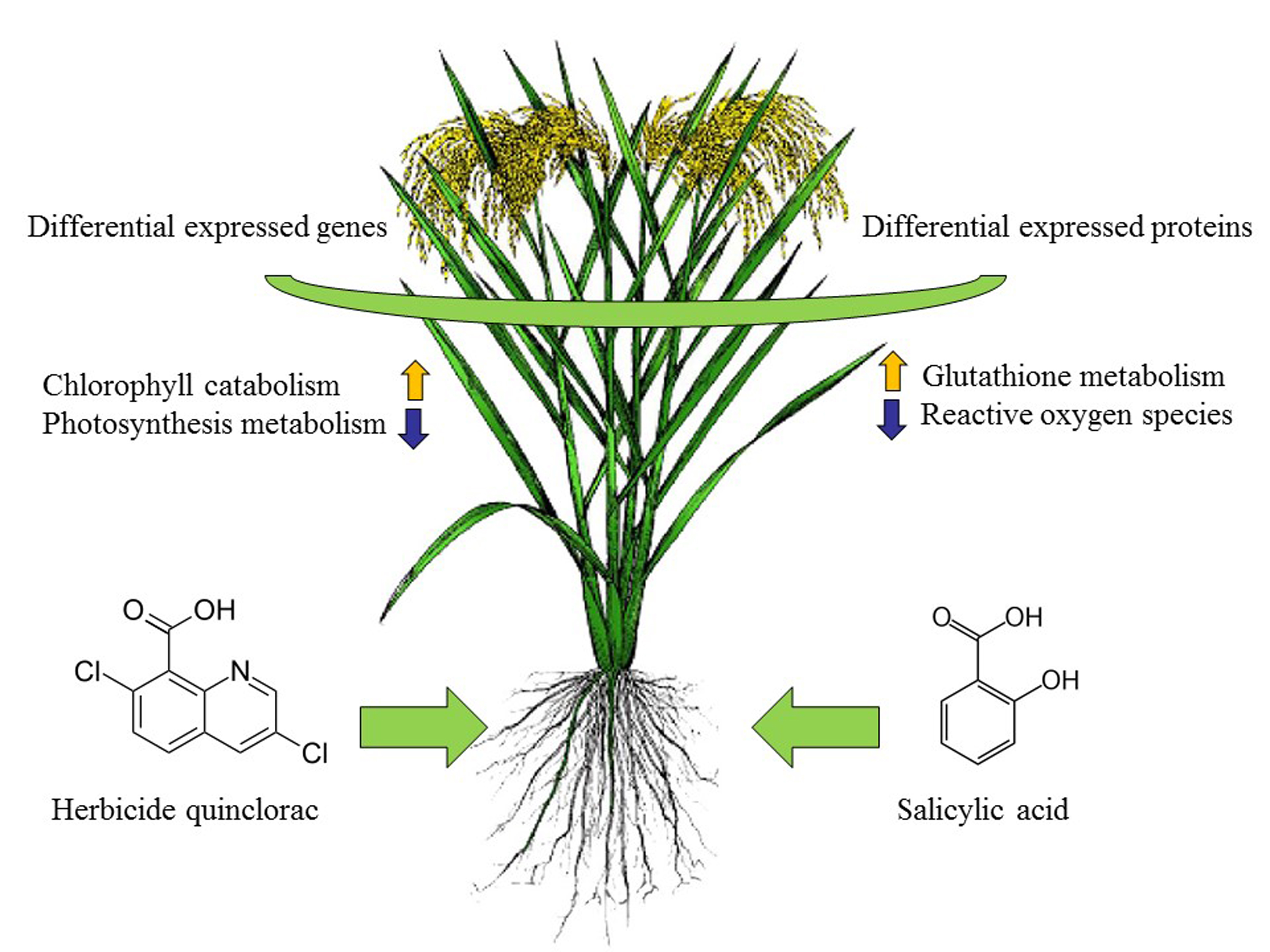
1. Introduction
2. Results
2.1. Primary Transcriptome Analysis
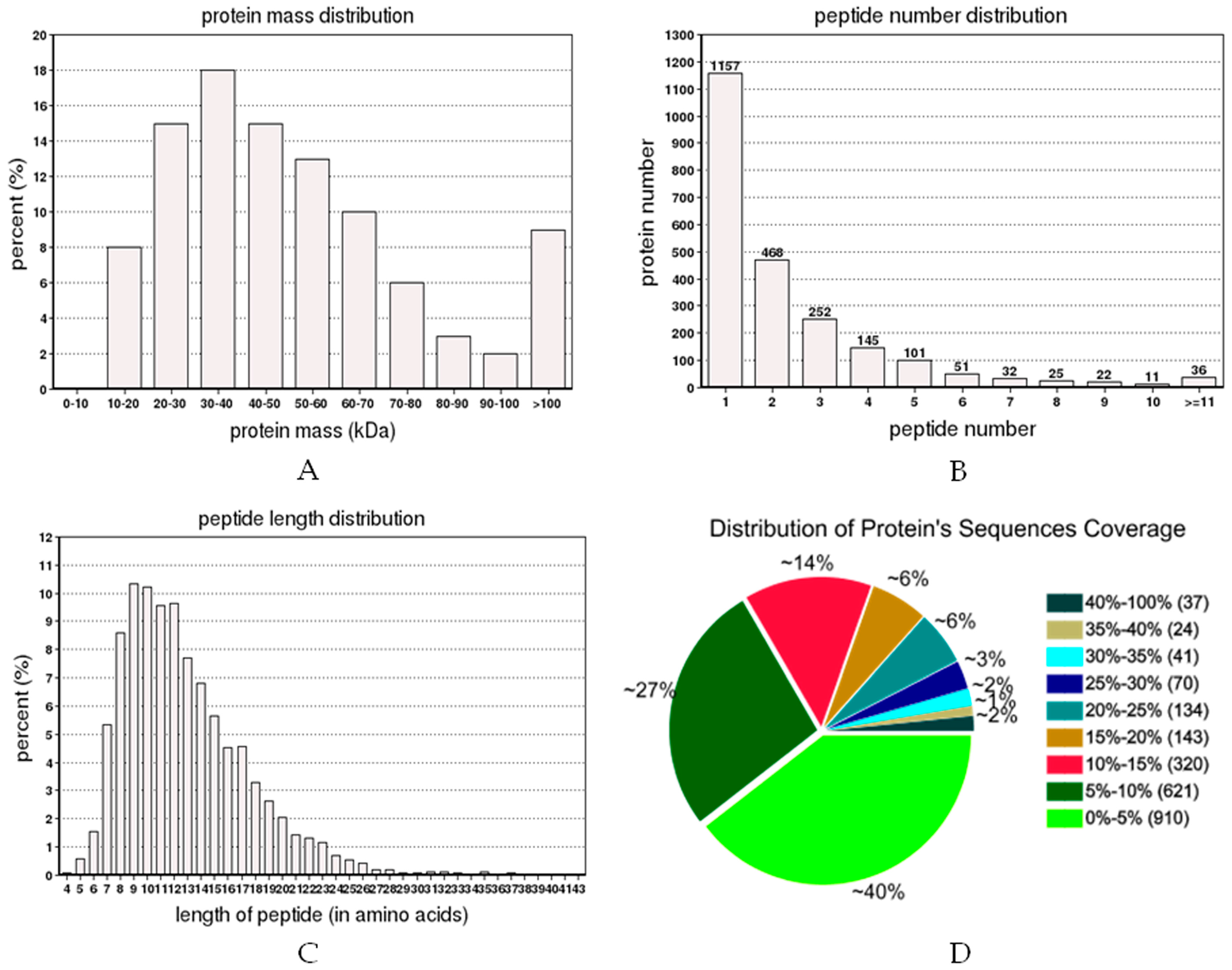
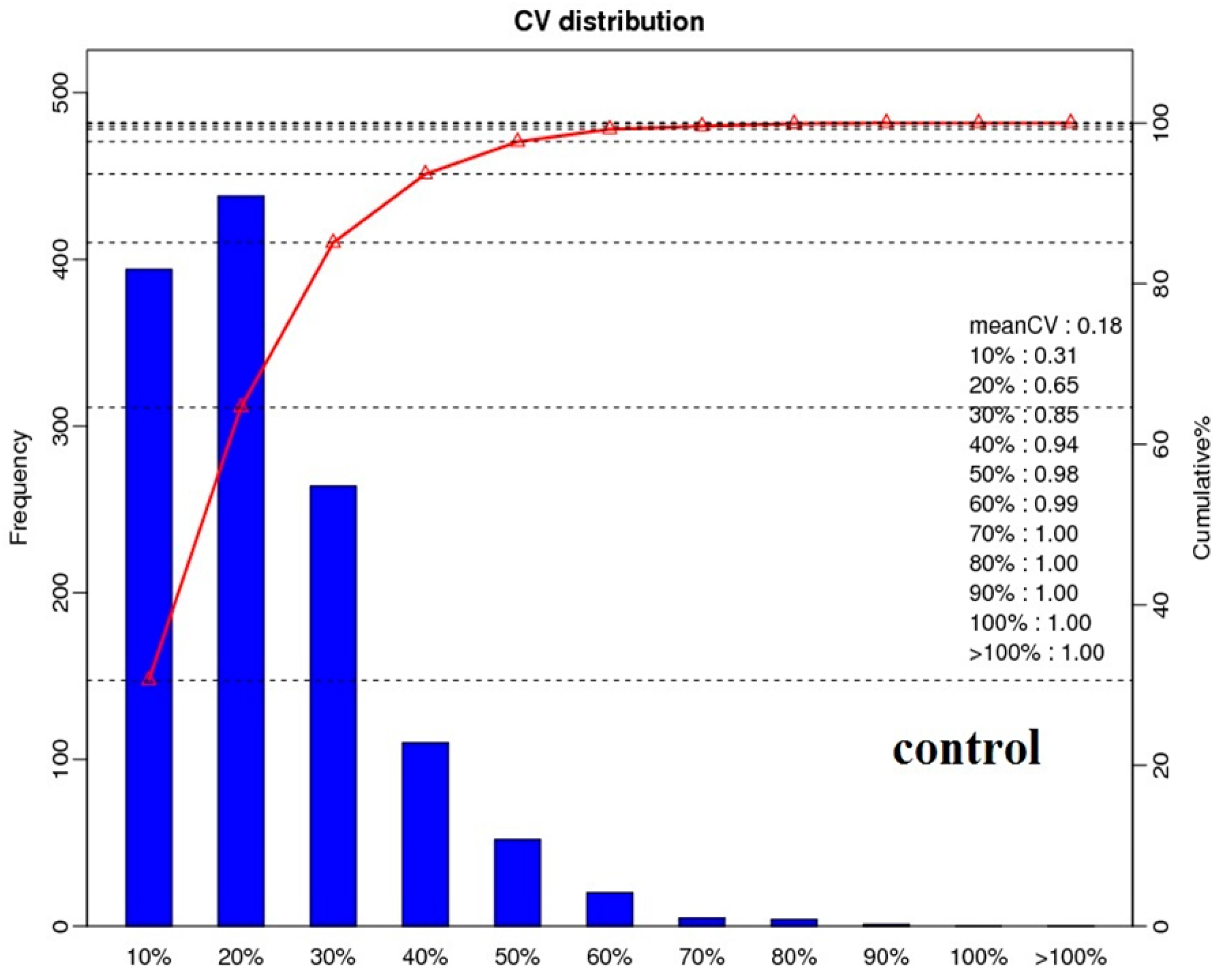
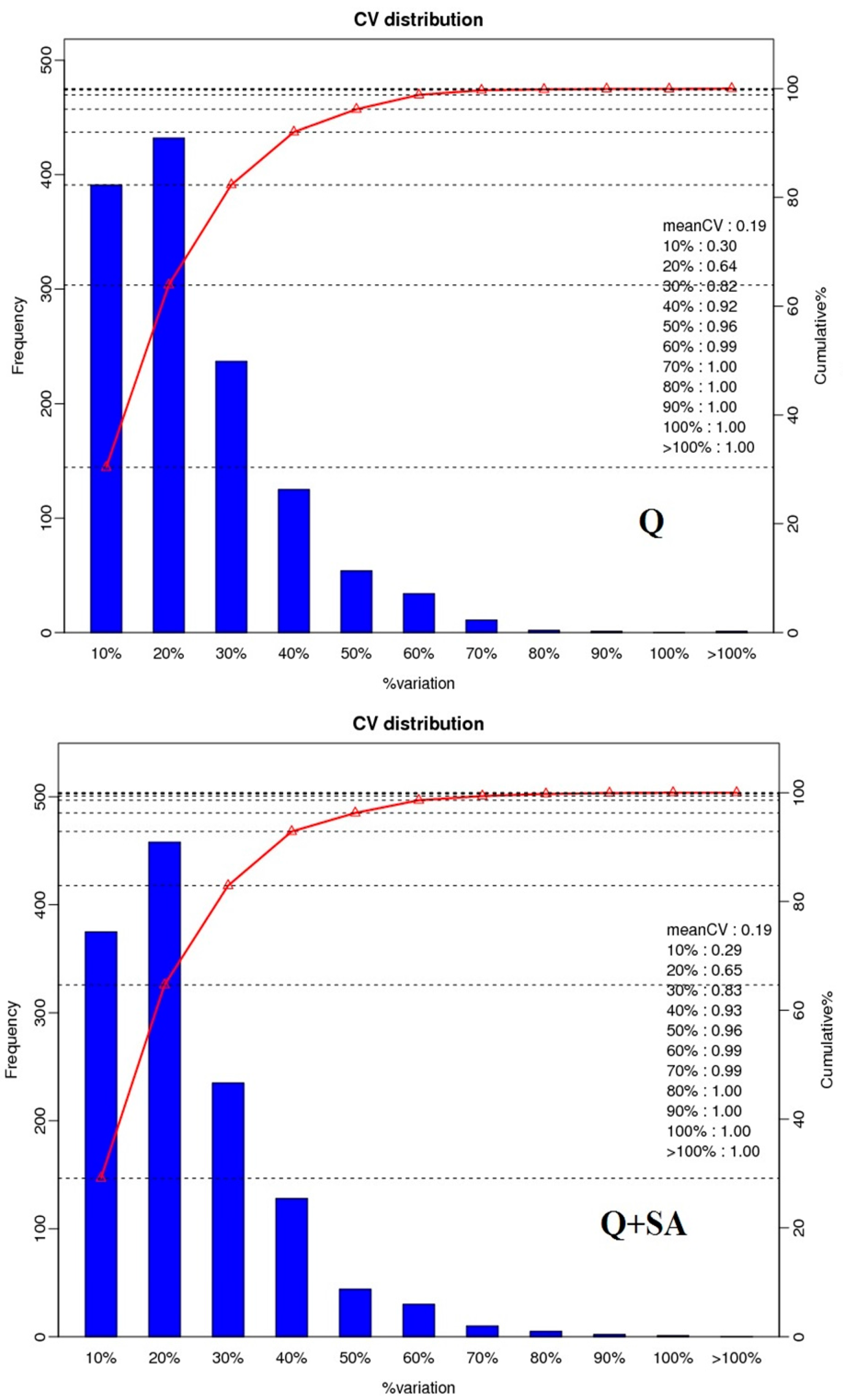
2.2. Protein Identification and Quantitation
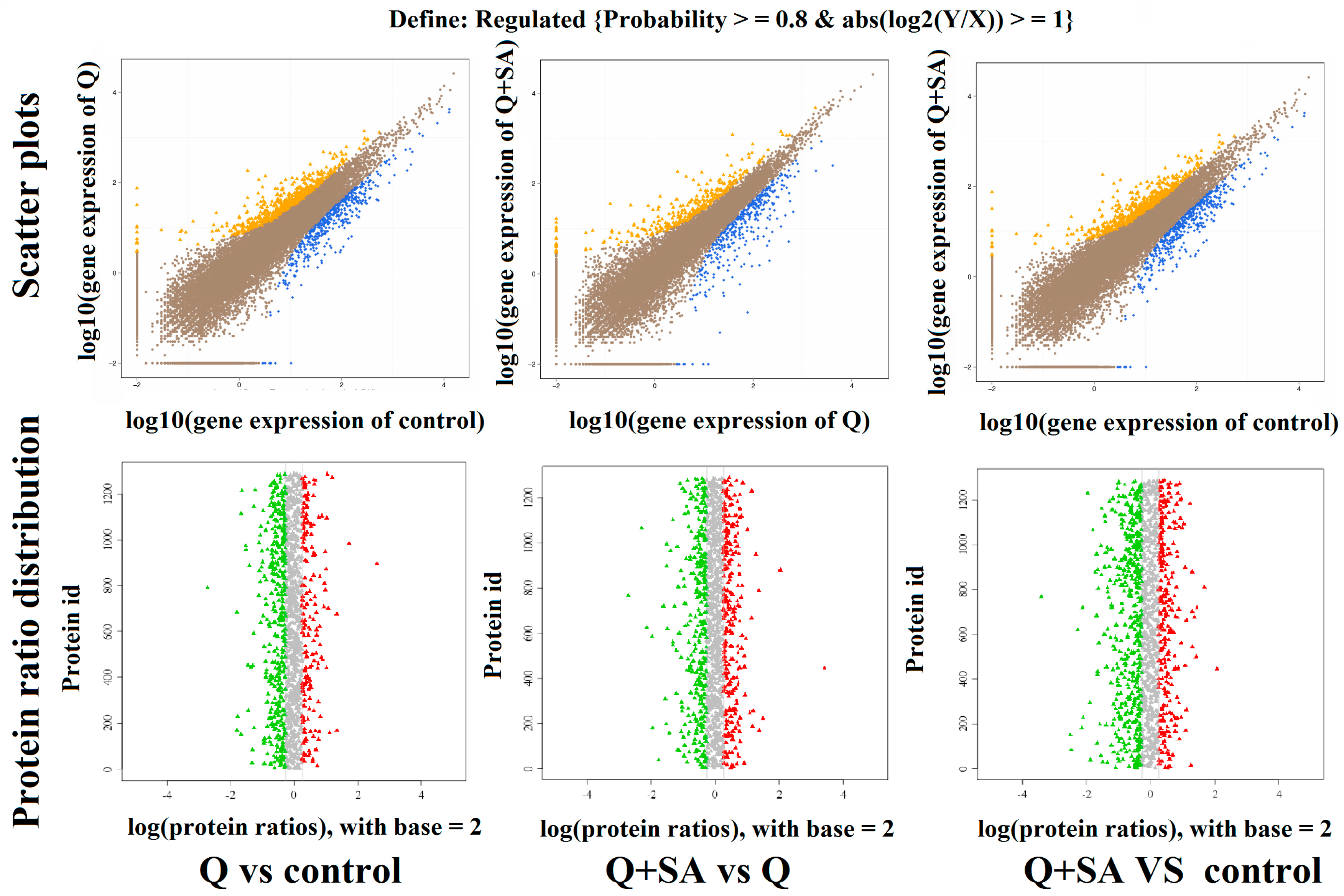
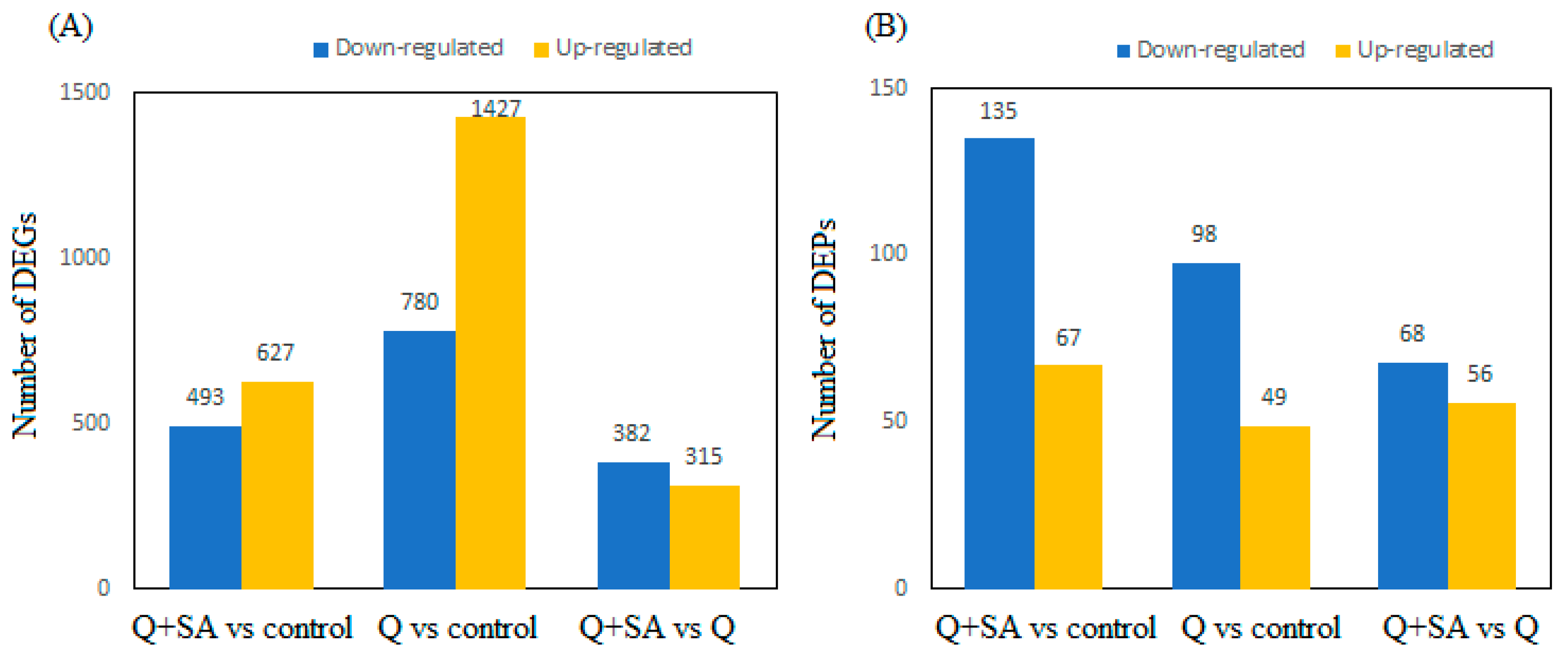
2.3. Screening of DEGs and DEPs
2.4. GO Functional Classification and Pathway Enrichment Analysis of DEGs
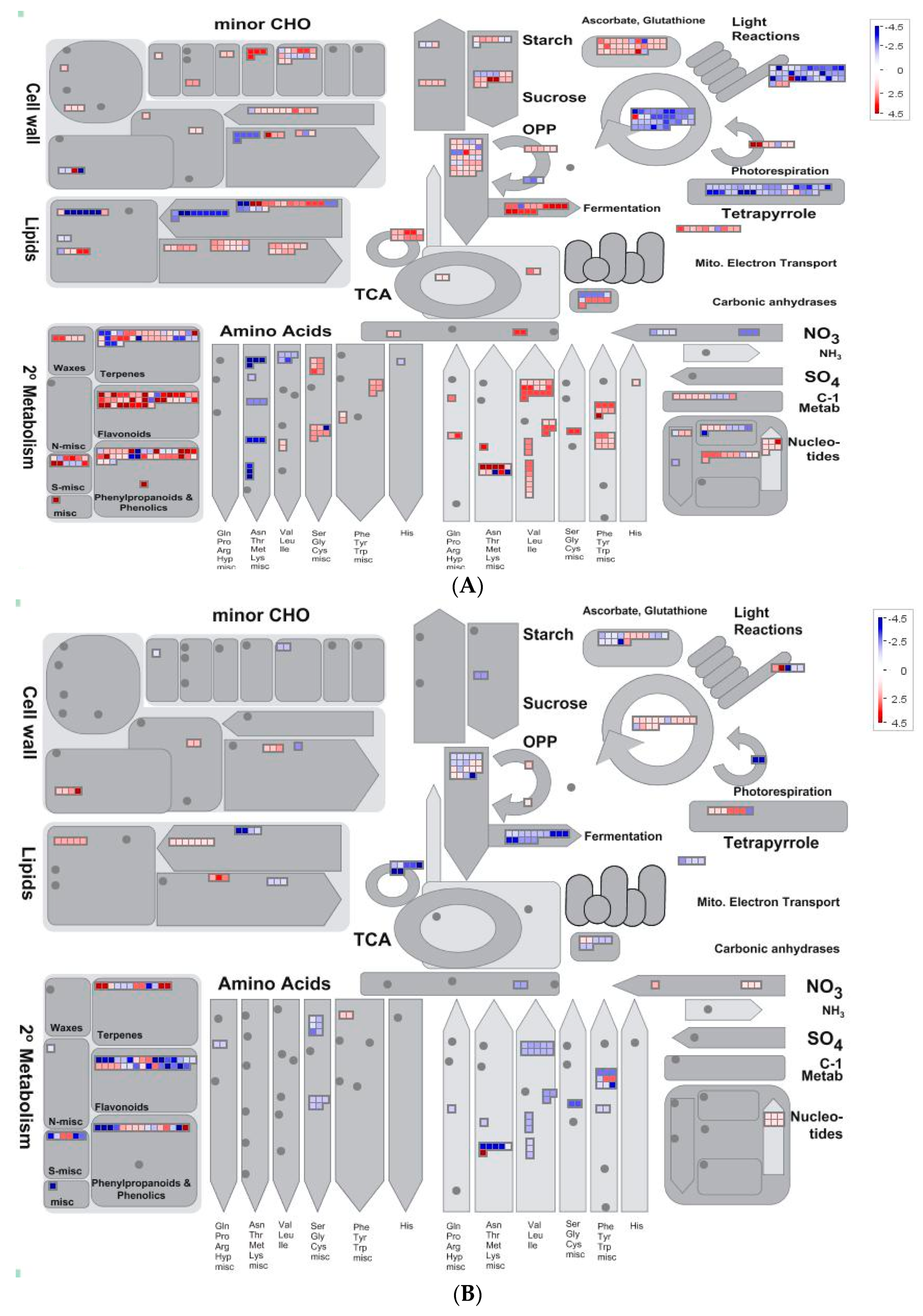
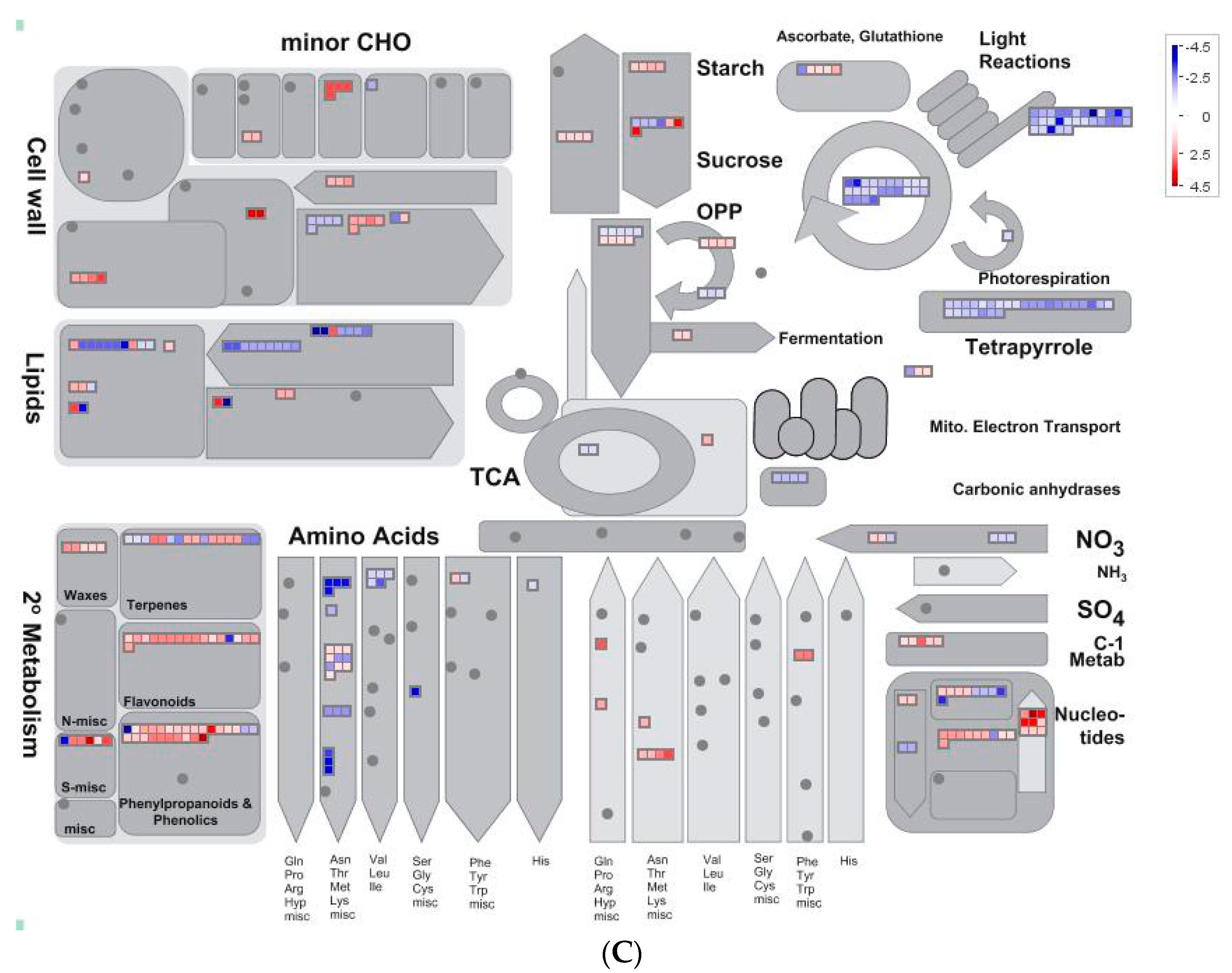
2.5. Overview of Metabolism by Using MapMan
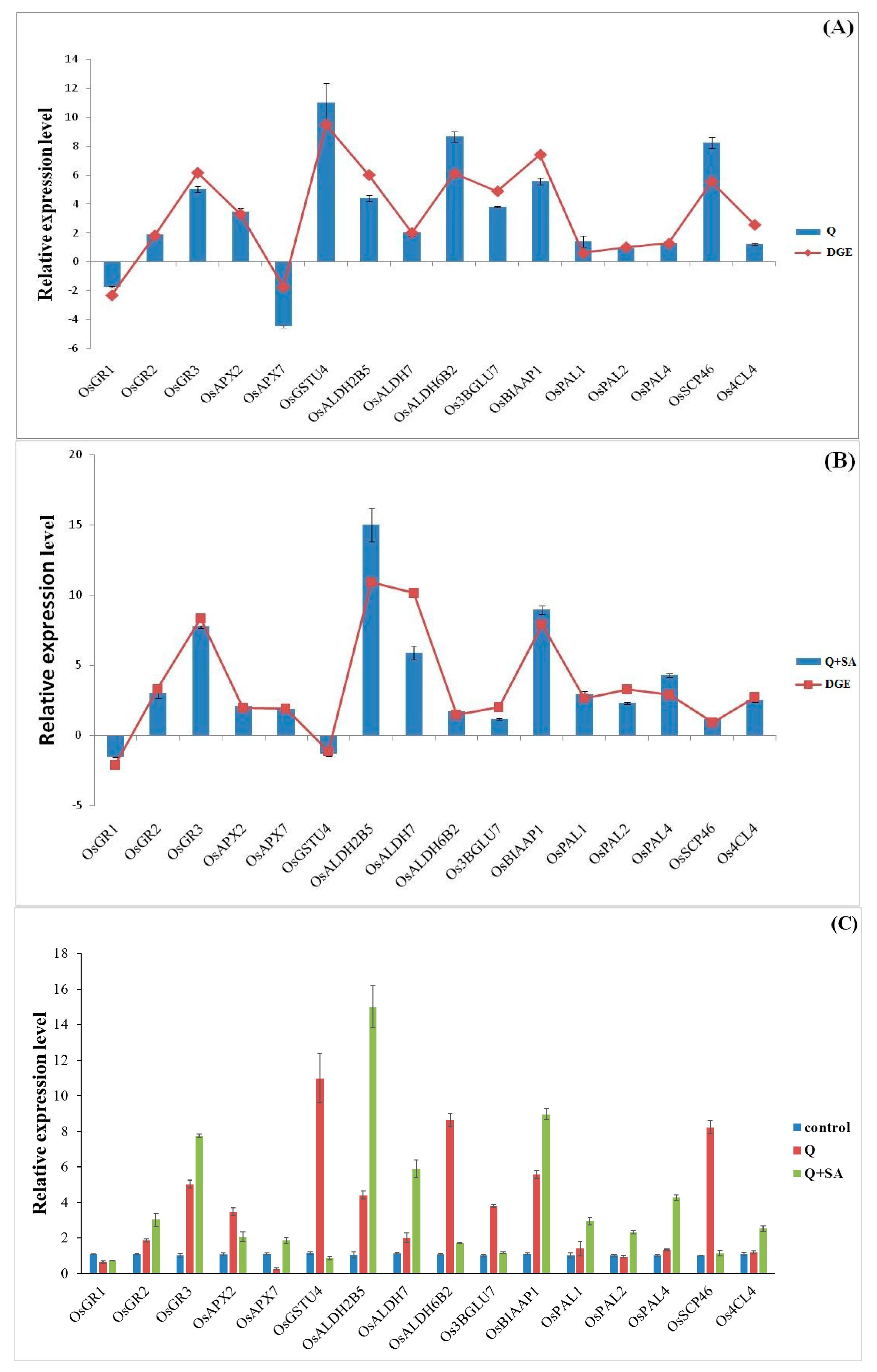
2.6. Quantitative PCR Analysis of Candidate Genes
2.7. Pathway Enrichment Analysis of DEPs
2.8. Transcription Factor Analysis
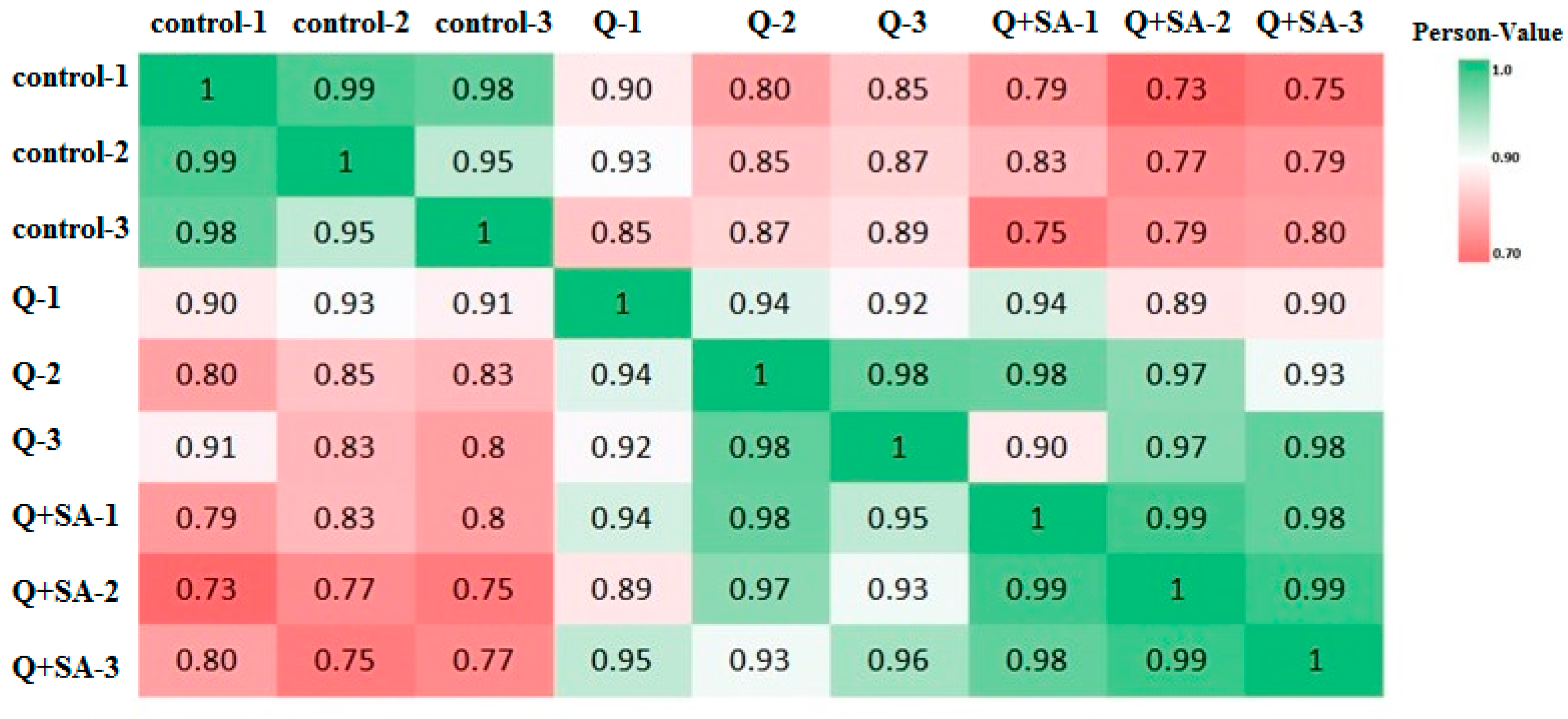
2.9. Correlation Analysis of Transcriptome and Proteome Data under Different Treatments
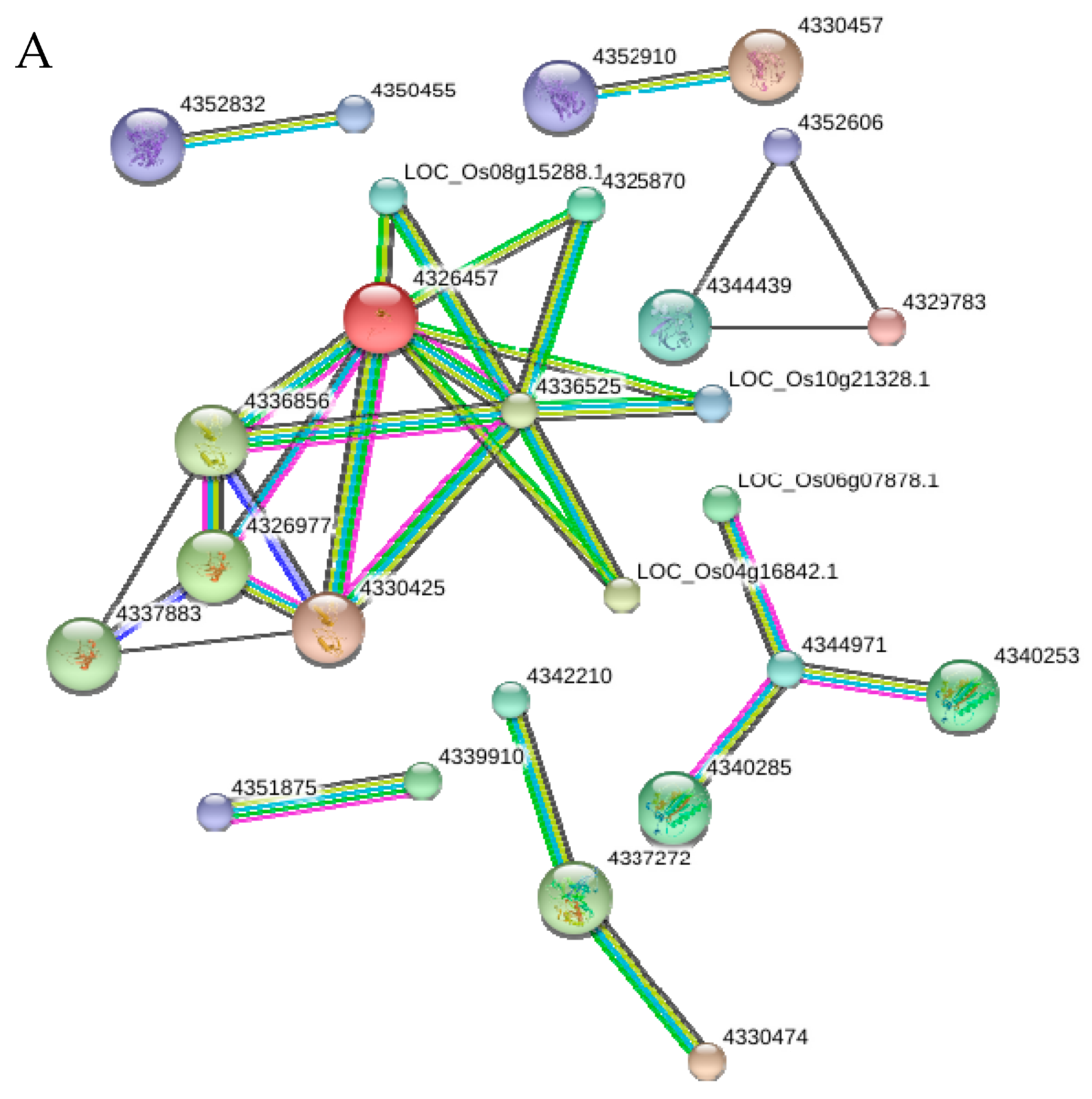
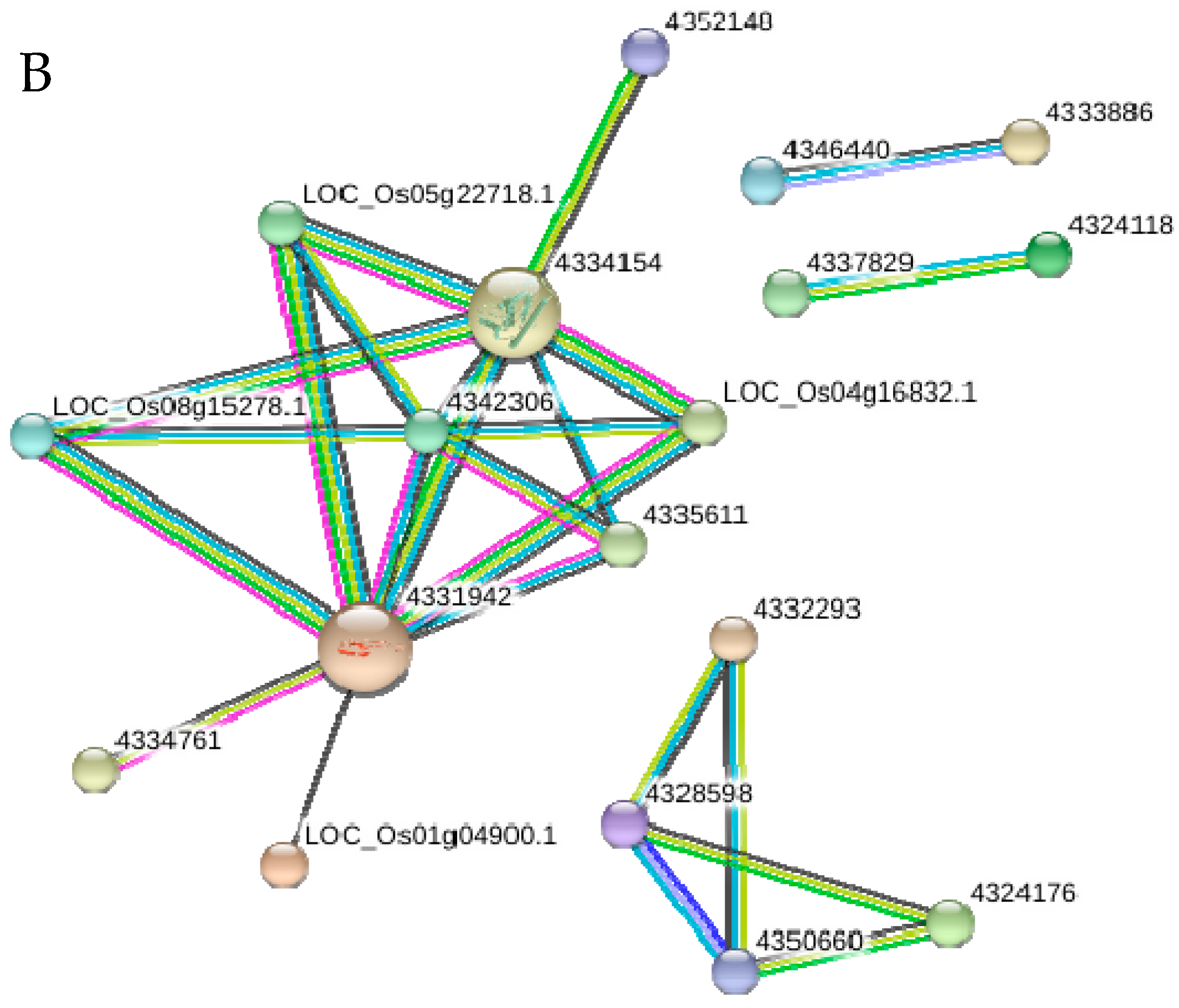
2.10. Functional Networks of the Selected Differentially Regulated Proteins
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Isolation and Library Preparation for Transcriptome Analysis
4.3. Analysis of RNA-Sequencing Data
4.4. qRT-PCR Analysis
4.5. Protein Extraction
4.6. Trypsin Digestion and iTRAQ Labeling
4.7. High-pH Reversed-Phase Chromatography
4.8. RPLC-MS/MS Analysis
4.9. Proteomic Data Analysis
4.10. Correlation Analysis between Transcriptome and Proteome
4.11. Protein-Protein Interaction Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author contributions
Conflicts of Interest
References
- National Bureau of Statistics of China. China Statistical Yearbook 2016. China Statistics Press. Available online: http://www.stats.gov.cn/tjsj/ndsj/2016/indexeh.htm (accessed on 7 September 2017).
- Chauhan, B.S.; Johnson, D.E. Opportunities to improve cultural approaches to manage weeds in direct-seeded rice. In Proceedings of the 17th Australasian Weeds Conference, Christchurch, New Zealand, 26–30 September 2010; New Zealand Plant Protection Society: Christchurch, New Zealand, 2010; pp. 40–43. [Google Scholar]
- Grossmann, K. Quinclorac belongs to a new class of highly selective auxin herbicides. Weed Sci. 1998, 46, 707–716. [Google Scholar]
- Li, G.; Xu, M.F.; Chen, L.P.; Cai, L.M.; Bai, L.Y.; Wu, C.X. A novel EcGH3 gene with a different expression pattern in quinclorac-resistant and susceptible barnyardgrass (Echinochloa crus-galli). Plant Gene 2016, 5, 65–70. [Google Scholar] [CrossRef]
- Hoffmann, O.L. Herbicide Antidotes: From Concept to Practice; National Meeting of the American Chemical Society: New Orleans, LA, USA, 1978; pp. 1–13. [Google Scholar]
- Chen, S.; Harmon, A.C. Advances in plant proteomics. Proteomics 2006, 6, 5504–5516. [Google Scholar] [CrossRef] [PubMed]
- Ekblom, R.; Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 2011, 107, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.S.; Tu, Z.J.; Cheung, F.; Xu, W.W.; Lamb, J.F.; Jung, H.-J.G.; Vance, C.P.; Gronwald, J.W. Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genom. 2011. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Zody, M.C. Advancing RNA-seq analysis. Nat. Biotechnol. 2010, 28, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Rakwal, R.; Agrawal, G.K. Rice proteomics: Current status and future perspectives. Electrophoresis 2003, 24, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, N.; Lee, D.; Alam, I.; Kim, P.J.; Lee, J.J.; Ahn, Y.O.; Kwak, S.S.; Lee, I.J.; Bahk, J.D.; Kang, K.Y.; et al. Comparative proteomic study of arsenic-induced differentially expressed proteins in rice roots reveals glutathione plays a central role during As stress. Proteomics 2009, 8, 3561–3576. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lv, M.T.; Islam, F.; Gill, R.A.; Yang, C.; Ali, B.; Yan, G.J.; Zhou, W.J. Salicylic acid mediates antioxidant defense system and ABA pathway related gene expression in Oryza sativa against quinclorac toxicity. Ecotoxicol. Environ. Saf. 2016, 133, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hu, L.Y.; Ali, B.; Islam, F.; Bai, Q.J.; Yun, X.P.; Yoneyama, K.; Zhou, W.J. Seed treatment with salicylic acid invokes defense mechanism of Helianthus annuus against Orobanche cumana. Ann. Appl. Biol. 2016, 169, 408–422. [Google Scholar] [CrossRef]
- Gill, R.A.; Zhang, N.; Ali, B.; Farooq, M.A.; Xu, J.X.; Gill, M.B.; Mao, B.Z.; Zhou, W.J. Role of exogenous salicylic acid in regulating physio-morphic and molecular changes under chromium toxicity in black- and yellow-seeded Brassica napus L. Environ. Sci. Pollut. Res. 2016, 23, 20483–20496. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Ali, S.; Farooq, M.A.; Wang, J.; Gill, R.A.; Zhu, J.; Zhou, W. Butachlor-induced alterations in ultrastructure, antioxidant, and stress-responsive gene regulations in rice cultivars. Clean Water Air Soil 2017. [Google Scholar] [CrossRef]
- Islam, F.; Farooq, M.A.; Gill, R.A.; Wang, J.; Chon, Y.; Ali, B.; Wang, G.-X.I.; Zhou, W.J. 2,4-D attenuates salinity-induced toxicity by mediating anatomical changes, antioxidant capacity and cation transporters in the roots of rice cultivars. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kaminaka, H.; Morita, S.; Nakajima, M.; Masumura, T.; Tanaka, K. Gene cloning and expression of cytosolic glutathione reductase in rice (Oryza sativa L.). Plant Cell Physiol. 1998, 39, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.Y.; Chao, Y.Y.; Yang, M.Y.; Cheng, S.Y.; Cho, S.C.; Kao, C.H. NaCl-induced expression of glutathione reductase in roots of rice (Oryza sativa L.) seedlings is mediated through hydrogen peroxide but not abscisic acid. Plant Soil 2009, 320, 103–115. [Google Scholar] [CrossRef]
- Wu, T.M.; Lin, W.R.; Kao, C.H.; Hong, C.Y. Gene knockout of glutathione reductase 3 results in increased sensitivity to salt stress in rice. Plant Mol. Biol. 2015, 87, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Dat, J.F.; Foyer, C.H.; Scott, I.M. Changes in salicylic acid and antioxidants during induced thermotolerance in mustard seedlings. Plant Physiol. 1998, 118, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Pappa, A.; Petersen, D.R. Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem. Biol. Interact. 2000, 129, 1–19. [Google Scholar] [CrossRef]
- Lindahl, R. Aldehyde dehydrogenases and their role in carcinogenesis. Crit. Rev. Biochem. Mol. 1992, 27, 283–335. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Rzhetsky, A.; Hsu, L.C.; Chang, C. Human aldehyde dehydrogenase gene family. Eur. J. Biochem. 1998, 251, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Kotchoni, S.O.; Bartels, D. Water stress induces the up-regulation of a specific set of genes in plants: Aldehyde dehydrogenase as an example. Bulg. J. Plant Physiol. 2003, 2003, 37–51. [Google Scholar]
- Kirch, H.H.; Bartels, D.; Wei, Y.; Schnable, P.S.; Wood, A.J. The ALDH gene superfamily of Arabidopsis. Trends Plant Sci. 2004, 9, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Meguro, N.; Suzuki, Y.; Tsutsumi, N.; Hirai, A.; Nakazono, M. Induction of mitochondrial aldehyde dehydrogenase by submergence facilitates oxidation of acetaldehyde during reaeration in rice. FEBS Lett. 2003, 546, 369–373. [Google Scholar] [CrossRef]
- Wu, C.; Su, S.; Peng, Y. Molecular cloning and differential expression of an aldehyde dehydrogenase gene in rice leaves in response to infection by blast fungus. Biologia 2007, 62, 523–528. [Google Scholar] [CrossRef]
- Shin, J.H.; Kim, S.R.; An, G. Rice aldehyde dehydrogenase7 is needed for seed maturation and viability. Plant Physiol. 2009, 149, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Han, B. Evolutionary and expression study of the aldehyde dehydrogenase (ALDH) gene superfamily in rice (Oryza sativa). Gene 2009, 431, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Bloom, A.J.; Queval, G.; Noctor, G. Photorespiratory metabolism: Genes, mutants, energetics, and redox signaling. Annu. Rev. Plant Biol. 2009, 60, 455–484. [Google Scholar] [CrossRef] [PubMed]
- Noctor, G.; Veljovic, J.S.; Driscoll, S.; Novitskaya, L.; Foyer, C.H. Drought and oxidative load in the leaves of C3 plants: A predominant role for photorespiration. Ann. Bot. 2002, 89, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Rizhsky, L.; Liang, H.; Mittler, R. The combined effect of drought stress and heat shock on gene expression in tobacco. Plant Physiol. 2002, 130, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Taler, D.; Galperin, M.; Benjamin, I.; Cohen, Y.; Kenigsbuch, D. Plant eR genes that encode photorespiratory enzymes confer resistance against disease. Plant Cell 2004, 16, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, Y.; Xie, Z.; Li, X.; He, Z.H.; Peng, X.X. Association–dissociation of glycolate oxidase with catalase in rice: A potential switch to modulate intracellular H2O2 levels. Mol. Plant 2016, 9, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tang, L.; Qiu, J.; Zhang, W.; Wang, Y.; Tong, X.; Wei, X.; Hou, Y.; Zhang, J. Serine carboxypeptidase 46 regulates grain filling and seed germination in rice (Oryza sativa L.). PLoS ONE 2016. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Shen, J.; Li, L. Functional characterization of evolutionarily divergent 4-coumarate: Coenzyme A ligases in rice. Plant Physiol. 2011, 157, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, X.; Zhang, H.; Song, F.M. Molecular characterization of a defense-related AMP-binding protein gene, OsBIABP1, from rice. J. Zhejiang Univ. Sci. B 2009, 10, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, Y.; Liu, Q.; Dubouzet, J.G.; Abe, H.; Shinozalri, K.; Yamaguci-Shinozaki, K. DNA binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem. Biophys. Res. Commun. 2002, 290, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Suzuki, K.; Fujimura, T.; Shin, S.H. Genome wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 2006, 140, 411–432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, Y.; Ma, Q.; Yang, C.; Mu, Y.; Suo, H.; Luo, L.; Nian, H. OsDREB2A, a rice transcription factor, significantly affects salt tolerance in transgenic soybean. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Zhang, W.; Zhang, Q.; Xu, Z.; Zhu, Z.; Duan, F.; Wu, R. Induced over-expression of the transcription factor OsDREB2A improves drought tolerance in rice. Plant Physiol. Biochem. 2011, 49, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, X.; Liu, J.; Ye, J.; Guo, Z. The rice ERF transcription factor OsERF922 negatively regulates resistance to Magnaporthe oryzae and salt tolerance. J. Exp. Bot. 2012, 63, 3899–3911. [Google Scholar] [CrossRef] [PubMed]
- Jisha, V.; Dampanaboina, L.; Vadassery, J.; Mithöfer, A.; Kappara, S.; Ramanan, R. Overexpression of an AP2/ERF type transcription factor OsEREBP1 confers biotic and abiotic stress tolerance in rice. PLoS ONE 2015, 10, e0127831. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.N.; Ernst, H.A.; Leggio, L.L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.S.; Kim, Y.S.; Baek, K.H.; Jung, H.; Ha, S.; Choi, Y.D.; Kim, M.; Reuzeau, C.; Kim, J. Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice under field drought conditions. Plant Physiol. 2010, 153, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Ogo, Y.; Nakanishi, I.R.; Nakanishi, H.; Kobayashi, T.; Takahashi, M.; Mori, S.; Nishizawa, N.K. The rice bHLH protein OsIRO2 is an essential regulator of the genes involved in Fe uptake under Fe-deficient conditions. Plant J. 2007, 51, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Ogo, Y.; Itai, R.N.; Kobayashi, T.; Aung, M.S.; Nakanishi, H.; Nishizawa, N.K. OsIRO2 is responsible for iron utilization in rice and improves growth and yield in calcareous soil. Plant Mol. Biol. 2011, 75, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, H.; Li, D.; Huang, L.; Hong, Y.; Ding, X.S.; Nelson, R.S.; Zhou, X.; Song, F. Functions of rice NAC transcriptional factors, ONAC122 and ONAC131, in defense responses against Magnaporthe grisea. Plant Mol. Biol. 2013, 81, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hong, Y.; Zhang, H.; Li, D.; Song, F. Rice NAC transcription factor ONAC095 plays opposite roles in drought and cold stress tolerance. BMC Plant Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.; Huang, J.; Zhao, W.; Peng, Y.; Chen, Z.; Kang, D. A salicylic acid-induced rice (Oryza sativa L.) transcription factor OsWRKY77 is involved in disease resistance of Arabidopsis thaliana. Plant Biol. 2013, 15, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Chujo, T.; Miyamoto, K.; Shimogawa, T.; Shimizu, T.; Otake, Y.; Yokotani, N.; Nishizawa, Y.; Shibuya, N.; Nojiri, H.; Yamane, H.; et al. OsWRKY28, a PAMP-responsive transrepressor, negatively regulates innate immune responses in rice against rice blast fungus. Plant Mol. Biol. 2013, 82, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Xiao, J.; Xie, W.; Liu, H.; Li, X.; Xiong, L.; Wang, S. Rice gene network inferred from expression profiling of plants overexpressing OsWRKY13, a positive regulator of disease resistance. Mol. Plant 2008, 1, 538–551. [Google Scholar] [PubMed]
- Tao, Z.; Kou, Y.; Liu, H.; Li, X.; Xiao, J.; Wang, S. OsWRKY45 alleles play different roles in abscisic acid signalling and salt stress tolerance but similar roles in drought and cold tolerance in rice. J. Exp. Bot. 2011, 62, 4863–4874. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.S.; Han, M.; Lee, S.K.; Cho, J.I.; Ryoo, N.; Heu, S.; Lee, Y.H.; Bhoo, S.H.; Wang, G.L.; Hahn, T.R.; et al. A comprehensive expression analysis of the WRKY gene superfamily in rice plants during defense response. Plant Cell Rep. 2006, 25, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Yokotani, N.; Sato, Y.; Tanabe, S.; Chujo, T.; Shimizu, T.; Okada, K.; Yamane, H.; Shimono, M.; Sugano, S.; Takatsuji, H.; et al. WRKY76 is a rice transcriptional repressor playing opposite roles in blast disease resistance and cold stress tolerance. J. Exp. Bot. 2013, 64, 5085–5097. [Google Scholar] [CrossRef] [PubMed]
- Caldana, C.; Degenkolbe, T.; Cuadros-Inostroza, A.; Klie, S.; Sulpice, R.; Leisse, A.; Steinhauser, D.; Fernie, A.R.; Willmitzer, L.; Hannah, M.A. High-density kinetic analysis of the metabolomic and transcriptomic response of Arabidopsis to eight environmental conditions. Plant J. 2011, 67, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.H.; Satoh, K.; Kikuchi, S.; Kim, S.C.; Ko, S.M.; Kang, H.G.; Jeon, J.S.; Kim, C.S.; Park, Y.I. Mitochondrial activity in illuminated leaves of chlorophyll-deficient mutant rice (OsCHLH) seedlings. Plant Biotechnol. Rep. 2010, 4, 281–291. [Google Scholar] [CrossRef]
- Jung, K.H.; Hur, J.; Ryu, C.H.; Choi, Y.; Chung, Y.Y.; Miyao, A.; Hirochika, H.; An, G. Characterization of a rice chlorophyll-deficient mutant using the T-DNA gene-trap system. Plant Cell Physiol. 2003, 44, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, Y.; Rahman, M.L.; Cho, S.H.; Kim, Y.S.; Koh, H.J.; Yoo, S.C.; Paek, N.C. The rice faded green leaf locus encodes protochlorophyllide oxidoreductase B and is essential for chlorophyll synthesis under high light conditions. Plant J. 2013, 74, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Xu, J.; Rao, Y.C.; Zeng, Y.J.; Liu, H.J.; Zheng, T.T.; Zhang, G.H.; Hu, J.; Guo, L.B.; Qian, Q.; et al. Cloning and functional analysis of pale-green leaf (PGL10) in rice (Oryza sativa L.). Plant Growth Regul. 2016, 78, 69–77. [Google Scholar] [CrossRef]
- Wu, Z.M.; Zhang, X.; He, B.; Diao, L.P.; Sheng, S.L.; Wang, J.L.; Guo, X.P.; Su, N.; Wang, L.F.; Jiang, L.; et al. A chlorophyll-deficient rice mutant with impaired chlorophyllide esterification in chlorophyll biosynthesis. Plant Physiol. 2007, 145, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, W.; Benz, J.; Guthoff, C. Detection and partial characterization of activity of chlorophyll synthetase in etioplast membranes. FEBS J. 1980, 109, 193–200. [Google Scholar] [CrossRef]
- Soll, J.; Schultz, G.; Rüdiger, W.; Benz, J. Hydrogenation of geranylgeraniol two pathways exist in spinach chloroplasts. Plant Physiol. 1983, 71, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, H.; Rümler, U.; Rüdiger, W. Reconstitution of pigment-containing complexes from light-harvesting chlorophyll a/b-binding protein overexpressed in Escherichia coli. Planta 1990, 181, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.A.; Ali, B.; Cui, P.; Shen, E.H.; Farooq, M.A.; Islam, F.; Ali, S.; Mao, B.Z.; Zhou, W.J. Comparative transcriptome profiling of two Brassica napus cultivars under chromium toxicity and its alleviation by reduced glutathione. BMC Genom. 2016. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Sonia, T.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar]
- Yang, C.; Xu, L.; Zhang, N.; Islam, F.; Song, W.J.; Hu, L.Y.; Liu, D.; Xie, X.N.; Zhou, W.J. iTRAQ-based proteomics of sunflower cultivars differing in resistance to parasitic weed Orobanche cumana. Proteomics 2017. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Total Reads | Quality Filtered Reads | Uniquely Mapped Reads | Genome Mapped Reads (%) | Gene Mapped Reads (%) |
|---|---|---|---|---|---|
| Control | 13,127,074 | 13,062,738 | 11,301,931 | 86.52 | 88.71 |
| Q | 13,126,991 | 13,047,776 | 11,277,174 | 86.43 | 89.12 |
| Q + SA | 13,127,032 | 13,028,062 | 11,223,009 | 86.15 | 89.83 |
| Average | 13,127,032 | 13,046,192 | 11,267,371 | 86.37 | 89.22 |
| Comparison | Pathway | Differential Proteins with Pathway Annotation (121) | All Proteins with Pathway Annotation (1871) | p Value | Pathway ID |
|---|---|---|---|---|---|
| Q vs. control | Cysteine and methionine metabolism | 9 (7.44%) | 39 (2.08%) | 0.0005958267 | ko00270 |
| Glyoxylate and dicarboxylate metabolism | 8 (6.61%) | 47 (2.51%) | 0.009008489 | ko00630 | |
| Phenylpropanoid biosynthesis | 9 (7.44%) | 58 (3.1%) | 0.01050140 | ko00940 | |
| Carbon metabolism | 18 (14.88%) | 159 (8.5%) | 0.01124338 | ko01200 | |
| C5-Branched dibasic acid metabolism | 2 (1.65%) | 5 (0.27%) | 0.03646180 | ko00660 | |
| Ascorbate and aldarate metabolism | 4 (3.31%) | 22 (1.18%) | 0.04913111 | ko00053 | |
| Metabolic pathways | 55 (45.45%) | 710 (37.95%) | 0.04918179 | ko01100 | |
| Q + SA vs. Q | Alanine, aspartate and glutamate metabolism | 5 (5.15%) | 28 (1.5%) | 0.01281717 | ko00250 |
| Riboflavin metabolism | 3 (3.09%) | 11 (0.59%) | 0.01644878 | ko00740 | |
| Arginine biosynthesis | 4 (4.12%) | 21 (1.12%) | 0.02056896 | ko00220 | |
| Terpenoid backbone biosynthesis | 3 (3.09%) | 12 (0.64%) | 0.02111914 | ko00900 | |
| 2-Oxocarboxylic acid metabolism | 5 (5.15%) | 32 (1.71%) | 0.02229215 | ko01210 | |
| Q + SA vs. control | Photosynthesis | 11 (6.29%) | 52 (2.78%) | 0.006954811 | ko00195 |
| Metabolic pathways | 82 (46.86%) | 710 (37.95%) | 0.007189333 | ko01100 | |
| Photosynthesis-antenna proteins | 4 (2.29%) | 13 (0.69%) | 0.02696767 | ko00196 | |
| Biosynthesis of amino acids | 18 (10.29%) | 123 (6.57%) | 0.03282929 | ko01230 | |
| Pyrimidine metabolism | 6 (3.43%) | 27 (1.44%) | 0.03444194 | ko00240 | |
| Valine, leucine and isoleucine biosynthesis | 3 (1.71%) | 9 (0.48%) | 0.04427594 | ko00290 | |
| Pentose and glucuronate interconversions | 4 (2.29%) | 15 (0.8%) | 0.04442334 | ko00040 |
| Comparison | Type | Proteins Number | Genes Number | Correlations Number |
|---|---|---|---|---|
| Q vs. control | Identification | 2300 | 27,342 | 2202 |
| Differ expressed | 147 | 2207 | 44 | |
| Q vs. Q+SA | Identification | 2300 | 27,342 | 2205 |
| Differ expressed | 124 | 697 | 5 | |
| Q+SA vs. control | Identification | 2300 | 27,342 | 2205 |
| Differ expressed | 202 | 1120 | 35 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Islam, F.; Li, L.; Long, M.; Yang, C.; Jin, X.; Ali, B.; Mao, B.; Zhou, W. Complementary RNA-Sequencing Based Transcriptomics and iTRAQ Proteomics Reveal the Mechanism of the Alleviation of Quinclorac Stress by Salicylic Acid in Oryza sativa ssp. japonica. Int. J. Mol. Sci. 2017, 18, 1975. https://doi.org/10.3390/ijms18091975
Wang J, Islam F, Li L, Long M, Yang C, Jin X, Ali B, Mao B, Zhou W. Complementary RNA-Sequencing Based Transcriptomics and iTRAQ Proteomics Reveal the Mechanism of the Alleviation of Quinclorac Stress by Salicylic Acid in Oryza sativa ssp. japonica. International Journal of Molecular Sciences. 2017; 18(9):1975. https://doi.org/10.3390/ijms18091975
Chicago/Turabian StyleWang, Jian, Faisal Islam, Lan Li, Meijuan Long, Chong Yang, Xiaoli Jin, Basharat Ali, Bizeng Mao, and Weijun Zhou. 2017. "Complementary RNA-Sequencing Based Transcriptomics and iTRAQ Proteomics Reveal the Mechanism of the Alleviation of Quinclorac Stress by Salicylic Acid in Oryza sativa ssp. japonica" International Journal of Molecular Sciences 18, no. 9: 1975. https://doi.org/10.3390/ijms18091975
APA StyleWang, J., Islam, F., Li, L., Long, M., Yang, C., Jin, X., Ali, B., Mao, B., & Zhou, W. (2017). Complementary RNA-Sequencing Based Transcriptomics and iTRAQ Proteomics Reveal the Mechanism of the Alleviation of Quinclorac Stress by Salicylic Acid in Oryza sativa ssp. japonica. International Journal of Molecular Sciences, 18(9), 1975. https://doi.org/10.3390/ijms18091975