Proangiogenic Effect of Metformin in Endothelial Cells Is via Upregulation of VEGFR1/2 and Their Signaling under Hyperglycemia-Hypoxia


 , ,
, ,
Abstract
:
1. Introduction
2. Results
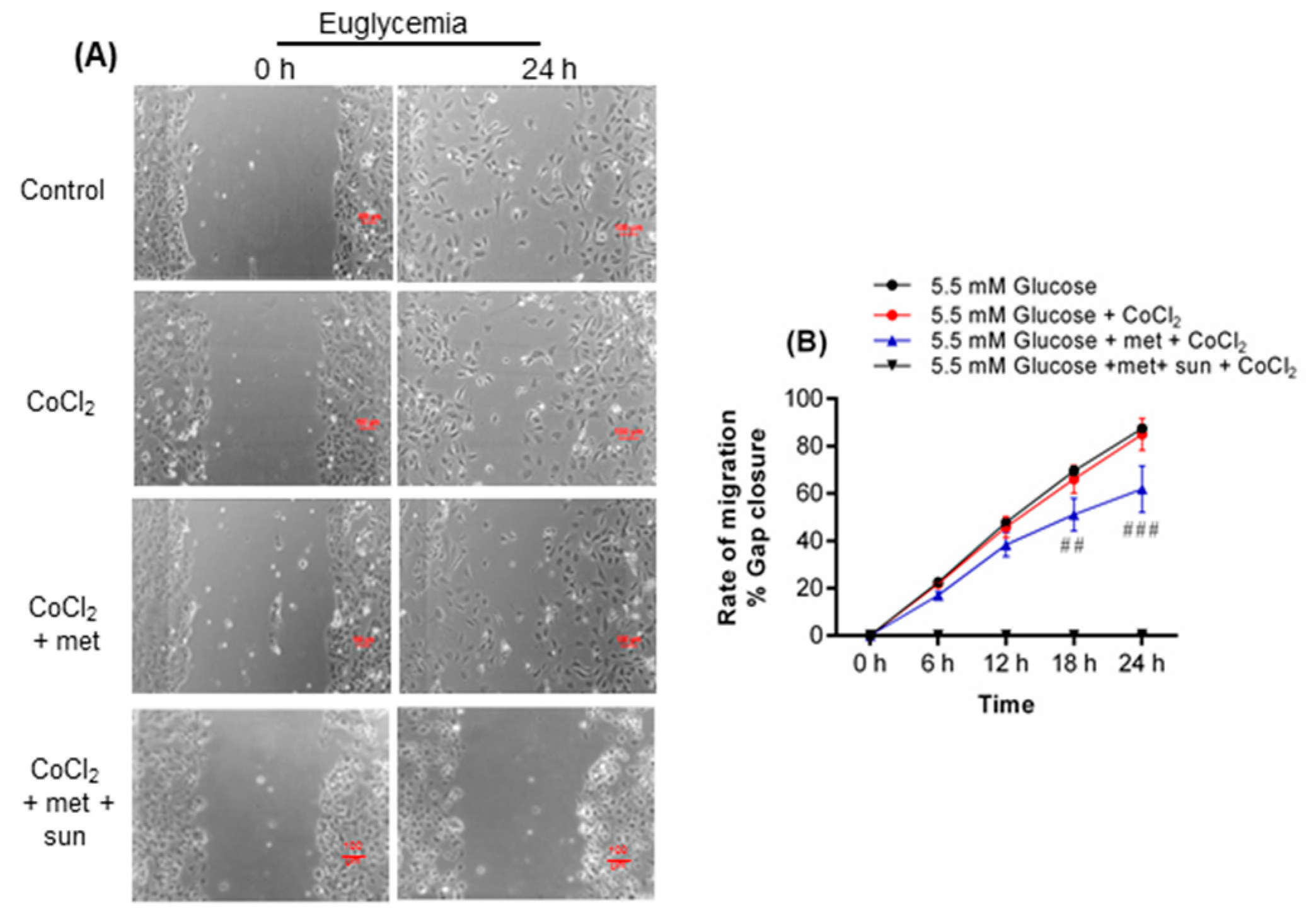
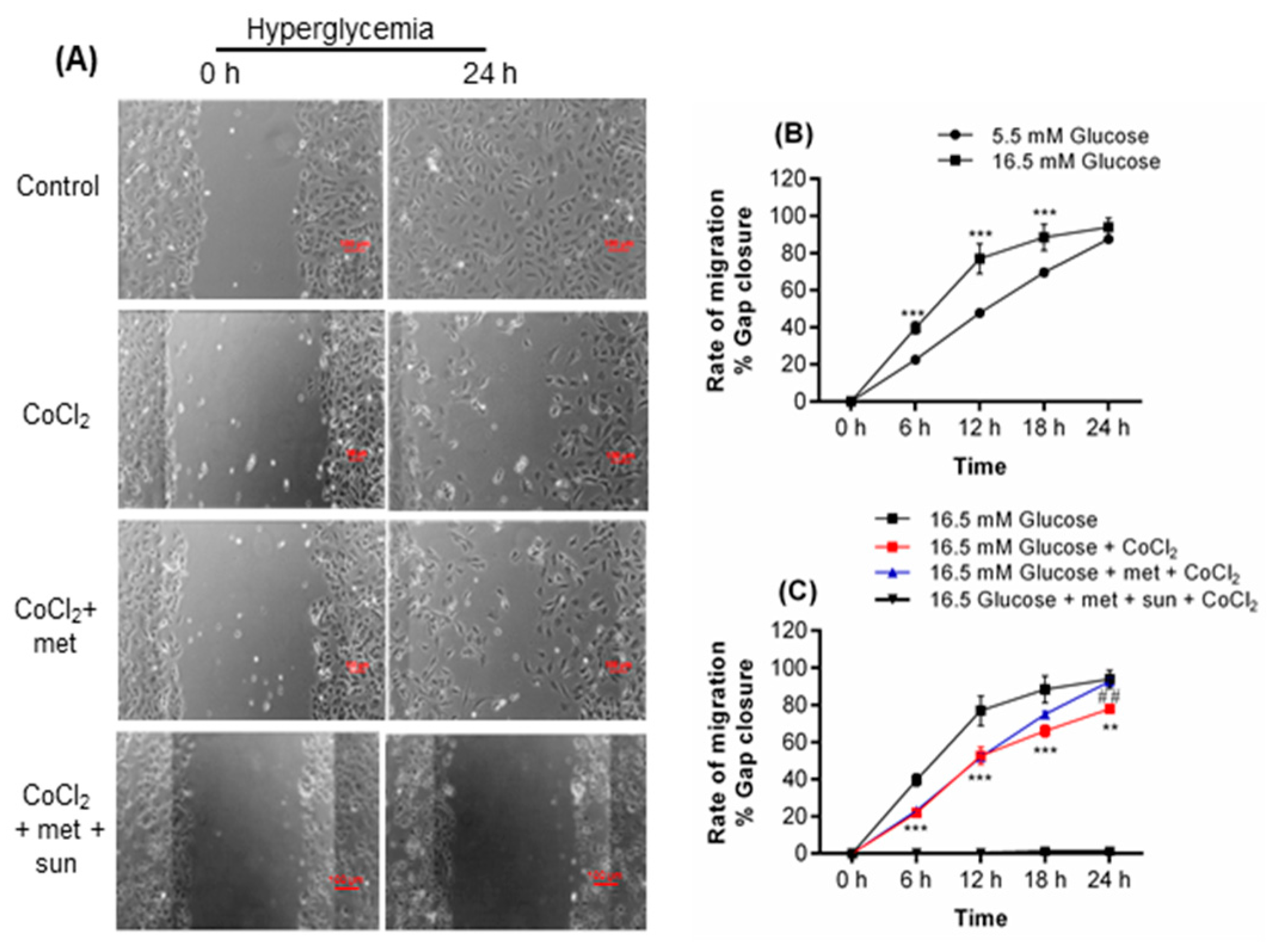
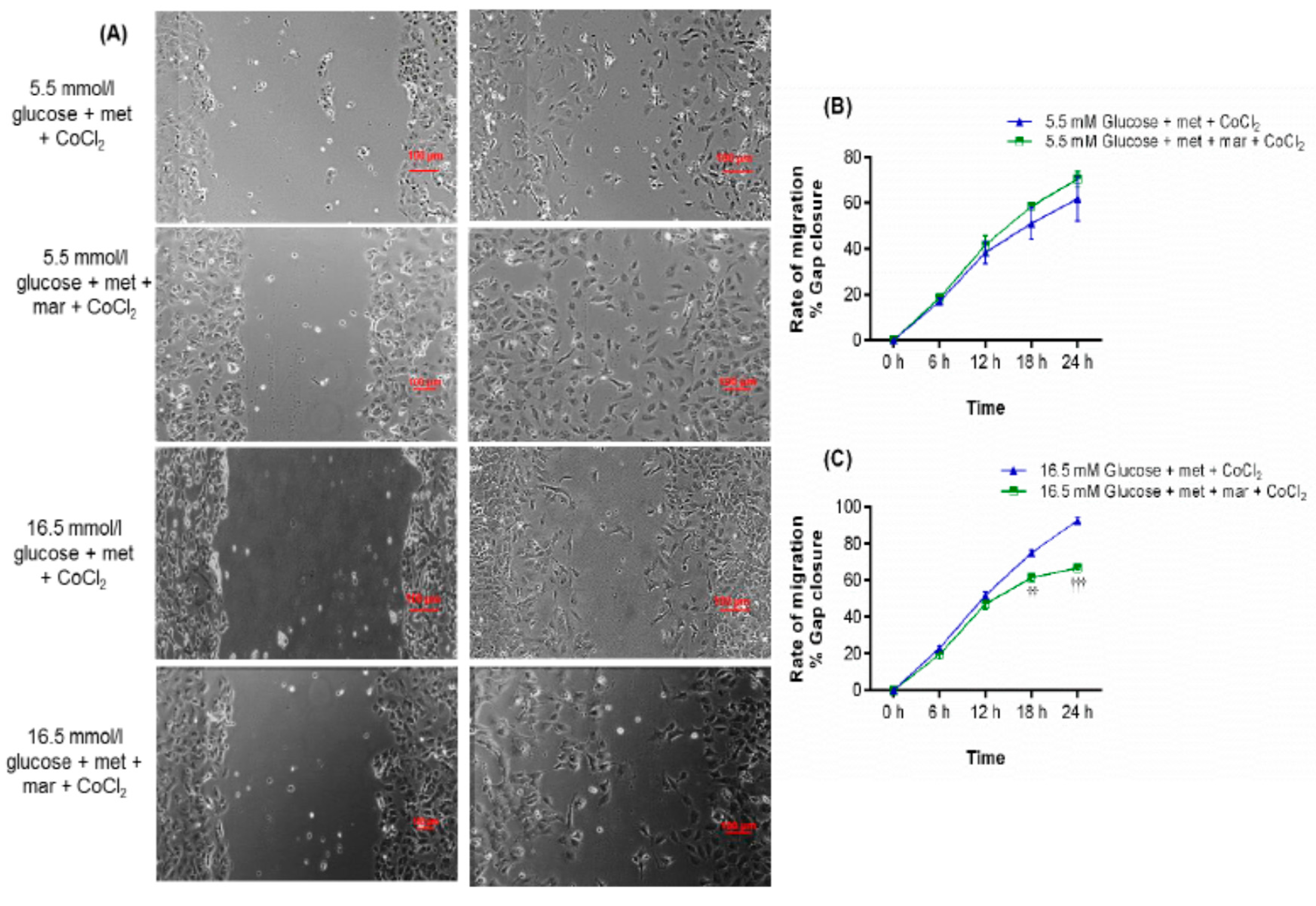
2.1. Metformin Promotes Endothelial Cell Migration under Hyperglycemia-CoCl2
2.2. Inhibition of Metformin Action Mediated by VEGFA in Cell Migration Assay
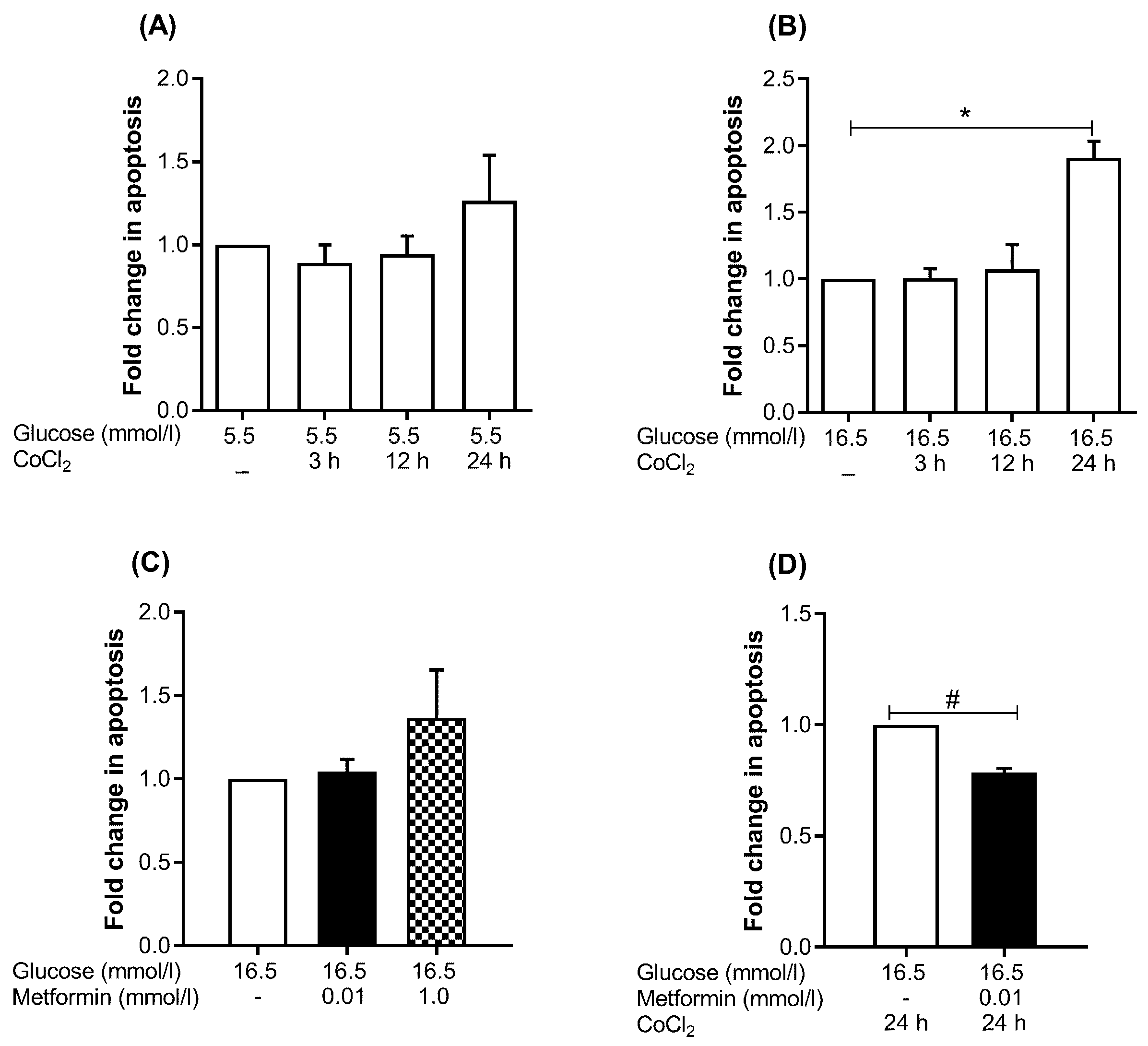
2.3. Metformin Reduces Apoptosis under Hyperglycemia-CoCl2
2.4. Microarray Gene Expression Profiling in HUVEC and Western Blot
2.5. Metformin Has No Effect on HIF-1α Gene/Protein Expression under Hyperglycemia-CoCl2
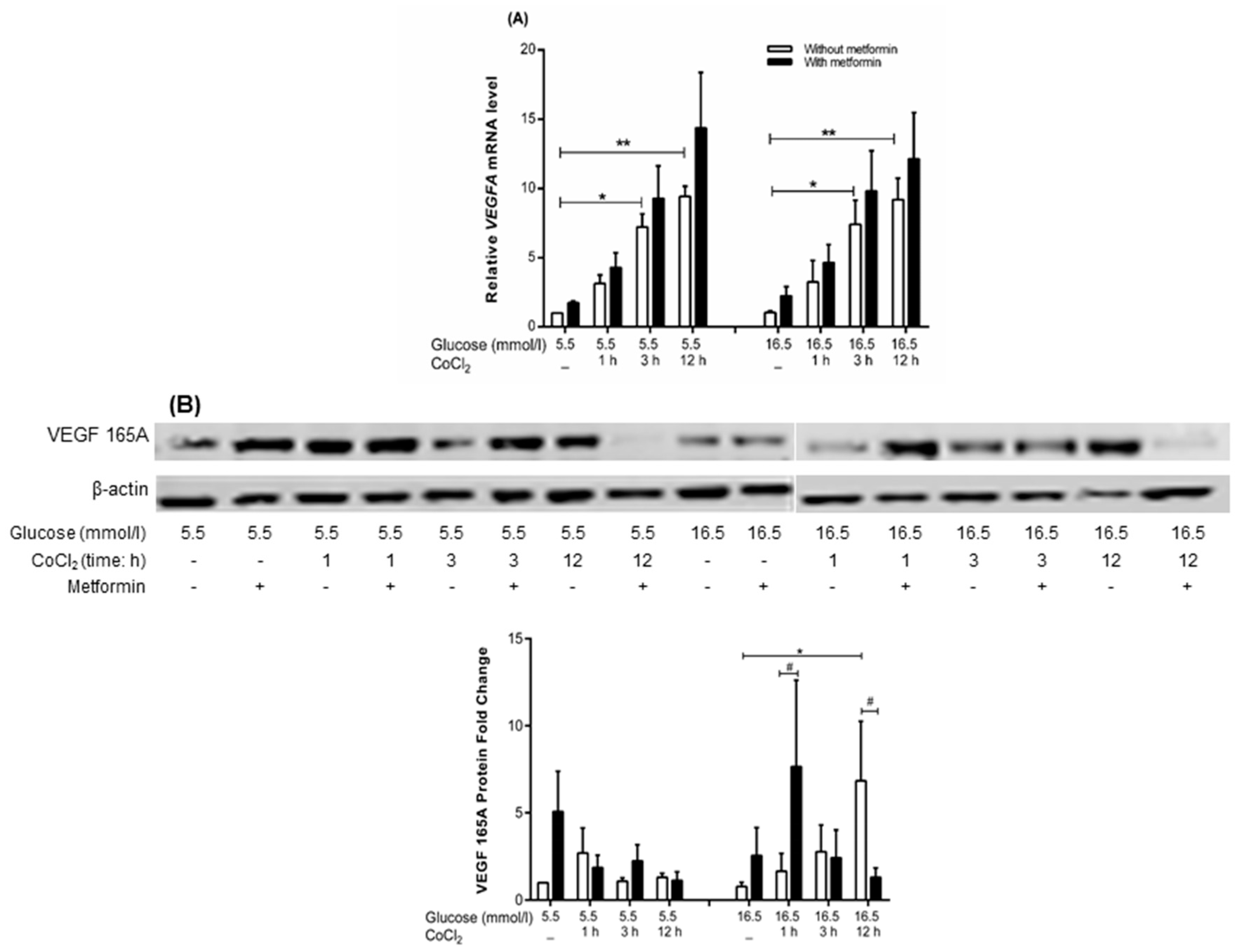
2.6. Metformin Has No Significant Effect on VEGFA Gene Expression but Increased Protein Expression at 1 h of CoCl2 under Hyperglycemia-CoCl2
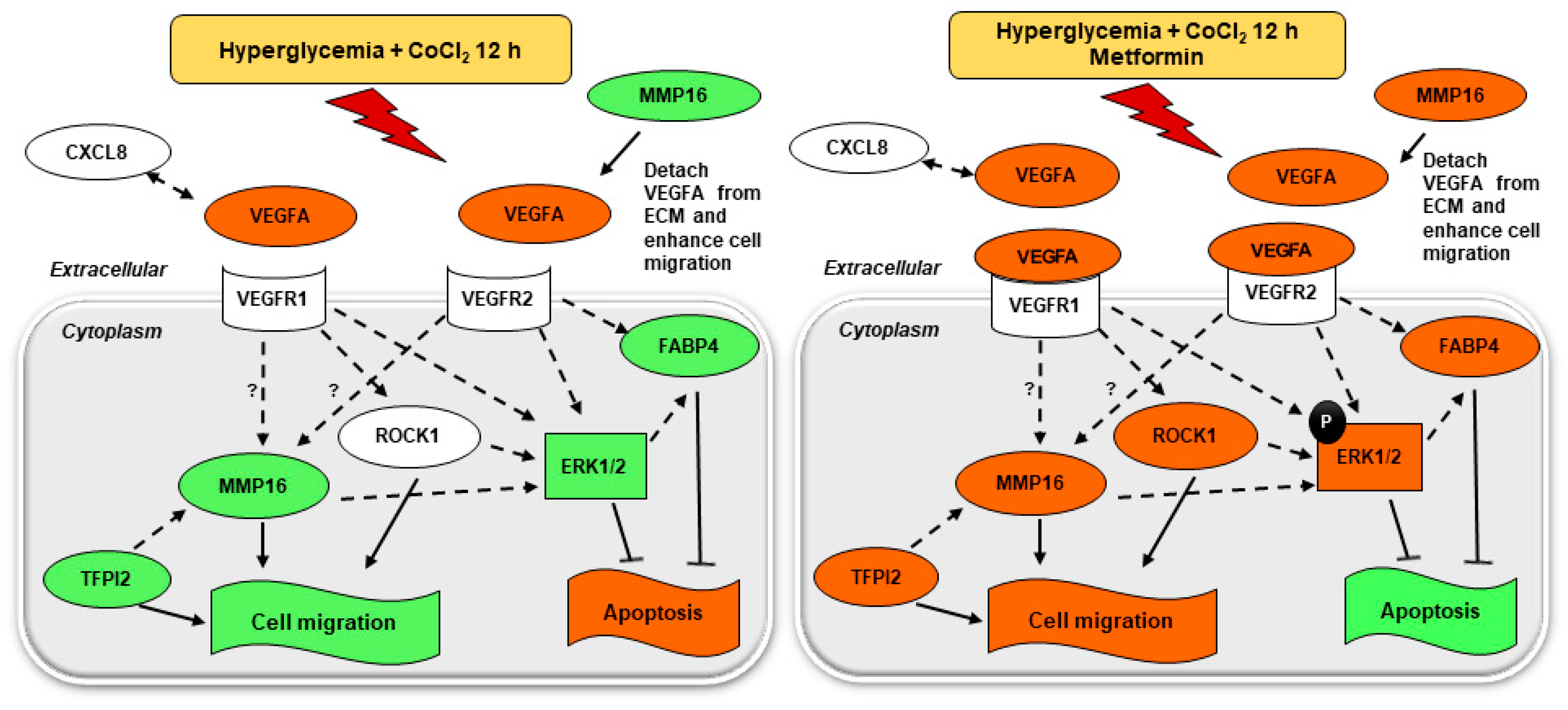
2.7. Metformin Activates VEGF Downstream Signaling
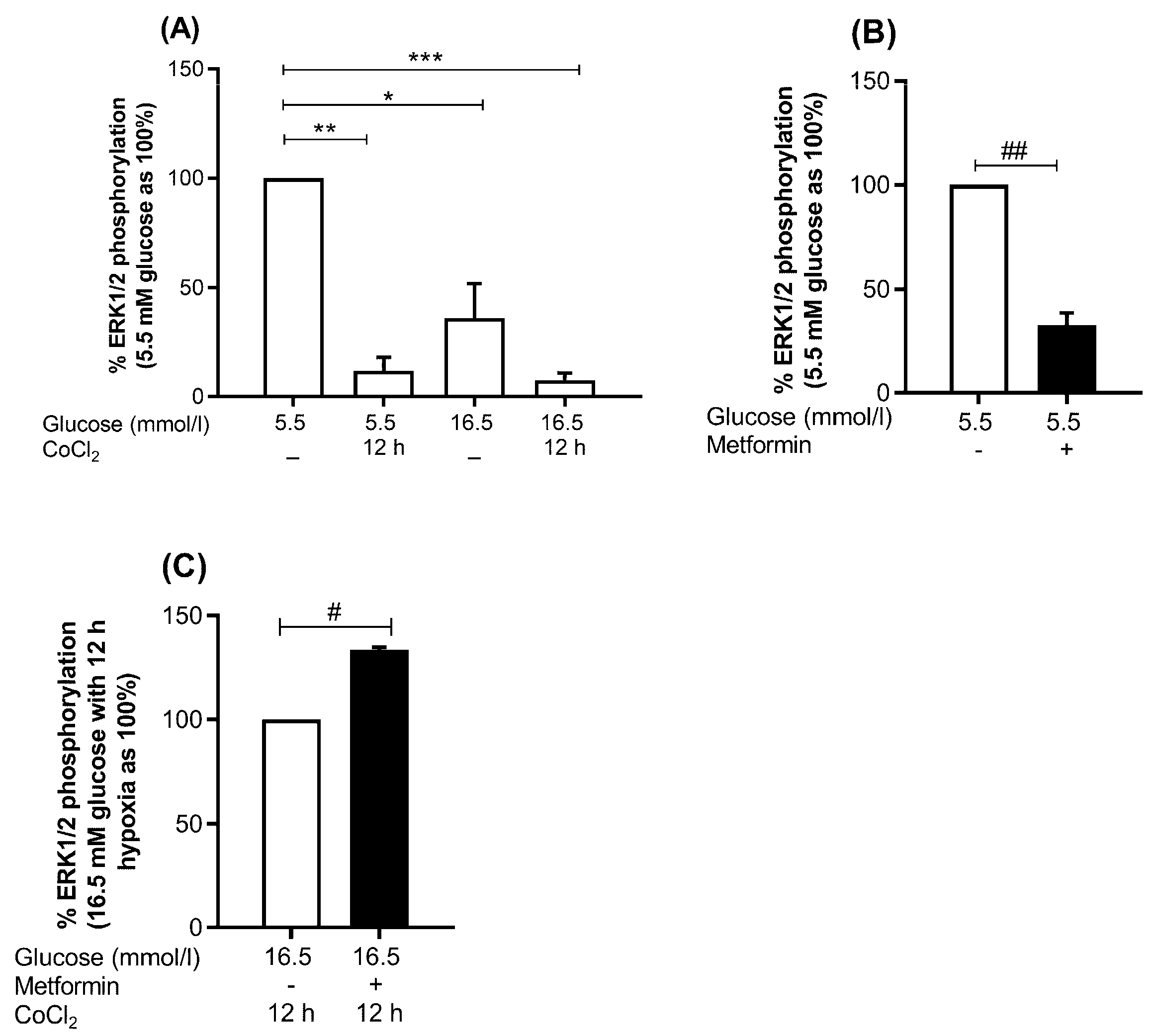
2.7.1. MAPK Pathway; Metformin Increases Phosphor-ERK1/2 to Total ERK Ratio under Hyperglycemia-CoCl2
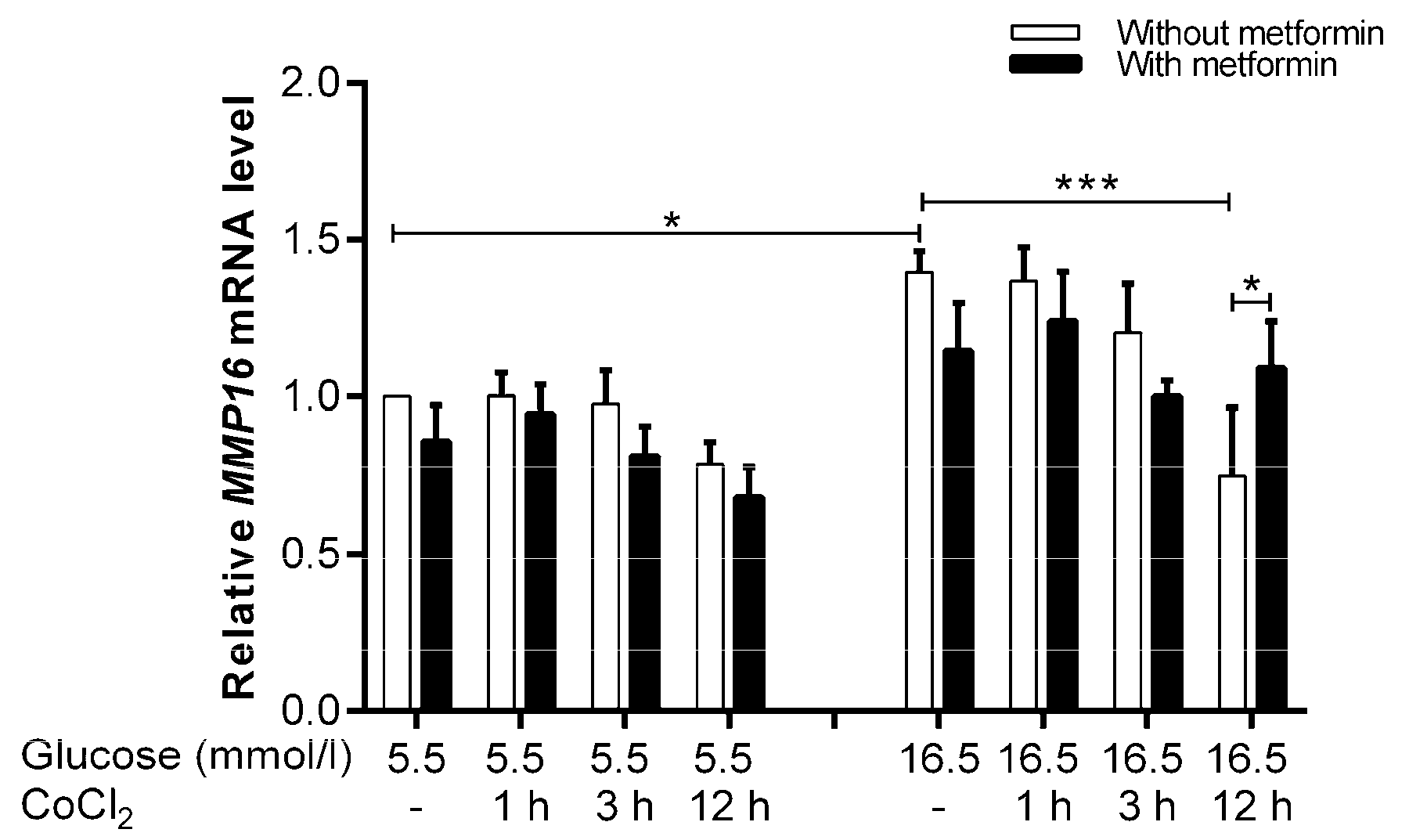
2.7.2. Metformin Increases the Expression of CXCL8, LY96, ROCK1, MMP16, TFPI2, and FABP4 under Hyperglycemia-CoCl2
2.8. Inhibition of Metformin Action Mediated by MMP16 in Cell Migration Assay
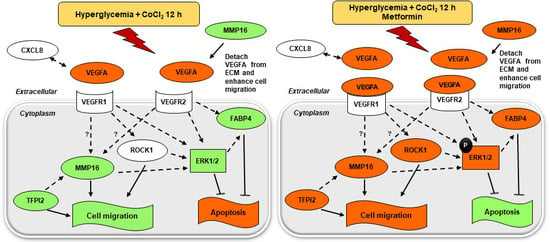
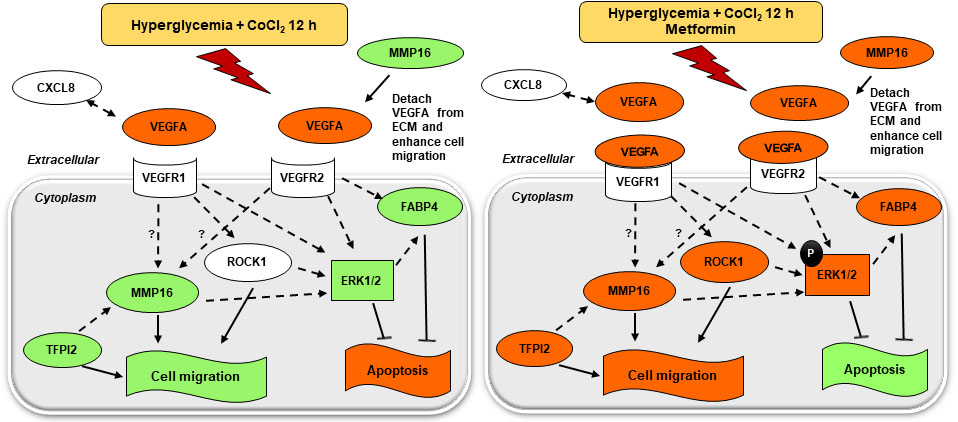
3. Discussion
4. Materials and Methods
4.1. HUVEC Cultures
4.2. In Vitro Scratch Assay (Cell Migration Assay)
4.3. Apoptosis Assay
4.4. Total RNA Extraction
4.5. Microarray Experiments and Gene Expression Analysis
4.6. Quantitative RT-PCR
4.7. Protein Studies
4.8. MAPK Activation Dual Detection Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CVD | Cardiovascular disease |
| CXCL8 | Chemokine (C-X-C Motif) Ligand 8 |
| DM | Diabetes mellitus |
| EC | Endothelial cell |
| ERK1/2 | Extracellular signal-regulated protein kinases 1 and 2 |
| FABP4 | Fatty acid binding protein 4 |
| LY96 | Lymphocyte antigen 96 |
| MAPK | Mitogen-activated protein kinase |
| MI | Myocardial infarction |
| MMP16 | Matrix metalloproteinase 16 |
| PI3K | Phosphatidylinositol 3′-kinase |
| ROCK1 | Rho-associated, coiled-coil containing protein kinase 1 |
| TFPI2 | Tissue factor pathway inhibitor 2 |
| VEGFA | Vascular endothelial growth factor A |
References
- Hanson, R.L.; Imperatore, G.; Bennett, P.H.; Knowler, W.C. Components of the “metabolic syndrome” and incidence of type 2 diabetes. Diabetes 2002, 51, 3120–3127. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Damm, P.; Prentki, M. Type 2 diabetes across generations: From pathophysiology to prevention and management. Lancet 2011, 378, 169–181. [Google Scholar] [CrossRef]
- Ekezue, B.F.; Laditka, S.B.; Laditka, J.N.; Studnicki, J.; Blanchette, C.M. Diabetes complications and adverse health outcomes after coronary revascularization. Diabetes Res. Clin. Pract. 2014, 103, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, R.; Galasso, G.; Iversen, A.Z.; Eitel, I.; Dominguez-Rodriguez, A.; Gu, Y.L.; de Smet, B.J.; Mahmoud, K.D.; Abreu-Gonzalez, P.; Trimarco, B.; et al. Effects of baseline coronary occlusion and diabetes mellitus in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Am. J. Cardiol. 2014, 114, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zou, M.H. Molecular insights and therapeutic targets for diabetic endothelial dysfunction. Circulation 2009, 120, 1266–1286. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar]
- Roussel, R.; Travert, F.; Pasquet, B.; Wilson, P.W.; Smith, S.C., Jr.; Goto, S.; Ravaud, P.; Marre, M.; Porath, A.; Bhatt, D.L.; et al. Reduction of Atherothrombosis for Continued Health Registry, I. Metformin use and mortality among patients with diabetes and atherothrombosis. Arch. Intern. Med. 2010, 170, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, J.P.; Zhao, X.Z.; Sun, X.J.; Xu, Z.W.; Song, S.J. Protective effect of metformin on myocardial injury in metabolic syndrome patients following percutaneous coronary intervention. Cardiology 2014, 127, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Lexis, C.P.; Wieringa, W.G.; Hiemstra, B.; van Deursen, V.M.; Lipsic, E.; van der Harst, P.; van Veldhuisen, D.J.; van der Horst, I.C. Chronic metformin treatment is associated with reduced myocardial infarct size in diabetic patients with ST-segment elevation myocardial infarction. Cardiovasc. Drugs Ther. 2014, 28, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Legtenberg, R.J.; Houston, R.J.; Oeseburg, B.; Smits, P. Metformin improves cardiac functional recovery after ischemia in rats. Horm. Metab. Res. 2002, 34, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Bhamra, G.S.; Hausenloy, D.J.; Davidson, S.M.; Carr, R.D.; Paiva, M.; Wynne, A.M.; Mocanu, M.M.; Yellon, D.M. Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res. Cardiol. 2008, 103, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Bakhashab, S.; Ahmed, F.W.; Schulten, H.J.; Bashir, A.; Karim, S.; Al-Malki, A.L.; Gari, M.A.; Abuzenadah, A.M.; Chaudhary, A.G.; Alqahtani, M.H.; et al. Metformin improves the angiogenic potential of human CD34(+) cells co-incident with downregulating CXCL10 and TIMP1 gene expression and increasing VEGFA under hyperglycemia and hypoxia within a therapeutic window for myocardial infarction. Cardiovasc. Diabetol. 2016, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201–207. [Google Scholar] [CrossRef]
- He, Y.; Wang, N.; Shen, Y.; Zheng, Z.; Xu, X. Inhibition of high glucose-induced apoptosis by uncoupling protein 2 in human umbilical vein endothelial cells. Int. J. Mol. Med. 2014, 33, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Tsuneki, H.; Sekizaki, N.; Suzuki, T.; Kobayashi, S.; Wada, T.; Okamoto, T.; Kimura, I.; Sasaoka, T. Coenzyme Q10 prevents high glucose-induced oxidative stress in human umbilical vein endothelial cells. Eur. J. Pharmacol. 2007, 566, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Altannavch, T.S.; Roubalova, K.; Kucera, P.; Andel, M. Effect of high glucose concentrations on expression of ELAM-1, VCAM-1 and ICAM-1 in HUVEC with and without cytokine activation. Physiol. Res. Acad. Sci. Bohemoslov. 2004, 53, 77–82. [Google Scholar]
- Bakhashab, S.; Lary, S.; Ahmed, F.; Schulten, H.J.; Bashir, A.; Ahmed, F.W.; Al-Malki, A.L.; Jamal, H.S.; Gari, M.A.; Weaver, J.U. Reference genes for expression studies in hypoxia and hyperglycemia models in human umbilical vein endothelial cells. G3 Genes Genomes Genet. 2014, 4, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Cohen, R.; Ullrich, V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothel. J. Endothel. Cell Res. 2004, 11, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Deng, Y.P.; Han, X.; Ren, G.F.; Cai, J.; Jiang, G.J. Metformin improves the angiogenic functions of endothelial progenitor cells via activating AMPK/eNOS pathway in diabetic mice. Cardiovasc. Diabetol. 2016, 15, 88. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, S.; Yamauchi, K.; Nakajima, K.; Iijima, S.; Aizawa, T.; Hashizume, K. D-Glucose and insulin stimulate migration and tubular formation of human endothelial cells in vitro. Am. J. Physiol. 1999, 277, E433–E438. [Google Scholar] [CrossRef] [PubMed]
- Esfahanian, N.; Shakiba, Y.; Nikbin, B.; Soraya, H.; Maleki-Dizaji, N.; Ghazi-Khansari, M.; Garjani, A. Effect of metformin on the proliferation, migration, and MMP-2 and -9 expression of human umbilical vein endothelial cells. Mol. Med. Rep. 2012, 5, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Bauters, C.; Pastore, C.; Kearney, M.; Rossow, S.; Bunting, S.; Ferrara, N.; Symes, J.F.; Isner, J.M. Local delivery of vascular endothelial growth factor accelerates reendothelialization and attenuates intimal hyperplasia in balloon-injured rat carotid artery. Circulation 1995, 91, 2793–2801. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Boil. 1996, 16, 4604–4613. [Google Scholar] [CrossRef]
- Namiki, A.; Brogi, E.; Kearney, M.; Kim, E.A.; Wu, T.; Couffinhal, T.; Varticovski, L.; Isner, J.M. Hypoxia induces vascular endothelial growth factor in cultured human endothelial cells. J. Biol. Chem. 1995, 270, 31189–31195. [Google Scholar] [CrossRef] [PubMed]
- Brogi, E.; Wu, T.; Namiki, A.; Isner, J.M. Indirect angiogenic cytokines upregulate VEGF and bFGF gene expression in vascular smooth muscle cells, whereas hypoxia upregulates VEGF expression only. Circulation 1994, 90, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Hotary, K.; Allen, E.; Punturieri, A.; Yana, I.; Weiss, S.J. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J. Cell Biol. 2000, 149, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Shofuda, K.I.; Hasenstab, D.; Kenagy, R.D.; Shofuda, T.; Li, Z.Y.; Lieber, A.; Clowes, A.W. Membrane-type matrix metalloproteinase-1 and -3 activity in primate smooth muscle cells. FASEB J. 2001, 15, 2010–2012. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Keller, G.A.; Ferrara, N. The vascular endothelial growth factor (VEGF) isoforms: Differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell 1993, 4, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Houck, K.A.; Ferrara, N.; Winer, J.; Cachianes, G.; Li, B.; Leung, D.W. The vascular endothelial growth factor family: Identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol. Endocrinol. 1991, 5, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Houck, K.A.; Leung, D.W.; Rowland, A.M.; Winer, J.; Ferrara, N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 1992, 267, 26031–26037. [Google Scholar] [PubMed]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [PubMed]
- Plaisier, M.; Kapiteijn, K.; Koolwijk, P.; Fijten, C.; Hanemaaijer, R.; Grimbergen, J.M.; Mulder-Stapel, A.; Quax, P.H.; Helmerhorst, F.M.; van Hinsbergh, V.W. Involvement of membrane-type matrix metalloproteinases (MT-MMPs) in capillary tube formation by human endometrial microvascular endothelial cells: Role of MT3-MMP. J. Clin. Endocrinol. Metab. 2004, 89, 5828–5836. [Google Scholar] [CrossRef] [PubMed]
- Elmasri, H.; Ghelfi, E.; Yu, C.W.; Traphagen, S.; Cernadas, M.; Cao, H.; Shi, G.P.; Plutzky, J.; Sahin, M.; Hotamisligil, G.; et al. Endothelial cell-fatty acid binding protein 4 promotes angiogenesis: Role of stem cell factor/c-kit pathway. Angiogenesis 2012, 15, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, B.E.; Worth, N.F.; World, C.J.; Campbell, J.H.; Campbell, G.R. Rho and vascular disease. Atherosclerosis 2005, 183, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.A.; Liu, Y.; Wang, B.; Li, R.; Sebti, S.M. Identification of novel ROCK inhibitors with anti-migratory and anti-invasive activities. Oncogene 2014, 33, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Galisteo, R.; Gutkind, J.S. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J. Boil. Chem. 2009, 284, 6038–6042. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Varney, M.L.; Valasek, J.; Godfrey, M.; Dave, B.J.; Singh, R.K. Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis. Angiogenesis 2005, 8, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, C.A.; Kisiel, W.; Mathewes, S.; Foster, D.C. Molecular cloning, expression, and partial characterization of a second human tissue-factor-pathway inhibitor. Proc. Natl. Acad. Sci. USA 1994, 91, 3353–3357. [Google Scholar] [CrossRef] [PubMed]
- Ivanciu, L.; Gerard, R.D.; Tang, H.; Lupu, F.; Lupu, C. Adenovirus-mediated expression of tissue factor pathway inhibitor-2 inhibits endothelial cell migration and angiogenesis. Arterioscler. Thromb. Vasc. Boil. 2007, 27, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Maiti, D.; Kisiel, W.; Duh, E.J. Tissue factor pathway inhibitor-2 is upregulated by vascular endothelial growth factor and suppresses growth factor-induced proliferation of endothelial cells. Arterioscler. Thromb. Vasc. Boil. 2006, 26, 2819–2825. [Google Scholar] [CrossRef] [PubMed]
- Kast, C.; Wang, M.; Whiteway, M. The ERK/MAPK pathway regulates the activity of the human tissue factor pathway inhibitor-2 promoter. J. Boil. Chem. 2003, 278, 6787–6794. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Kshirsagar, S.; Li, W.; Gui, L.; Ramakrishnan, S.; Gupta, P.; Law, P.Y.; Hebbel, R.P. VEGF prevents apoptosis of human microvascular endothelial cells via opposing effects on MAPK/ERK and SAPK/JNK signaling. Exp. Cell Res. 1999, 247, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhang, Y.; Chen, X.; Lam, P.Y.; Yang, H.; Xu, Q.; Yu, A.C. Activation of Erk1/2 and Akt in astrocytes under ischemia. Biochem. Biophys. Res. Commun. 2002, 294, 726–733. [Google Scholar] [CrossRef]
- Yue, T.L.; Wang, C.; Gu, J.L.; Ma, X.L.; Kumar, S.; Lee, J.C.; Feuerstein, G.Z.; Thomas, H.; Maleeff, B.; Ohlstein, E.H. Inhibition of extracellular signal-regulated kinase enhances Ischemia/Reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ. Res. 2000, 86, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Lange, J.; Kranz, A. Vascular endothelial growth factor-A-induced chemotaxis of monocytes is attenuated in patients with diabetes mellitus: A potential predictor for the individual capacity to develop collaterals. Circulation 2000, 102, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Torella, D.; Carbonara, O.; Ellison, G.M.; Torella, M.; Scardone, M.; Marra, C.; Nasti, R.; Marfella, R.; Cozzolino, D.; et al. Increased vascular endothelial growth factor expression but impaired vascular endothelial growth factor receptor signaling in the myocardium of type 2 diabetic patients with chronic coronary heart disease. J. Am. Coll. Cardiol. 2005, 46, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Zaitone, S.A.; Abo-Gresha, N.M. Rosuvastatin promotes angiogenesis and reverses isoproterenol-induced acute myocardial infarction in rats: Role of iNOS and VEGF. Eur. J. Pharmacol. 2012, 691, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Preiss, D.; Lloyd, S.M.; Ford, I.; McMurray, J.J.; Holman, R.R.; Welsh, P.; Fisher, M.; Packard, C.J.; Sattar, N. Metformin for non-diabetic patients with coronary heart disease (the CAMERA study): A randomised controlled trial. Lancet Diabetes Endocrinol. 2014, 2, 116–124. [Google Scholar] [CrossRef]
- Lexis, C.P.; van der Horst, I.C.; Lipsic, E.; Wieringa, W.G.; de Boer, R.A.; van den Heuvel, A.F.; van der Werf, H.W.; Schurer, R.A.; Pundziute, G.; Tan, E.S.; et al. Effect of metformin on left ventricular function after acute myocardial infarction in patients without diabetes: The GIPS-III randomized clinical trial. JAMA 2014, 311, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Sultana, C.; Shen, Y.; Johnson, C.; Kalra, V.K. Cobalt chloride-induced signaling in endothelium leading to the augmented adherence of sickle red blood cells and transendothelial migration of monocyte-like HL-60 cells is blocked by PAF-receptor antagonist. J. Cell. Physiol. 1999, 179, 67–78. [Google Scholar] [CrossRef]
- Teng, R.J.; Du, J.; Afolayan, A.J.; Eis, A.; Shi, Y.; Konduri, G.G. AMP kinase activation improves angiogenesis in pulmonary artery endothelial cells with in utero pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L29–L42. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.A.; Wood, E.J.; Karran, E.H. A matrix metalloproteinase inhibitor which prevents fibroblast-mediated collagen lattice contraction. FEBS Lett. 1998, 441, 137–140. [Google Scholar] [CrossRef]
- Huang, D.; Ding, Y.; Li, Y.; Luo, W.M.; Zhang, Z.F.; Snider, J.; Vandenbeldt, K.; Qian, C.N.; Teh, B.T. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res. 2010, 70, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Merdad, A.; Karim, S.; Schulten, H.J.; Dallol, A.; Buhmeida, A.; Al-Thubaity, F.; Gari, M.A.; Chaudhary, A.G.; Abuzenadah, A.M.; Al-Qahtani, M.H. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer: MMP-9 as a potential biomarker for cancer invasion and metastasis. Anticancer Res. 2014, 34, 1355–1366. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene Symbol | p-Value | FC Hyperglycemia-CoCl2 3 h + Metformin | p-Value | FC Hyperglycemia-CoCl2 12 h + Metformin |
|---|---|---|---|---|---|
| small nucleolar RNA, H/ACA box 20 | SNORA20 | 5.45 × 10−1 | −1.28 | 1.23 × 10−3 | 4.01 |
| small nucleolar RNA, C/D box 45C | SNORD45C | 3.62 × 10−1 | −1.50 | 1.26 × 10−2 | 3.16 |
| metastasis associated lung adenocarcinoma transcript 1 (non-protein) | MALAT1 | 2.88 × 10−2 | 2.38 | 8.78 × 10−3 | 2.87 |
| Rho-associated, coiled-coil containing protein kinase 1 | ROCK1 | 9.38 × 10−2 | 1.90 | 1.15 × 10−2 | 2.68 |
| cerebellar degeneration-related protein 1, 34 kDa | CDR1 | 5.47 × 10−3 | 2.80 | 1.20 × 10−2 | 2.52 |
| Chemokine (C-X-C Motif) ligand 8 | CXCL8 | 1.49 × 10−1 | 1.54 | 2.84 × 10−2 | 1.94 |
| cytochrome c oxidase subunit VIIb | COX7B | 4.23 × 10−1 | 1.17 | 5.53 × 10−3 | 1.76 |
| matrix metallopeptidase 16 (membrane-inserted) | MMP16 | 8.78 × 10−1 | −1.04 | 2.34 × 10−2 | 1.73 |
| fatty acid binding protein 4 | FABP4 | 2.92 × 10−2 | 1.58 | 2.53 × 10−2 | 1.60 |
| lymphocyte antigen 96 | LY96 | 2.29 × 10−1 | 1.18 | 2.01 × 10−3 | 1.57 |
| tissue factor pathway inhibitor 2 | TFPI2 | 6.17 × 10−1 | 1.06 | 2.07 × 10−4 | 1.55 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakhashab, S.; Ahmed, F.; Schulten, H.-J.; Ahmed, F.W.; Glanville, M.; Al-Qahtani, M.H.; Weaver, J.U. Proangiogenic Effect of Metformin in Endothelial Cells Is via Upregulation of VEGFR1/2 and Their Signaling under Hyperglycemia-Hypoxia. Int. J. Mol. Sci. 2018, 19, 293. https://doi.org/10.3390/ijms19010293
Bakhashab S, Ahmed F, Schulten H-J, Ahmed FW, Glanville M, Al-Qahtani MH, Weaver JU. Proangiogenic Effect of Metformin in Endothelial Cells Is via Upregulation of VEGFR1/2 and Their Signaling under Hyperglycemia-Hypoxia. International Journal of Molecular Sciences. 2018; 19(1):293. https://doi.org/10.3390/ijms19010293
Chicago/Turabian StyleBakhashab, Sherin, Farid Ahmed, Hans-Juergen Schulten, Fahad W. Ahmed, Michael Glanville, Mohammed H. Al-Qahtani, and Jolanta U. Weaver. 2018. "Proangiogenic Effect of Metformin in Endothelial Cells Is via Upregulation of VEGFR1/2 and Their Signaling under Hyperglycemia-Hypoxia" International Journal of Molecular Sciences 19, no. 1: 293. https://doi.org/10.3390/ijms19010293
APA StyleBakhashab, S., Ahmed, F., Schulten, H. -J., Ahmed, F. W., Glanville, M., Al-Qahtani, M. H., & Weaver, J. U. (2018). Proangiogenic Effect of Metformin in Endothelial Cells Is via Upregulation of VEGFR1/2 and Their Signaling under Hyperglycemia-Hypoxia. International Journal of Molecular Sciences, 19(1), 293. https://doi.org/10.3390/ijms19010293