1. Introduction
During the past decades, fluorescent proteins (FPs) have been widely used as tools in fluorescence microscopy to address a broad array of biological and biomedical questions. At the heart of this success is the non-invasive and sensitive nature of fluorescence microscopy, combined with the outstanding selectivity and labeling properties of the fluorophores [
1]. The widespread success of fluorescent proteins in imaging has led to an impressive toolbox of FPs that covers a gamut of possible colors and encompasses a large range of photo-physical properties [
2,
3].
The discovery and development of photoresponsive FPs has been of particular interest [
1,
4]. This class of “smart” fluorescent proteins possesses controllable dynamic fluorescence emission, which enables their use for several sub-diffraction imaging techniques such as (fluorescence) photoactivated localization microscopy ((f)PALM) [
5,
6], reversible saturable optical linear fluorescence transitions (RESOLFT) [
7,
8,
9], non-linear structured illumination microscopy (NL-SIM) [
10,
11], and photochromic stochastic optical fluctuation imaging (pcSOFI) [
12,
13]. These sub-diffraction techniques rely heavily on the performance of the fluorophores [
14]. Consequently, plenty of effort has been devoted to creating more optimized photoresponsive FPs, as well as to achieving a fundamental understanding of the mechanisms through which this “smart” behavior is defined [
15,
16,
17,
18,
19,
20,
21].
A recent example is the development of the rsGreen series of reversibly switchable fluorescent proteins (RSFPs), based on the enhanced green fluorescent protein (EGFP) [
22]. The development of these and many other fluorescent proteins has established that the immediate chromophore microenvironment crucially affects the spectroscopy and photochemistry of these labels [
2,
22]. Intriguingly, this dependence on the protein structure is not just limited to the residues closest to the chromophore, as different research groups have shown that the spectroscopic properties of FPs can be manipulated through interactions with other molecules, often proteins, further removed from the chromophore environment. The postulated mechanism for these modulators often relies on an influence on protein dynamics and rearrangements which indirectly change the chromophore environment, thereby affecting the photo-physical properties [
23,
24,
25,
26,
27,
28]. Nanobodies, for example, tightly bind GFP-like fluorescent proteins, and have been shown to modulate the spectral properties and pH sensitivity of these labels in doing so. One of these, the so-called “Enhancer” nanobody, is of particular interest as it results in increased fluorescence and improved pH stability of wild type (wt)GFP, superfolder (sf)GFP and EGFP upon binding [
23,
29]. In addition, the nanobody could be fused directly to the fluorescent protein itself, leading to a single label with improved spectroscopic properties that can directly benefit live cell imaging [
29].
We hypothesized that such effects could similarly lead to altered properties in RSFPs that may improve their performance in advanced fluorescence imaging. In this work we therefore set out to examine the effect of the Enhancer nanobody on RSFPs from the rsGreen series.
3. Discussion
We set out to determine whether the direct fusion of the Enhancer nanobody to the rsGreen series of photochromic fluorescent proteins resulted in similar advantages as described previously for non-photochromic FPs [
23,
29]. While in vitro fusion indeed resulted in analogous spectroscopic improvements in terms of molecular brightness and pH stability, a decrease in fluorescence signal was observed in living cells. We attribute this reduction in apparent brightness in situ to altered expression, degradation and/or maturation of the protein in the complex cellular environment, as is well known to occur for fluorescent proteins [
22].
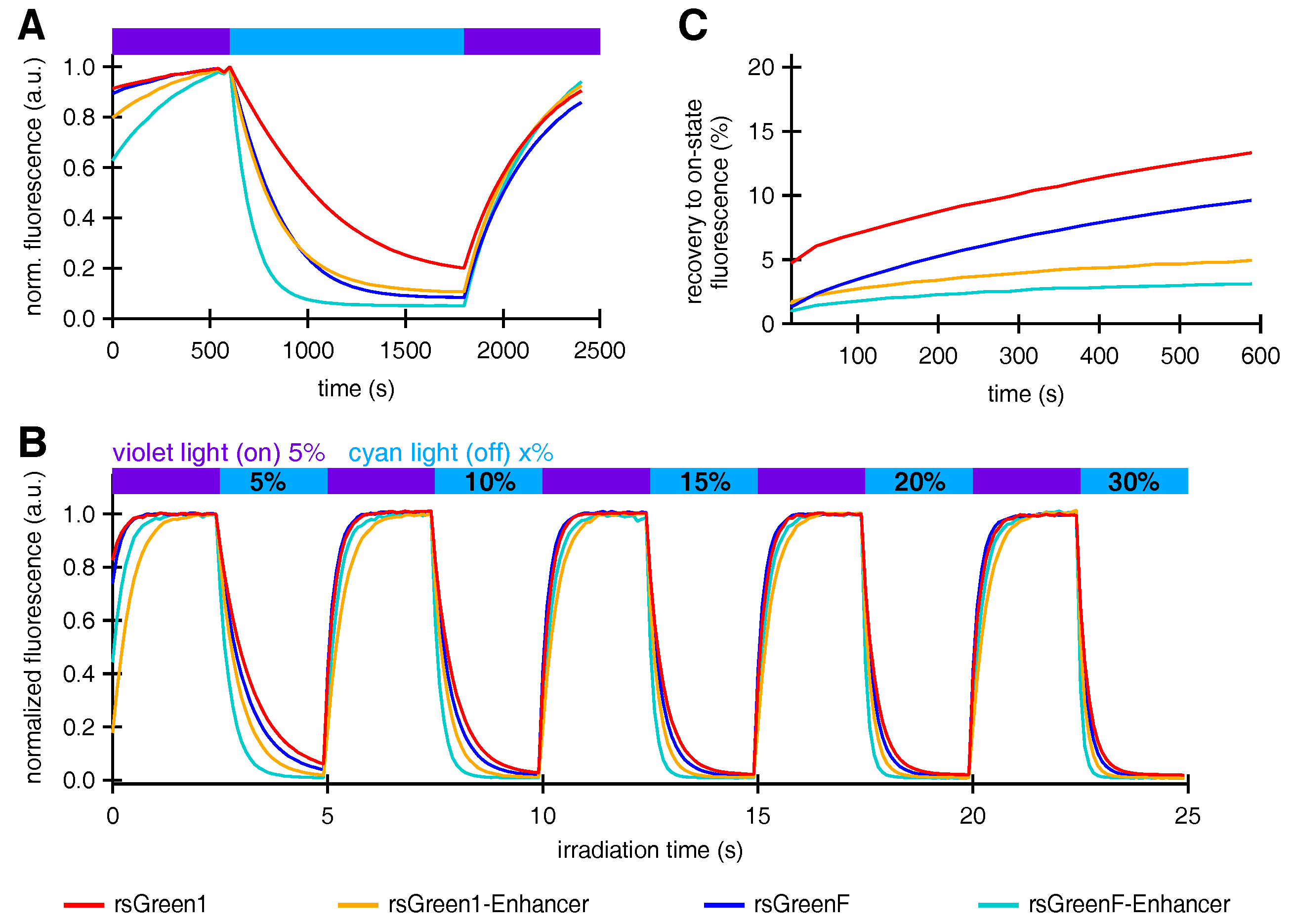
Interestingly, nanobody fusion also affected the photochromism of the labels, increasing the off-switching rate of both rsGreen1 and rsGreenF. This potentially makes the rsGreen-Enhancer fusions superior candidates for RESOLFT sub-diffraction imaging, since fast off-switching allows for short pixel dwell times, which in turn lead to increased temporal resolution [
32,
33]. To test this hypothesis, future experiments should probe the performance of Enhancer-bound rsGreens in RESOLFT-type imaging compared to the unbound rsGreens and other existing labels. Additionally, the fact that binding of a protein partner changes the switching kinetics opens up the possibility for Enhancer binding to serve as a contrast mechanism for advanced imaging techniques which can discern fluorescent labels with different photochromic behavior, such as lock-in detection [
34,
35,
36], multitau (mt)-pcSOFI [
37] or
-RESOLFT [
38]. A highly interesting prospect in this direction is that modulation of photochromism through binding with interaction partners could revolutionize biosensor design by providing a (ratiometric and super-resolution) readout mechanism, which is easy to multiplex with other fluorescence measurements due to limited usage of the visible spectrum [
39,
40]. A similar modulation of photochromic behavior has recently been published in a biosensor design that enables the visualization of protein kinase A activity with sub-diffraction resolution [
28].
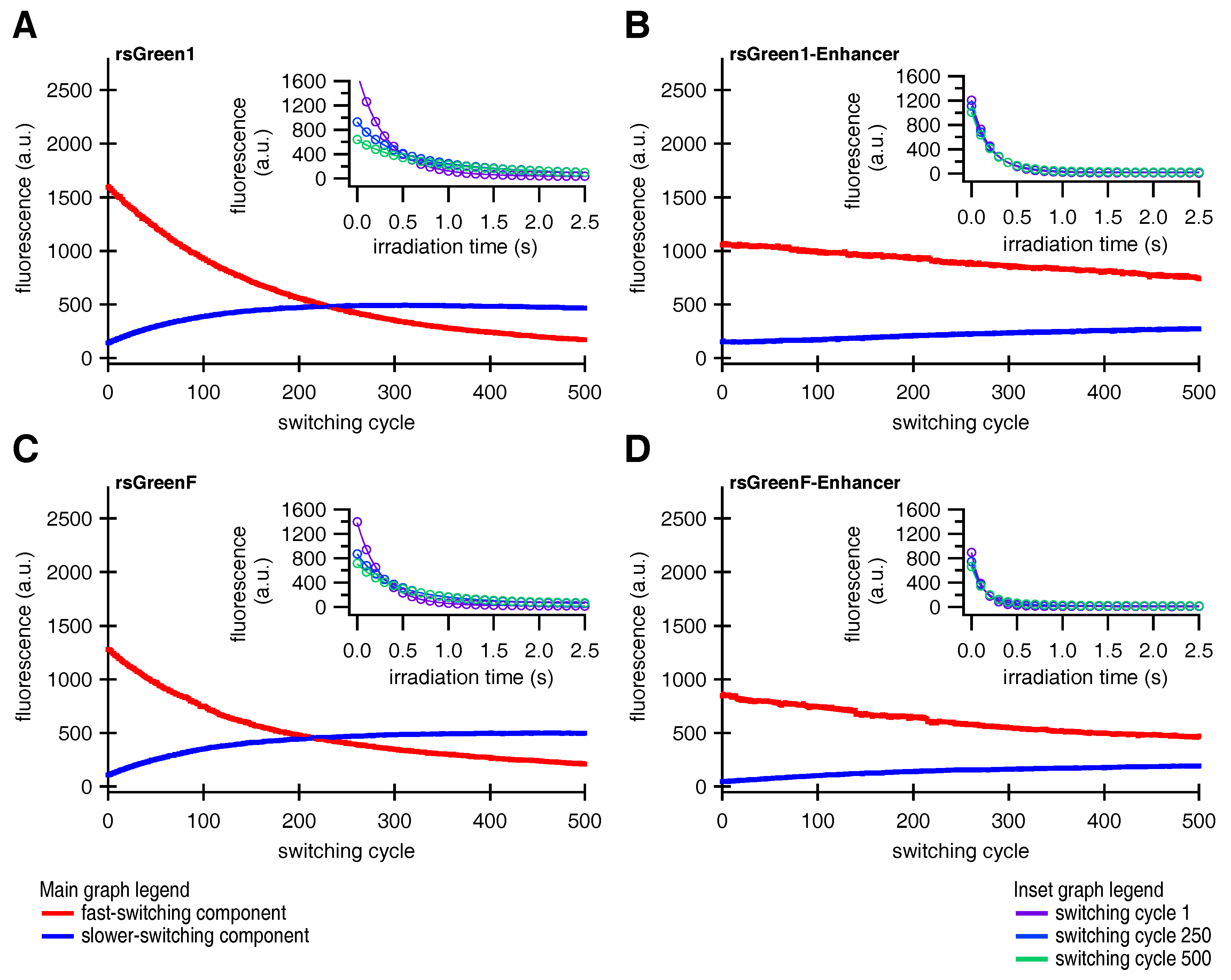
Fusions of Enhancer to rsGreen1 and rsGreenF also showed a remarkable increase in their resistance to photoswitching fatigue, as seen by the increase in the amount of achievable switching cycles. This effect was considerably stronger than expected simply based on the increase in photoswitching rate, indicating an intrinsic increase in stability. The reduced photoswitching fatigue could directly benefit any imaging technique that makes use of these labels, since it means more images can be obtained before photodestruction makes the sample unusable. For most imaging techniques this results in the ability to measure the same sample for a longer time. This is illustrated by GMars-Q and rsEGFP2, two reversibly switchable fluorescent proteins that allow prolonged RESOLFT-type imaging owing to their increased fatigue resistance [
32,
33]. In SOFI, additionally, more recorded images result in an increased signal to noise ratio [
41], making it possible to achieve higher resolutions [
42,
43].
Finally, close examination of the photoswitching behavior suggested that the fluorescent rsGreens and nanobody fusions exist as two spectroscopically different and interconverting molecular species, each with different photochromic behavior. Furthermore, the Enhancer nanobody appears to influence both species, changing their spectroscopic properties while preferentially stabilizing the fast-switching state. This might in turn explain the diminished conversion to the slower-switching species upon repeated photoswitching. The generation of a slower-switching species during repetitive photoswitching also provided an explanation for the increasing baseline fluorescence, which is sometimes observed when using RSFPs, without the need for an additional non-photoswitchable species [
33].
Since the forms discovered in this work readily interconvert during the course of a (photoswitching) experiment, caution is warranted when interpreting the findings of such experiments. For instance, it is possible that species B is responsible for most of the signal in RESOLFT and pcSOFI measurements while the in vitro characterization is preformed on a mixture consisting mainly of species A. This information should be taken into account when correlating the performance of the measurement to the molecular parameters, which raises the question whether the presence of several species with distinct switching behavior is a general feature of (green) RSFPs. Likewise, if the technique needs to be tuned to a particular kinetic behavior, this tuning can become progressively worse over the duration of the experiment. Moreover, some applications might favor the use of one state over the other, depending if fast and complete switching (e.g., RESOLFT) or slower switching with a significant on-fraction (e.g., pcSOFI) is preferred. Luckily, since binding of the Enhancer exerts an effect on the occupancy of both species it seems reasonable to assume that one type of photoswitching behavior can be selected for by changing the environment of the chromophore. This could be achieved by either the development of novel RSFPs, through interaction of an existing RSFP with a (suitably modified) version of the Enhancer, or by a combination of both approaches. Shifting the occupancy of chromophore states with different photochromism through binding by a modified Enhancer further intensifies the idea of it serving as a contrast mechanism, as was mentioned above. In any case, a detailed characterization of both species at the molecular level should provide useful information to guide future developments [
15,
16,
17,
18,
19,
20,
21].
In summary, five distinct effects of binding by the Enhancer where detected: a drop in the
, a pH independent increase of extinction coefficient, an increase in off-switching speed (
Appendix A), a slower thermal recovery from the off-state and an increased stability of the fast-switching species (
Appendix C). Therefore we hypothesize that additional structural changes are induced by Enhancer binding. Crystal structure determination of on- and off-states of rsGreen0.7 [
22], rsEGFP2 and rsFolders [
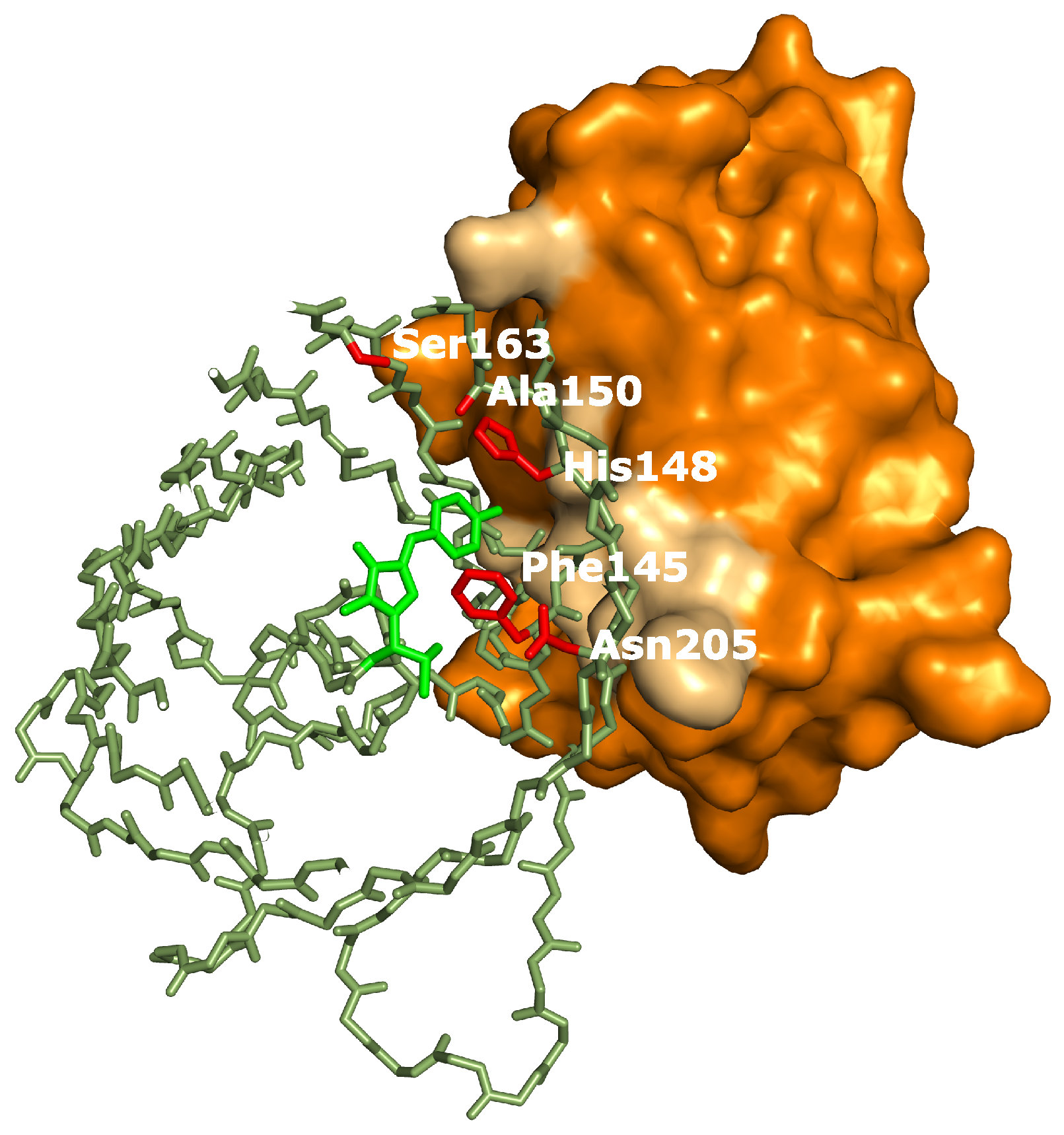
19], all closely related to the RSFPs used in this work, identified several key amino acids affecting the photoswitching behavior. Interestingly, the positions occupied by these amino acids correspond very well with the binding site of the Enhancer nanobody [
23], as is apparent in
Figure 6. The identity of residue 145, for example, was found to influence the photochromism by affecting the chromophore flexibility and is directly involved in Enhancer binding [
22]. Additionally, Enhancer binding induces slight structural changes within loop region 142 to 148, shifting His148 into close proximity with Arg168. This might facilitate proton abstraction from the chromophore, therefore favoring the anionic state of the chromophore which in turn affects the photoswitching behavior [
44]. Lastly, the Enhancer nanobody is involved in non-polar interactions with a hydrophobic patch on GFP that neighbors residue 205, which was found to be involved in a backbone shift facilitating
cis-trans isomerization. The bound Enhancer possibly affects this structural change, potentially promoting this backbone shift and thus the photochromic behavior.
The close relationship between the amino acids important for photoswitching and nanobody binding might be a good starting point for understanding the change in photoswitching behavior seen upon enhancer binding. However, to acquire deeper insight into the mechanism by which the Enhancer influences photochromism, the effect of the nanobody on other GFP-based RSFPs should be studied in detail. Additionally, acquiring crystal structures for Enhancer-bound rsGreen1 and rsGreenF will provide a clearer view on the structural rearrangements induced by nanobody binding.
Interestingly, several other proteins exist that selectively bind to fluorescent proteins which could potentially also serve to influence the photochromic behavior of these labels [
24,
28,
45]. For instance, the same work that reported the Enhancer also disclosed a second GFP-binding nanobody that influences spectroscopic properties [
23]. This “Minimizer” nanobody has an opposite effect, stabilizing the neutral chromophore state instead of the anionic form, thereby reducing the brightness upon 488 nm excitation. As both the neutral and anionic chromophore states play an important part in the photoswitching mechanism, it is reasonable to expect that binding of the Minimizer nanobody will also induce a change in the photoswitching kinetics of rsGreens.
Taken together, our work demonstrates a novel way to affect the photochromism properties of RSFPs through binding of selected protein motifs, which is a promising prospect regarding a wide variety of advanced fluorescence imaging applications and future protein design.
4. Materials and Methods
4.1. Cloning, Expression and Purification
The amino acid sequence for Enhancer nanobody was adapted from the Protein Data Bank (Accession number 3K1K, chain A. Deposition authors: Kirchhofer et al. [
23]), translated into a Homo Sapiens codon optimized DNA sequence using the Integrated DNA Technologies (IDT, Haasrode, Belgium) codon optimization tool (Web-based tool to be found at
https://eu.idtdna.com/CodonOpt) and was ordered in the form of a gBlock
® Gene Fragment from IDT (for full amino acid sequence, see
Supplementary Materials). The genes encoding rsGreen1 and rsGreenF in the expression vector pRSETb were spliced directly to the Enhancer nanobody by polymerase chain reaction (PCR) driven splicing by overlap extension (SOE) [
46]. Final PCR-amplified Enhancer-fusion cDNA contained a
BamHI restriction site at the 5
end and an
EcoRI restriction site at the 3
end for insertion into pRSETb in frame with a polyhistidine sequence and into a vector for mammalian expression (pcDNA3). All the used PCR primers are listed in
Table 2.
All plasmid transformations were performed by a sonoporation protocol. Chemocompetent E. coli JM109(DE3) and DH5 cells (Promega, Leiden, The Netherlands) were incubated with 5 L plasmid DNA for 5–15 min on ice, followed by sonoporation in a Branson 2210 ultrasonic cleaner for 15 s and incubation with SOC medium for 20 min at 37 C and were finally plated on bacterial growth plates supplemented with a suitable selection antibiotic.
Expression was conducted by E. coli JM109(DE3) strains through leak expression of the T7 promoter of the inserted genes within the pRSETb expression vector. A fresh colony was inoculated in 200–300 mL LB (Luria Bertani) medium supplemented with 100 g/mL ampicillin and incubated for three successive days (60–72 h) at 23 C in a thoroughly shaking incubator. Bacterial suspensions were centrifuged for 15 min at 4 C at 5000 rpm (Sorvall Evolution RC with SLA-1500 rotor). E. coli cells were lysed using a french pressure cell press, followed by centrifugation of the cell-debris for 10 min at 4 C at 8000 rpm with a Biofuge primoR (DJB Labcare ltd, Buckinghamshire, UK). The supernatant containing the crude protein extract was incubated on ice with 2 to 3 mL Ni-nitrilotriacetic acid (Ni-NTA) agarose resin (Qiagen, Antwerp, Belgium) and was allowed to bind the protein for 30 min while regularly shaking. This suspension was transferred to a 2 mL polystyrene column (Thermo Fisher Scientific, Merelbeke, Belgium) and washed with excess TN buffer (100 mM Tris-HCl, 300 mM NaCl, pH 7.4). Proteins were eluted with TN buffer supplemented with 100 mM imidazole. Buffer exchange to fresh TN buffer was performed with a PD-10 desalting column (GE Healthcare, London, UK) and protein solutions were stably stored at 4 C for the duration of the experiments.
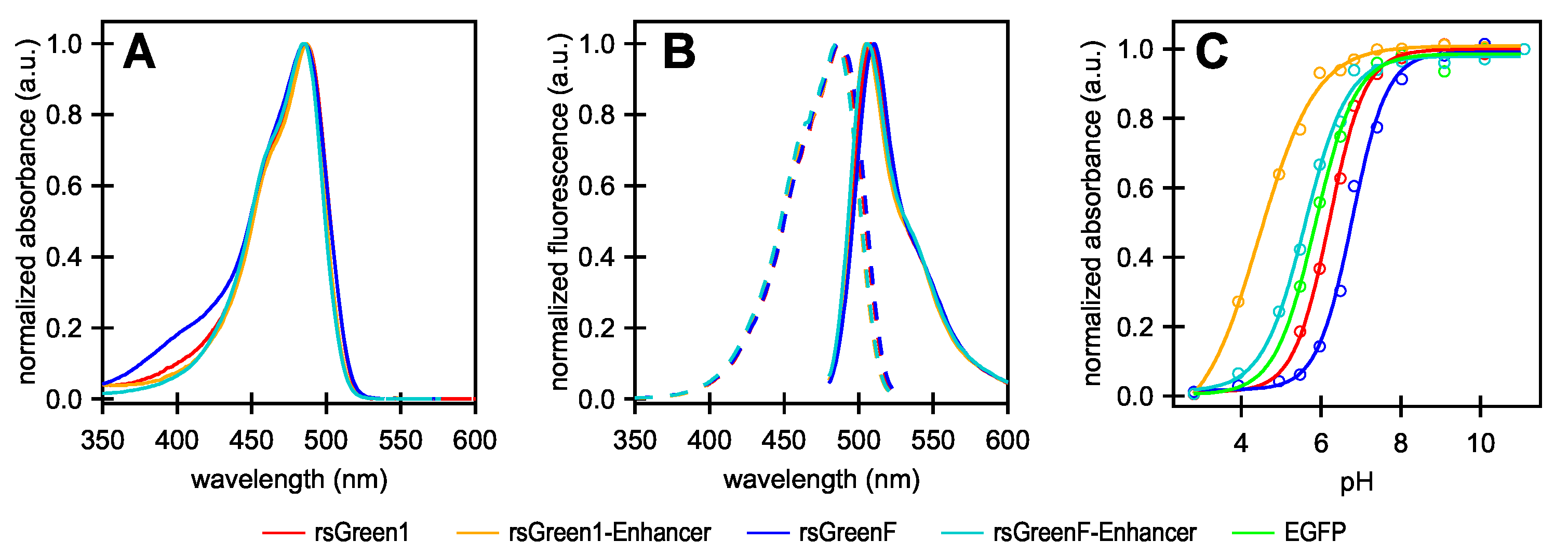
4.2. In-Vitro Characterization: Spectra, Molecular Brightness and pH Dependence
Absorption spectra of purified FPs were recorded using a Shimadzu UV-1650PC spectrophotometer (Shimadzu GmbH, Duisburg, Germany), while excitation and emission spectra were acquired with an Edinburgh FLS980 spectrometer (Edinburgh Instruments Ltd., Livingston, UK) with a slit widening set at 2 nm. Spectra were measured in TN buffer (pH 7.4).
Extinction coefficients (
) were determined according to Ward’s method [
47], using the literature value of EGFP as a reference [
30]. Extinction coefficients (
) were extrapolated to the extinction coefficient of the deprotonated state (
) by using the Henderson-Hasselbalch equation. Quantum yield (QY) was determined relative to EGFP (=0.60 [
30]). Molecular brightness was defined as the product of
and
, scaled to 100 for rsGreen1.
pH sensitivity measurements were conducted in PBS buffer supplemented with 50 mM citric acid (monoḣydrate), 50 mM KHPO and 50 mM glycine, adjusted to the desired pH. The was determined as the inflection point of the sigmoidal curve that was fitted to the absorption maxima of the anionic form at pH values ranging from 3 to 11.
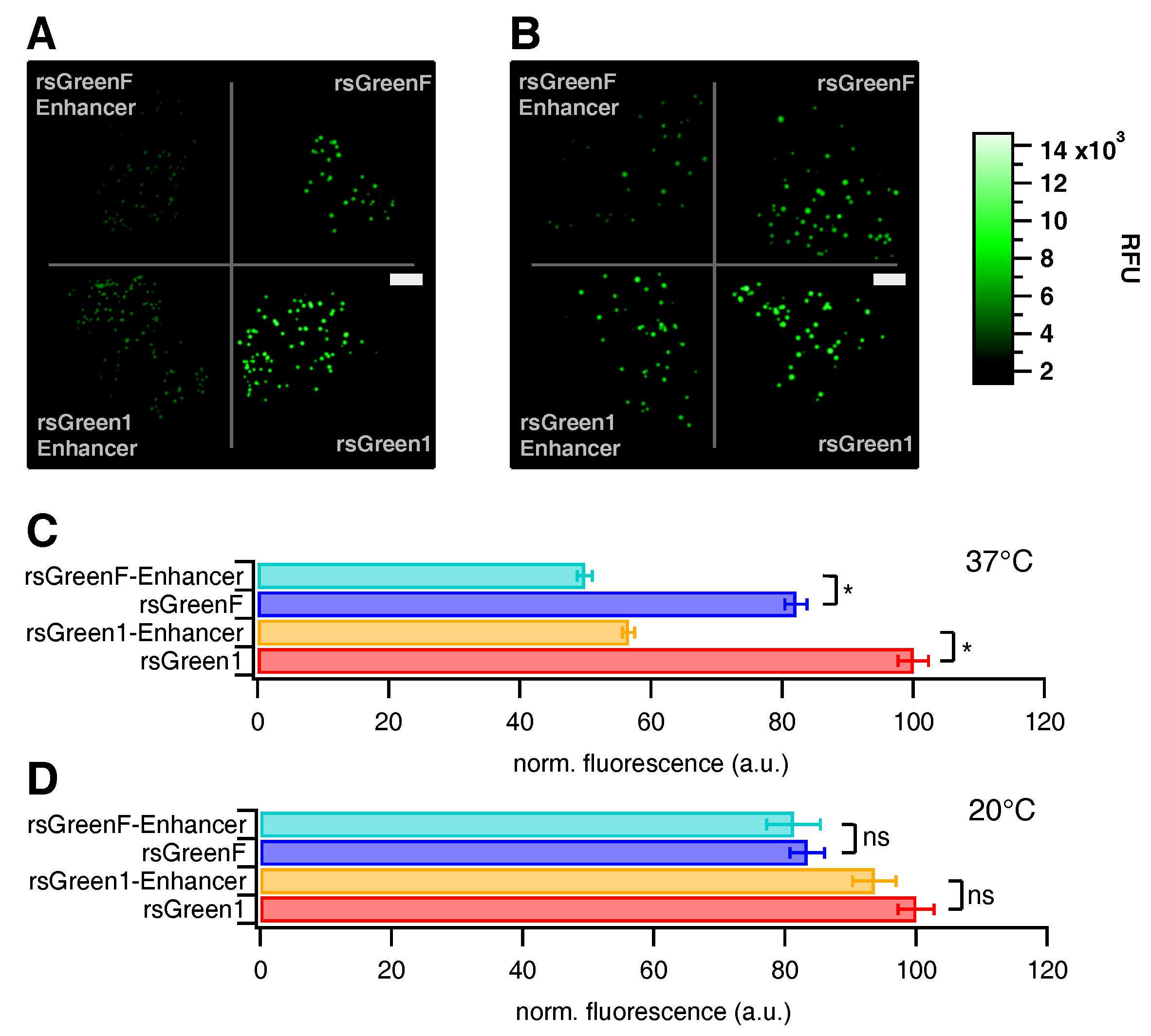
4.3. In Situ Characterization: Brightness and Photoswitching
Transformed E. coli JM109(DE3) cells expressing the different FPs were plated on equally sized surfaces with 15 L bacterial suspension and incubated for 72 h at 20 C or 24 h at 37 C. Growth plates were illuminated with a MAX-302 xenon lamp (Asahi Spectra, Tokyo, Japan) and fluorescence was detected with a Cascade 512B CCD camera (Photometrics, Tuscon, AZ, USA). Excitation wavelengths for off-switching and on-switching were selected by a 480/40 nm and 400/30 nm band pass filter, respectively. Emission wavelengths were selected using a 530/40 nm bandpass filter.
Photoswitching of E. coli colonies was conducted by submitting growth plates to one full switching cycle comprising 20 periods on-switching, 40 periods off-switching and again 20 periods on-switching, each period lasting 30 s and the emission recorded between periods with a camera exposure time of 0.5 s.
Acquired images were analyzed using Igor Pro 7 (Wavemetrics, Portland, OR, USA). A threshold was set to select all fluorescent colonies and an average intensity trace over the duration of the experiment was calculated and normalized to the point of complete on-switching. The maximum brightness of each separate colony was determined and used to calculate an average brightness value. The maximum fluorescence of every colony was chosen over the average fluorescence to minimize thresholding artifacts.
HeLa cells were cultured in DMEM supplemented with 10% (v/v) FBS, 1% (v/v) glutaMAX and 0.1% (v/v) gentamicin (all Gibco, Merelbeke, Belgium). Prior to transfection, 250,000–500,000 cells were seeded in 35 mm glass bottom dishes (MatTek, Ashland, MA, USA). The transfection was performed using the FuGene 6 (Promega, Leiden, The Netherlands) transfection protocol. Briefly, 3 L FuGene 6 was thoroughly mixed with 1 g DNA (pcDNA3) to a total volume of 100 L and incubated at room temperature for 20 min. The DNA mixture was then added in dropwise fashion to the medium on top of the cells. After incubation for 20–24 h, cells were washed twice with 2 mL PBS (pH 7.4) and finally supplied with 2 mL HBSS (Gibco, Merelbeke, Belgium) prior to imaging.
High-power photoswitching in HeLa cells was measured on a setup comprising an Olympus IX 71 inverted microscope (Olympus, Berchem, Belgium) coupled to a Spectra X Light Engine (Lumencor, Beaverton, OR, USA), equipped with a objective (UplanSApo, Olympus, Berchem, Belgium), a dichroic turret wheel mounting a ZT488RDC (Chroma, Olching, Germany) dichroic filter with a 535/30 bandpass emission filter. Fluorescence images were recorded with an iXon Ultra 897 EMCCD camera (Andor, Belfast, UK) operating at −70 C and were processed and analyzed using Igor Pro 7. Images were acquired over 0.05 s during illumination with 1% cyan excitation light. Proteins were submitted to multiple switching cycles comprising 25 periods on-switching with violet light, followed by 25 periods of off-switching with cyan light, each period lasting 100 ms.
Optimal power settings that could be applied for all FPs in a consequent fashion were assessed by recording multiple switching cycles while varying the cyan light source output from 5% to 30%. Twenty percent was determined as the optimal power setting. The decrease in on-state fluorescence was assessed over the course of 500 switching cycles using 7% violet and 20% cyan light. The average on-state fluorescence of the first switching cycle was used as a measure for brightness in HeLa cells as reported in
Table 1.
For the measurement of light-independent recovery from the off-state, proteins were submitted to a slightly different illumination scheme consisting of two cycles comprising 5 periods of on- and off switching during 500 ms, followed by acquisition every 30 s during the course of 10 min.
Global fitting was done according to
Appendix B using a build-in function of Igor Pro 7.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





