Juvenile Moyamoya and Craniosynostosis in a Child with Deletion 1p32p31: Expanding the Clinical Spectrum of 1p32p31 Deletion Syndrome and a Review of the Literature

 ,
,  ,
, 
 and
and
Abstract
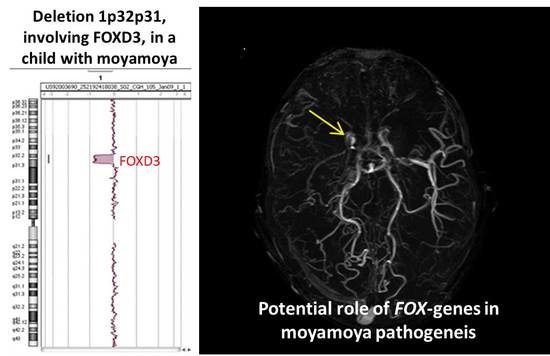
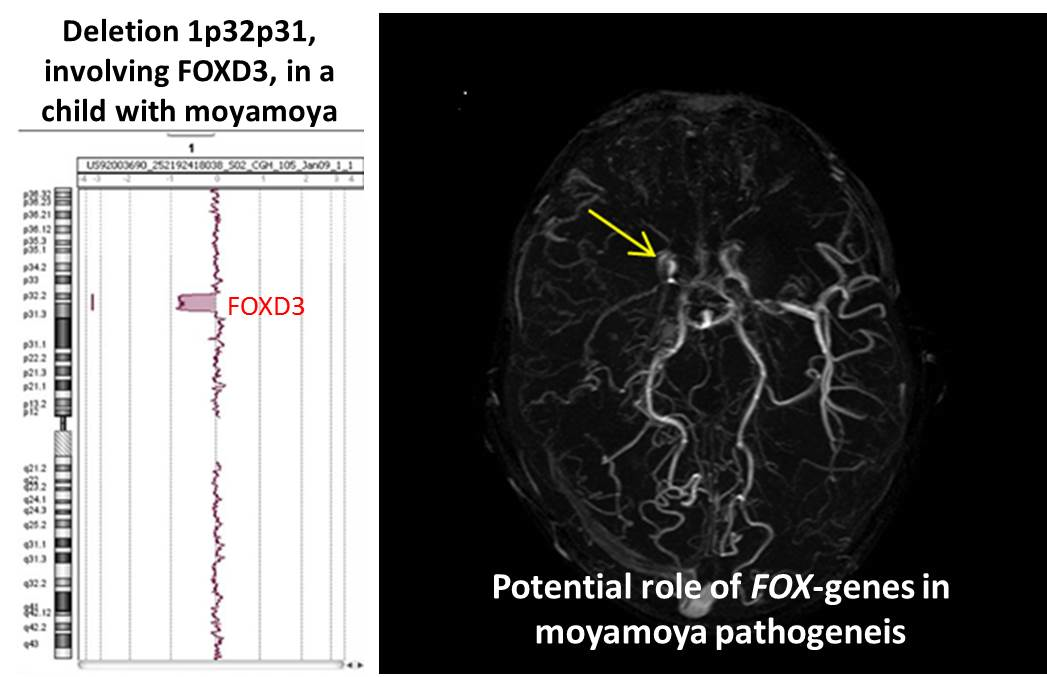
:
1. Introduction
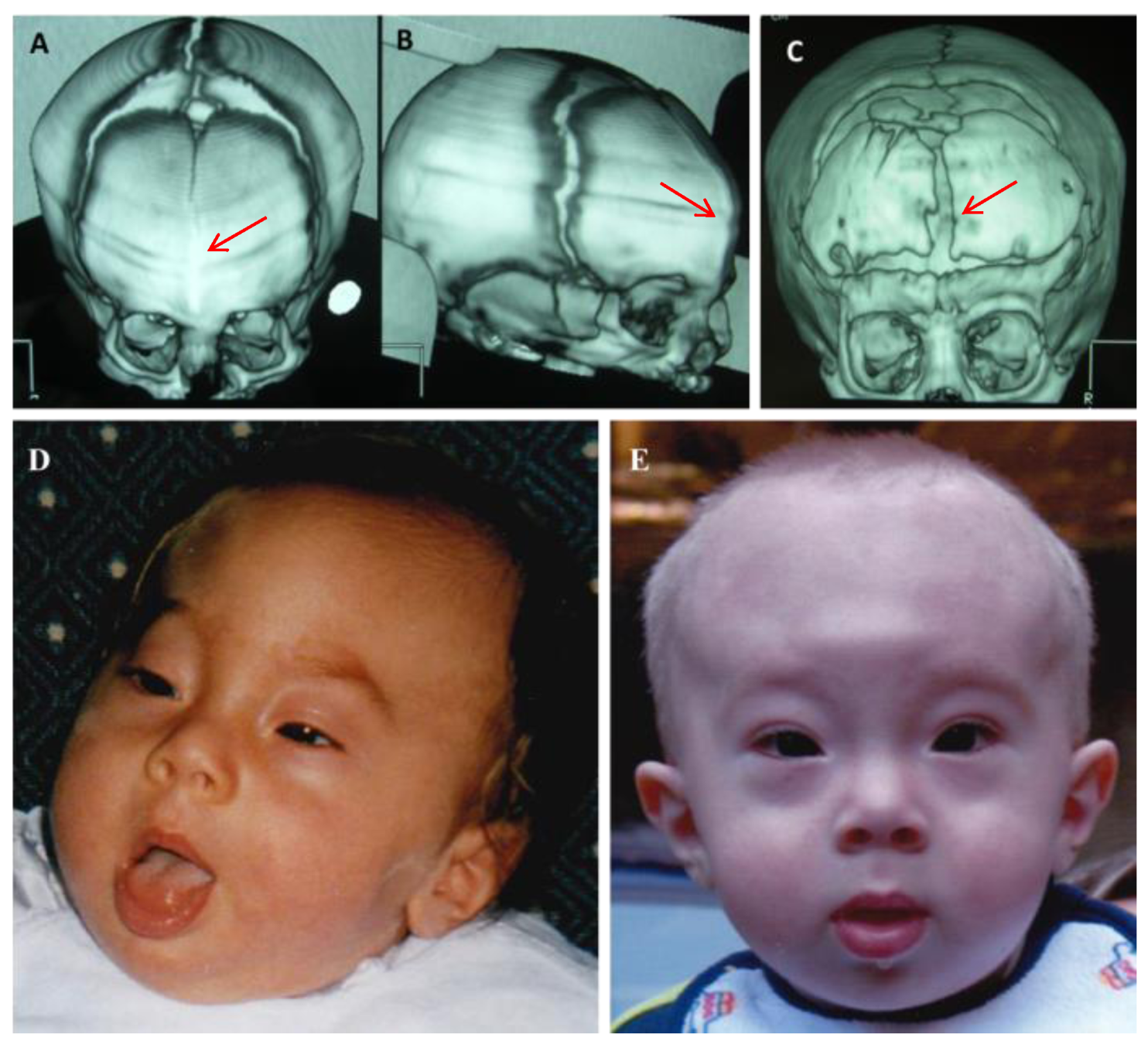
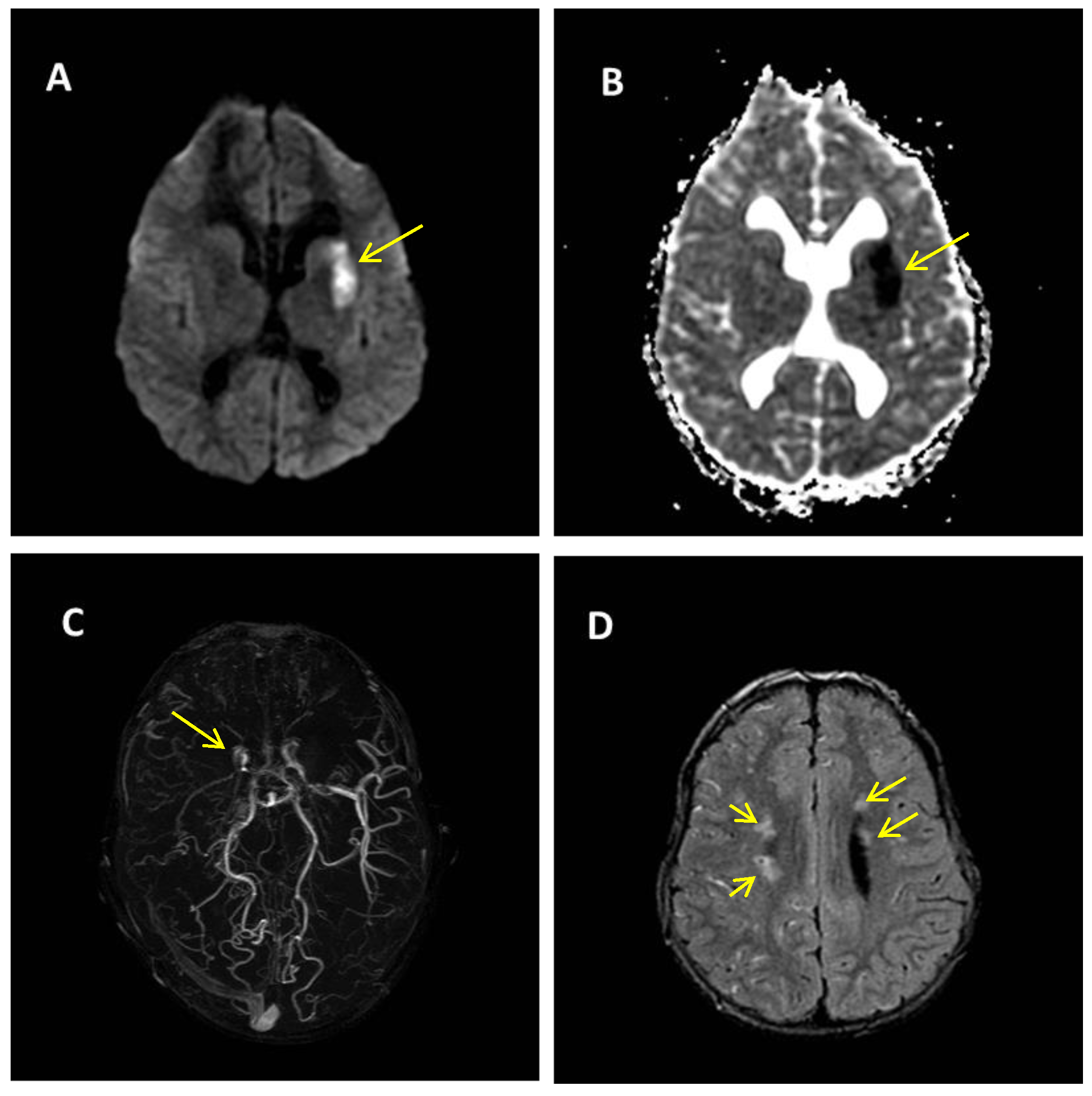
2. Case Report
Patient Presentation
3. Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Suzuki, J.; Takaku, A. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessel in base brain. Arch. Neurol. 1969, 20, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Guey, S.; Kraemer, M.; Hervé, D.; Ludwig, T.; Kossorotoff, M.; Bergametti, F.; Schwitalla, J.C.; Choi, S.; Broseus, L.; Callebaut, I.; et al. Rare RNF213 variants in the C-terminal region encompassing the RING-finger domain are associated with moyamoya angiopathy in Caucasians. Eur. J. Hum. Genet. 2017, 25, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Kainth, D.S.; Chaudhry, S.A.; Kainth, H.S.; Suri, F.K.; Qureshi, A.I. Prevalence and characteristics of concurrent down syndrome in patients with moyamoya disease. Neurosurgery 2013, 72, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Spengos, K.; Kosmaidou-Aravidou, Z.; Tsivgoulis, G.; Vassilopoulou, S.; Grigori-Kostaraki, P.; Zis, V. Moyamoya syndrome in a Caucasian woman with Turner's syndrome. Eur. J. Neurol. 2006, 13, e7–e8. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.E.; Egan, M.; Rodgers, S.; Harter, D.; Burnside, R.D.; Milla, S.; Pappas, J. Complex chromosome rearrangement of 6p25.3->p23 and 12q24.32->qter in a child with Moyamoya. Pediatrics 2013, 131, e1996–e2001. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, V.; Smith, E.R. Moyamoya: Defining current knowledge gaps. Dev. Med. Child Neurol. 2015, 57. [Google Scholar] [CrossRef] [PubMed]
- Janczar, S.; Fogtman, A.; Koblowska, M.; Baranska, D.; Pastorczak, A.; Wegner, O.; Kostrzewska, M.; Laguna, P.; Borowiec, M.; Mlynarski, W. Novel severe hemophilia A and moyamoya (SHAM) syndrome caused by Xq28 deletions encompassing F8 and BRCC3 genes. Blood 2014, 123, 4002–4004. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Mendoza-Londono, R.; Vlangos, C.N.; Dupuis, L.; Nowak, N.J.; Bunyan, D.J.; Hatchwell, E.; Elsea, S.H. Smith-Magenis syndrome and Moyamoya disease in a patient with del(17)(p11.2p13.1). Am. J. Med. Genet. A 2007, 143, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.O.; Baek, H.J.; Woo, Y.J.; Choi, Y.Y.; Chung, T.W. Moyamoya syndrome in a child with trisomy 12p syndrome. Pediatr. Neurol. 2006, 35, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Di Rocco, F.; Kossorotoff, M.; Zerah, M.; Boddaert, N.; Calmon, R.; Vidaud, D.; Cirillo, M.; Cinalli, G.; Mirone, G.; et al. Moyamoya syndrome in children with neurofibromatosis type 1: Italian-French experience. Am. J. Med. Genet. A 2017, 173, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Choudhri, O.A.; Feroze, A.H.; Do, H.M.; Grant, G.A.; Steinberg, G.K. Management of moyamoya syndrome in patients with Noonan syndrome. J. Clin. Neurosci. 2016, 28, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Shiihara, T.; Kato, M.; Mitsuhashi, Y.; Hayasaka, K. Costello syndrome showing moyamoya-like vasculopathy. Pediatr. Neurol. 2005, 32, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.; Soro, I.; Leitão, A.; Silva, M.L.; Leão, M. Moyamoya vascular pattern in Alagille syndrome. Pediatr. Neurol. 2012, 47, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Yokote, H.; Tsuura, M.; Nakai, K.; Ohshima, A.; Itakura, T. Marfan syndrome associated with moyamoya phenomenon and aortic dissection. Acta Neurochir. 1999, 141, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xu, R.; Porras, J.L.; Takemoto, C.M.; Khalid, S.; Garzon-Muvdi, T.; Caplan, J.M.; Colby, G.P.; Coon, A.L.; Tamargo, R.J.; et al. Effectiveness of surgical revascularization for stroke prevention in pediatric patients with sickle cell disease and moyamoya syndrome. J. Neurosurg. Pediatr. 2017, 20, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Hervé, D.; Philippi, A.; Belbouab, R.; Zerah, M.; Chabrier, S.; Collardeau-Frachon, S.; Bergametti, F.; Essongue, A.; Berrou, E.; Krivosic, V.; et al. Loss of α1β1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am. J. Hum. Genet. 2014, 94, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Xin, B.; Jones, S.; Puffenberger, E.G.; Hinze, C.; Bright, A.; Tan, H.; Zhou, A.; Wu, G.; Vargus-Adams, J.; Agamanolis, D.; et al. Homozygous mutation in SAMHD1 gene causes cerebral vasculopathy and early onset stroke. Proc. Natl. Acad. Sci. USA 2011, 108, 5372–5377. [Google Scholar] [CrossRef] [PubMed]
- Kılıç, E.; Utine, E.; Unal, S.; Haliloğlu, G.; Oğuz, K.K.; Cetin, M.; Boduroğlu, K.; Alanay, Y. Medical management of moyamoya disease and recurrent stroke in an infant with Majewski osteodysplastic primordial dwarfism type II (MOPD II). Eur. J. Pediatr. 2012, 171, 1567–1571. [Google Scholar] [CrossRef] [PubMed]
- Codd, P.J.; Scott, R.M.; Smith, E.R. Seckel syndrome and moyamoya. J. Neurosurg. Pediatr. 2009, 3, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Kamada, F.; Aoki, Y.; Narisawa, A.; Abe, Y.; Komatsuzaki, S.; Kikuchi, A.; Kanno, J.; Niihori, T.; Ono, M.; Ishii, N.; et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum. Genet. 2011, 56, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Bersano, A.; Guey, S.; Bedini, G.; Nava, S.; Hervé, D.; Vajkoczy, P.; Tatlisumak, T.; Sareela, M.; van der Zwan, A.; Klijn, C.J.; et al. Research Progresses in Understanding the Pathophysiology of Moyamoya Disease. Cerebrovasc. Dis. 2016, 41, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Miskinyte, S.; Butler, M.G.; Hervé, D.; Sarret, C.; Nicolino, M.; Petralia, J.D.; Bergametti, F.; Arnould, M.; Pham, V.N.; Gore, A.V.; et al. Loss of BRCC3 deubiquitinating enzyme leads to abnormal angiogenesis and is associated with syndromic moyamoya. Am. J. Hum. Genet. 2011, 88, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Kono, S.; Sueishi, K.; Ikezaki, K. Moyamoya disease. Neuropathology 2000, 20, S61–S64. [Google Scholar] [CrossRef] [PubMed]
- Tokita, M.J.; Chow, P.M.; Mirzaa, G.; Dikow, N.; Maas, B.; Isidor, B.; Le Caignec, C.; Penney, L.S.; Mazzotta, G.; Bernardini, L.; et al. Five children with deletions of 1p34.3 encompassing AGO1 and AGO3. Eur. J. Hum. Genet. 2015, 23, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Prontera, P.; Bartocci, A.; Ottaviani, V.; Isidori, I.; Rogaia, D.; Ardisia, C.; Guercini, G.; Mencarelli, A.; Donti, E. Aicardi syndrome associated with autosomal genomic imbalance: Coincidence or evidence for autosomal inheritance with sex-limited expression? Mol. Syndromol. 2013, 4, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Quintero-Rivera, F.; Fan, Y.; Alkuraya, F.S.; Donovan, D.J.; Xi, Q.; Turbe-Doan, A.; Li, Q.G.; Campbell, C.G.; Shanske, A.L.; et al. NFIA haploinsufficiency is associated with a CNS malformation syndrome and urinary tract defects. PLoS Genet. 2007, 3, e80. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.P.; Su, Y.N.; Chen, Y.Y.; Chern, S.R.; Liu, Y.P.; Wu, P.C.; Lee, C.C.; Chen, Y.T.; Wang, W. Chromosome 1p32-p31 deletion syndrome: Prenatal diagnosis by array comparative genomic hybridization using uncultured amniocytes and association with NFIA haploinsufficiency, ventriculomegaly, corpus callosum hypogenesis, abnormal external genitalia, and intrauterine growth restriction. Taiwan J. Obstet. Gynecol. 2011, 50, 345–352. [Google Scholar] [PubMed]
- Wellcome Trust Sanger Institute. Available online: http://decipher.sanger.ac.uk (accessed on 1 August 2017).
- Dottori, M.; Gross, M.K.; Labosky, P.; Goulding, M. The winged-helix transcription factor Foxd3 suppresses interneuron differentiation and promotes neural crest cell fate. Development 2001, 128, 4127–4138. [Google Scholar] [PubMed]
- Mundell, N.A.; Labosky, P.A. Neural crest stem cell multipotency requires Foxd3 to maintain neural potential and repress mesenchymal fates. Development 2011, 138, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Couly, G.; Coltey, P.; Le Douarin, N. The triple origin of skull in higher vertebrates: A study in quail-chick chimeras. Development 1993, 117, 409–429. [Google Scholar] [PubMed]
- Mouse Genome Informatics. Available online: http://www.informatics.jax.org (accessed on 1 August 2017).
- Dollfus, H.; Biswas, P.; Kumaramanickavel, G.; Stoetzel, C.; Quillet, R.; Biswas, J.; Lajeunie, E.; Renier, D.; Perrin-Schmitt, F. Saethre-Chotzen syndrome: Notable intrafamilial phenotypic variability in a large family with Q28X TWIST mutation. Am. J. Med. Genet. 2002, 109, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Horie, N.; Suyama, K.; Nagata, I. Clinical features and long-term follow-up of quasi-moyamoya disease in children. Pediatr. Neurosurg. 2011, 47, 15–21. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
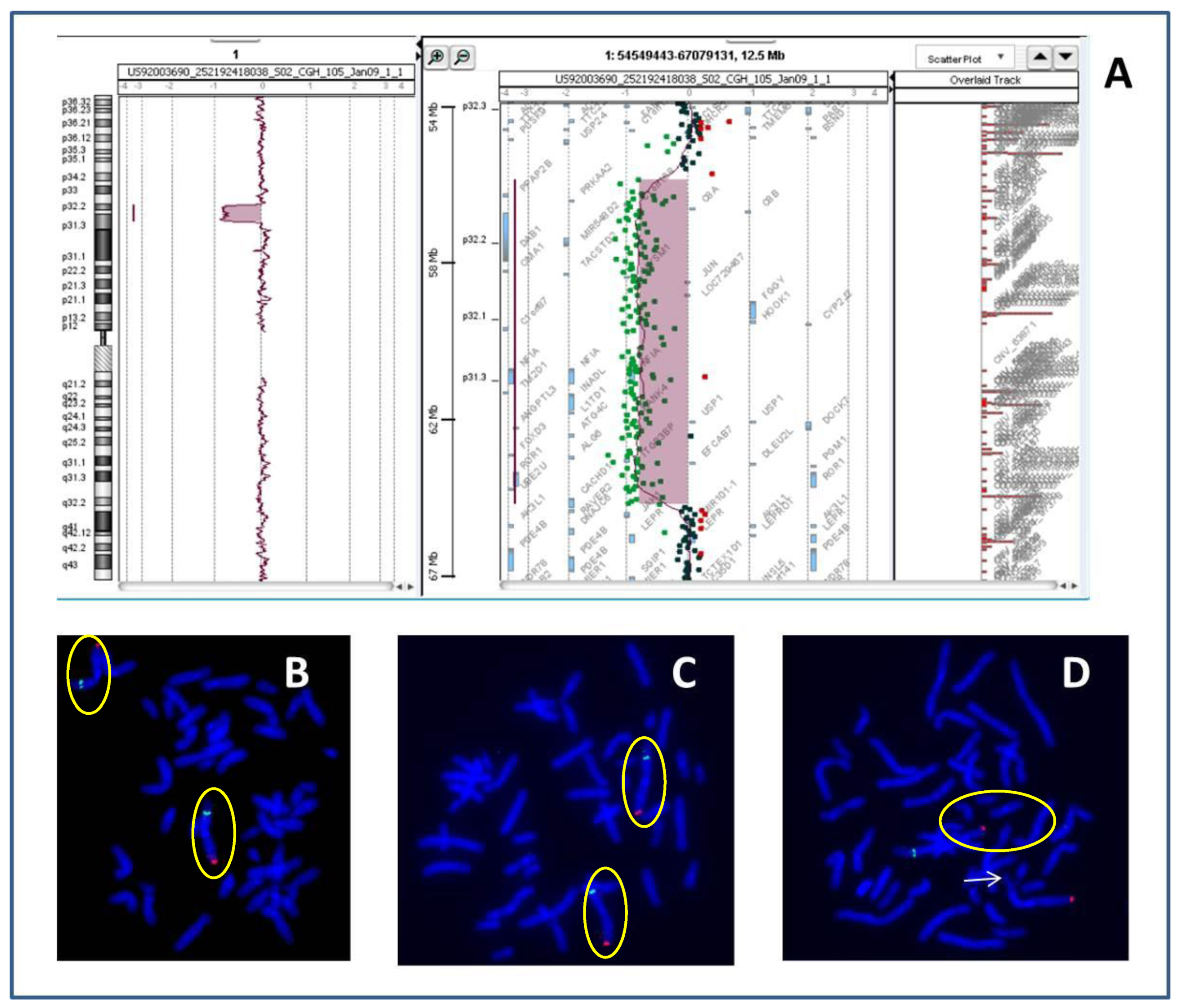{kind=link}
| Chromosomic Disorders | Chromosome Involved |
| Down syndrome (complete or mosaic form) [3] | 21 |
| Turner syndrome (complete or mosaic form) [4] | X |
| Genomic Disorders | Gene/s Involved |
| 6p25.3-p23 del/dup and 12q24.32-qter dup [5] | On 6p region: IRF4 and other 51 OMIM genes including FOXC1; On 12q region: 22 OMIM genes not associated with genetic disorders. |
| 15q13.3 duplication (Decipher ID: 263336) [2,6] | CHRNA7, OTUD7A |
| Xq28 deletion [7] | F8 (exon 1-6), FUNDC2, MTCP1NB, MTCP1, BRCC3 |
| Smith-Magenis syndrome (del 17p11.2-p13.1) [8] | More than 25 genes including: RAI1; MED9; RASD1; FLCN; PMP22; COX10; ELAC2; ZNF18; MYH1 |
| Trisomy 12p [9] | Genes included in the region of the rearrangement: 46, XX, rec(12)dup(12p)inv(12)(p11.2q24.3)mat |
| 1p32p31 deletion (present report) | OMIM genes included in the region of the rearrangement: C8A; C8B; TACSTD2; ANGPTL3; FOXD3; ALG6; PGM1 |
| Monogenic Disorders | Gene/s Involved |
| Type 1 Neurofibromatosis [10] | NF1 |
| Noonan syndrome [11] | PTPN11, SOS1, RAF1 and, more rarely: KRAS; NRAS; BRAF, and; MAP2K1 |
| Costello syndrome [12] | HRAS |
| Alagille syndrome [13] | JAG1, NOTCH2 |
| Marfan syndrome [14] | FBN1 |
| Sickle cell disease [15] | HBB |
| Moyamoya disease-6 with achalasia [16] | GUCY1A3 |
| SAMHD1-related disorders [17] | SAMHD1 |
| MOPD2/Majewski syndrome [18] | PCNT |
| Seckel syndrome (microcephalic primordial dwarfism) [19] | ATR, RBBP8, CENPJ, CEP152, CEP63, NIN |
| Clinical Features | Present Case | Frequency on Reported Cases (Literature, Decipher, Present Case) | Total | References |
|---|---|---|---|---|
| Brain malformations | + | 12/20 | 60% | Decipher ID: 288170; Literature: [16,17,18,19,20,21] |
| Corpus callosum defects | + | 11/20 | 55% | Decipher ID: 251391-4638; Literature: [16,17,18,19,20,21] |
| Facial dysmorphisms | + | 9/20 | 45% | Decipher ID: 264827-285848; Literature: [16,17,18,19,20,21] |
| Genito-urinary defects | + | 7/20 | 35% | Literature: [16,18,19,20,21] |
| Developmental delay | − | 6/20 | 30% | Decipher ID: 264827-285848; Literature: [16,17,19,20] |
| Intellectual disability | + | 5/20 | 25% | Decipher ID: 251391-288170-4638 |
| Hypotonia | + | 5/20 | 25% | Decipher ID: 285848; Literature: [16,17,19,20] |
| Language disabilities | + | 3/20 | 15% | Decipher ID: 276512 |
| Craniosynostosis | + | 2/20 | 10% | Decipher ID: 251391 |
| Moyamoya disease | + | 1/20 | 5% | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prontera, P.; Rogaia, D.; Mencarelli, A.; Ottaviani, V.; Sallicandro, E.; Guercini, G.; Esposito, S.; Bersano, A.; Merla, G.; Stangoni, G. Juvenile Moyamoya and Craniosynostosis in a Child with Deletion 1p32p31: Expanding the Clinical Spectrum of 1p32p31 Deletion Syndrome and a Review of the Literature. Int. J. Mol. Sci. 2017, 18, 1998. https://doi.org/10.3390/ijms18091998
Prontera P, Rogaia D, Mencarelli A, Ottaviani V, Sallicandro E, Guercini G, Esposito S, Bersano A, Merla G, Stangoni G. Juvenile Moyamoya and Craniosynostosis in a Child with Deletion 1p32p31: Expanding the Clinical Spectrum of 1p32p31 Deletion Syndrome and a Review of the Literature. International Journal of Molecular Sciences. 2017; 18(9):1998. https://doi.org/10.3390/ijms18091998
Chicago/Turabian StyleProntera, Paolo, Daniela Rogaia, Amedea Mencarelli, Valentina Ottaviani, Ester Sallicandro, Giorgio Guercini, Susanna Esposito, Anna Bersano, Giuseppe Merla, and Gabriela Stangoni. 2017. "Juvenile Moyamoya and Craniosynostosis in a Child with Deletion 1p32p31: Expanding the Clinical Spectrum of 1p32p31 Deletion Syndrome and a Review of the Literature" International Journal of Molecular Sciences 18, no. 9: 1998. https://doi.org/10.3390/ijms18091998
APA StyleProntera, P., Rogaia, D., Mencarelli, A., Ottaviani, V., Sallicandro, E., Guercini, G., Esposito, S., Bersano, A., Merla, G., & Stangoni, G. (2017). Juvenile Moyamoya and Craniosynostosis in a Child with Deletion 1p32p31: Expanding the Clinical Spectrum of 1p32p31 Deletion Syndrome and a Review of the Literature. International Journal of Molecular Sciences, 18(9), 1998. https://doi.org/10.3390/ijms18091998