Human MHC-II with Shared Epitope Motifs Are Optimal Epstein-Barr Virus Glycoprotein 42 Ligands—Relation to Rheumatoid Arthritis

Abstract
:1. Introduction
1.1. Rheumatoid Arthritis
1.2. Rheumatoid Arthritis and Genetic Risk Factors
1.3. Rheumatoid Arthritis and Environmental Risk Factors
1.4. Epstein-Barr Virus
1.4.1. Glycoprotein 42, Characteristics and Interactions
1.4.2. Epstein-Barr Virus as a Contributor to Initiation of Rheumatoid Arthritis
2. Discussion
HLA-DR1 and Gp42 Interaction as a Mediator or EBV Entry and Ultimately Onset of SE-Positive Rheumatoid Arthritis
Author Contributions
Conflicts of Interest
Abbreviations
| ACPA | Anti-citrullinated protein antibodies |
| CD | Cluster of differentiation |
| CTLA | Cytotoxic T lymphocyte-associated antigen |
| CTLD | C-type lectin domain |
| EBV | Epstein-Barr virus |
| Gp | Glycoprotein |
| HLA | Human leukocyte antigen |
| IRF | Interferon-regulating factor |
| MHC | Major histocompability complex |
| PTPN22 | Protein tyrosine phosphatase, non-receptor type 22 |
| PAD | Peptidyl arginine deiminase |
| RA | Rheumatoid arthritis |
| RF | Rheumatoid factor |
| SE | Shared epitope |
| SLE | Systemic lupus erythematosus |
| STAT | Signal transducer and activator of transcription |
| TRAF | Tumor necrosis factor receptor-associated factor |
References
- Cooles, F.A.; Isaacs, J.D. Pathophysiology of rheumatoid arthritis. Curr. Opin. Rheumatol. 2011, 23, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W. Rheumatoid arthritis. Lancet 2010, 376, 1094–1108. [Google Scholar] [CrossRef]
- Cooper, N.J. Economic burden of rheumatoid arthritis: A systematic review. Rheumatology 2000, 39, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, J.; Cobo, T.; Balsa, A.; Descalzo, M.A.; Carmona, L.; Group, S.S. The incidence of rheumatoid arthritis in spain: Results from a nationwide primary care registry. Rheumatology 2008, 47, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.K.; Kjaer, N.K.; Svendsen, A.J.; Horslev-Petersen, K. Incidence of rheumatoid arthritis from 1995 to 2001: Impact of ascertainment from multiple sources. Rheumatol. Int. 2009, 29, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Sangha, O. Epidemiology of rheumatic diseases. Rheumatology 2000, 39, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Symmons, D.; Turner, G.; Webb, R.; Asten, P.; Barrett, E.; Lunt, M.; Scott, D.; Silman, A. The prevalence of rheumatoid arthritis in the united kingdom: New estimates for a new century. Rheumatology 2002, 41, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An American college of rheumatology/european league against rheumatism collaborative initiative. Arthritis Rheumatol. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Dorner, T.; Egerer, K.; Feist, E.; Burmester, G.R. Rheumatoid factor revisited. Curr. Opin. Rheumatol. 2004, 16, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Sugiyama, D.; Kogata, Y.; Tsuji, G.; Nakazawa, T.; Kawano, S.; Saigo, K.; Morinobu, A.; Koshiba, M.; Kuntz, K.M.; et al. Meta-analysis: Diagnostic accuracy of anti-cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis. Ann. Intern. Med. 2007, 146, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Shmerling, R.H.; Delbanco, T.L. The rheumatoid factor: An analysis of clinical utility. Am. J. Med. 1991, 91, 528–534. [Google Scholar] [CrossRef]
- Rantapaa-Dahlqvist, S.; de Jong, B.A.; Berglin, E.; Hallmans, G.; Wadell, G.; Stenlund, H.; Sundin, U.; van Venrooij, W.J. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheumatol. 2003, 48, 2741–2749. [Google Scholar] [CrossRef] [PubMed]
- Ronnelid, J.; Wick, M.C.; Lampa, J.; Lindblad, S.; Nordmark, B.; Klareskog, L.; van Vollenhoven, R.F. Longitudinal analysis of citrullinated protein/peptide antibodies (anti-cp) during 5 year follow up in early rheumatoid arthritis: Anti-cp status predicts worse disease activity and greater radiological progression. Ann. Rheum. Dis. 2005, 64, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, G.A.; Visser, H.; de Jong, B.A.; van den Hoogen, F.H.; Hazes, J.M.; Breedveld, F.C.; van Venrooij, W.J. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheumatol. 2000, 43, 155–163. [Google Scholar] [CrossRef]
- Van Gaalen, F.A.; Linn-Rasker, S.P.; van Venrooij, W.J.; de Jong, B.A.; Breedveld, F.C.; Verweij, C.L.; Toes, R.E.; Huizinga, T.W. Autoantibodies to cyclic citrullinated peptides predict progression to rheumatoid arthritis in patients with undifferentiated arthritis: A prospective cohort study. Arthritis Rheumatol. 2004, 50, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Nell, V.P.; Machold, K.P.; Stamm, T.A.; Eberl, G.; Heinzl, H.; Uffmann, M.; Smolen, J.S.; Steiner, G. Autoantibody profiling as early diagnostic and prognostic tool for rheumatoid arthritis. Ann. Rheum. Dis. 2005, 64, 1731–1736. [Google Scholar] [CrossRef] [PubMed]
- Payet, J.; Goulvestre, C.; Biale, L.; Avouac, J.; Wipff, J.; Job-Deslandre, C.; Batteux, F.; Dougados, M.; Kahan, A.; Allanore, Y. Anticyclic citrullinated peptide antibodies in rheumatoid and nonrheumatoid rheumatic disorders: Experience with 1162 patients. J. Rheumatol. 2014, 41, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Peene, I.; Union, A.; Meheus, L.; Sebbag, M.; Serre, G.; Veys, E.M.; de Keyser, F. Specific presence of intracellular citrullinated proteins in rheumatoid arthritis synovium: Relevance to antifilaggrin autoantibodies. Arthritis Rheumatol. 2001, 44, 2255–2262. [Google Scholar] [CrossRef]
- Snir, O.; Widhe, M.; Hermansson, M.; von Spee, C.; Lindberg, J.; Hensen, S.; Lundberg, K.; Engstrom, A.; Venables, P.J.; Toes, R.E.; et al. Antibodies to several citrullinated antigens are enriched in the joints of rheumatoid arthritis patients. Arthritis Rheumatol. 2010, 62, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Tarcsa, E.; Marekov, L.N.; Mei, G.; Melino, G.; Lee, S.C.; Steinert, P.M. Protein unfolding by peptidylarginine deiminase. Substrate specificity and structural relationships of the natural substrates trichohyalin and filaggrin. J. Biol. Chem. 1996, 271, 30709–30716. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Toth, E.; Tarcsa, E.; Falus, A.; Buzas, E.I. Citrullination: A posttranslational modification in health and disease. Int. J. Biochem. Cell Biol. 2006, 38, 1662–1677. [Google Scholar] [CrossRef] [PubMed]
- Reparon-Schuijt, C.C.; van Esch, W.J.; van Kooten, C.; Schellekens, G.A.; de Jong, B.A.; van Venrooij, W.J.; Breedveld, F.C.; Verweij, C.L. Secretion of anti-citrulline-containing peptide antibody by b lymphocytes in rheumatoid arthritis. Arthritis Rheumatol. 2001, 44, 41–47. [Google Scholar] [CrossRef]
- Kuhn, K.A.; Kulik, L.; Tomooka, B.; Braschler, K.J.; Arend, W.P.; Robinson, W.H.; Holers, V.M. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J. Clin. Investig. 2006, 116, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Van der Helm-van Mil, A.H.; Verpoort, K.N.; Breedveld, F.C.; Toes, R.E.; Huizinga, T.W. Antibodies to citrullinated proteins and differences in clinical progression of rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R949–R958. [Google Scholar] [CrossRef] [PubMed]
- Van der Woude, D.; Young, A.; Jayakumar, K.; Mertens, B.J.; Toes, R.E.; van der Heijde, D.; Huizinga, T.W.; van der Helm-van Mil, A.H. Prevalence of and predictive factors for sustained disease-modifying antirheumatic drug-free remission in rheumatoid arthritis: Results from two large early arthritis cohorts. Arthritis Rheumatol. 2009, 60, 2262–2271. [Google Scholar] [CrossRef] [PubMed]
- Verpoort, K.N.; van Gaalen, F.A.; van der Helm-van Mil, A.H.; Schreuder, G.M.; Breedveld, F.C.; Huizinga, T.W.; de Vries, R.R.; Toes, R.E. Association of HLA-DR3 with anti-cyclic citrullinated peptide antibody-negative rheumatoid arthritis. Arthritis Rheumatol. 2005, 52, 3058–3062. [Google Scholar] [CrossRef] [PubMed]
- Berglin, E.; Johansson, T.; Sundin, U.; Jidell, E.; Wadell, G.; Hallmans, G.; Rantapaa-Dahlqvist, S. Radiological outcome in rheumatoid arthritis is predicted by presence of antibodies against cyclic citrullinated peptide before and at disease onset, and by iga-rf at disease onset. Ann. Rheum. Dis. 2006, 65, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Machold, K.P.; Stamm, T.A.; Nell, V.P.; Pflugbeil, S.; Aletaha, D.; Steiner, G.; Uffmann, M.; Smolen, J.S. Very recent onset rheumatoid arthritis: Clinical and serological patient characteristics associated with radiographic progression over the first years of disease. Rheumatology 2007, 46, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.A.; Gough, A.K.; Green, M.J.; Devlin, J.; Hensor, E.M.; Greenstein, A.; Fraser, A.; Emery, P. Anti-ccp antibodies measured at disease onset help identify seronegative rheumatoid arthritis and predict radiological and functional outcome. Rheumatology 2006, 45, 478–480. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, A.J.; Snieder, H.; Rigby, A.S.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheumatol. 2000, 43, 30–37. [Google Scholar] [CrossRef]
- Zanelli, E.; Breedveld, F.C.; de Vries, R.R. Hla class II association with rheumatoid arthritis: Facts and interpretations. Hum. Immunol. 2000, 61, 1254–1261. [Google Scholar] [CrossRef]
- Gourraud, P.A.; Boyer, J.F.; Barnetche, T.; Abbal, M.; Cambon-Thomsen, A.; Cantagrel, A.; Constantin, A. A new classification of HLA-DRB1 alleles differentiates predisposing and protective alleles for rheumatoid arthritis structural severity. Arthritis Rheumatol. 2006, 54, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Du Montcel, S.T.; Michou, L.; Petit-Teixeira, E.; Osorio, J.; Lemaire, I.; Lasbleiz, S.; Pierlot, C.; Quillet, P.; Bardin, T.; Prum, B.; et al. New classification of HLA-DRB1 alleles supports the shared epitope hypothesis of rheumatoid arthritis susceptibility. Arthritis Rheumatol. 2005, 52, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Stastny, P. Mixed lymphocyte cultures in rheumatoid arthritis. J. Clin. Investig. 1976, 57, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Holoshitz, J. The rheumatoid arthritis HLA-DRB1 shared epitope. Curr. Opin. Rheumatol. 2010, 22, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Mattey, D.L.; Dawes, P.T.; Gonzalez-Gay, M.A.; Garcia-Porrua, C.; Thomson, W.; Hajeer, A.H.; Ollier, W.E. HLA-DRB1 alleles encoding an aspartic acid at position 70 protect against development of rheumatoid arthritis. J. Rheumatol. 2001, 28, 232–239. [Google Scholar] [PubMed]
- Gao, X.; Gazit, E.; Livneh, A.; Stastny, P. Rheumatoid arthritis in Israeli Jews: Shared sequences in the third hypervariable region of DRB1 alleles are associated with susceptibility. J. Rheumatol. 1991, 18, 801–803. [Google Scholar] [PubMed]
- Michou, L.; Croiseau, P.; Petit-Teixeira, E.; du Montcel, S.T.; Lemaire, I.; Pierlot, C.; Osorio, J.; Frigui, W.; Lasbleiz, S.; Quillet, P.; et al. Validation of the reshaped shared epitope HLA-DRB1 classification in rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R79. [Google Scholar] [CrossRef] [PubMed]
- Carrier, N.; Cossette, P.; Daniel, C.; de Brum-Fernandes, A.; Liang, P.; Menard, H.A.; Boire, G. The deraa HLA-DR alleles in patients with early polyarthritis: Protection against severe disease and lack of association with rheumatoid arthritis autoantibodies. Arthritis Rheumatol. 2009, 60, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Mackie, S.L.; Taylor, J.C.; Martin, S.G.; Consortium, Y.; Consortium, U.; Wordsworth, P.; Steer, S.; Wilson, A.G.; Worthington, J.; Emery, P.; et al. A spectrum of susceptibility to rheumatoid arthritis within HLA-DRB1: Stratification by autoantibody status in a large UK population. Genes Immun. 2012, 13, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.W.; Thomson, W.; Martin, S.G.; Yorkshire Early Arthritis Register Consortium; Carter, A.M.; Consortium, U.K.R.A.G.; Erlich, H.A.; Barton, A.; Hocking, L.; Reid, D.M.; et al. Reevaluation of the interaction between HLA-DRB1 shared epitope alleles, PTPN22, and smoking in determining susceptibility to autoantibody-positive and autoantibody-negative rheumatoid arthritis in a large UK caucasian population. Arthritis Rheumatol. 2009, 60, 2565–2576. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Sandor, C.; Stahl, E.A.; Freudenberg, J.; Lee, H.S.; Jia, X.; Alfredsson, L.; Padyukov, L.; Klareskog, L.; Worthington, J.; et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012, 44, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Mattey, D.L.; Thomson, W.; Ollier, W.E.; Batley, M.; Davies, P.G.; Gough, A.K.; Devlin, J.; Prouse, P.; James, D.W.; Williams, P.L.; et al. Association of drb1 shared epitope genotypes with early mortality in rheumatoid arthritis: Results of eighteen years of followup from the early rheumatoid arthritis study. Arthritis Rheumatol. 2007, 56, 1408–1416. [Google Scholar] [CrossRef] [PubMed]
- Wagner, U.; Kaltenhauser, S.; Sauer, H.; Arnold, S.; Seidel, W.; Hantzschel, H.; Kalden, J.R.; Wassmuth, R. HLA markers and prediction of clinical course and outcome in rheumatoid arthritis. Arthritis Rheumatol. 1997, 40, 341–351. [Google Scholar] [CrossRef]
- Auger, I.; Toussirot, E.; Roudier, J. Molecular mechanisms involved in the association of HLA-DR4 and rheumatoid arthritis. Immunol. Res. 1997, 16, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Bhayani, H.R.; Hedrick, S.M. The role of polymorphic amino acids of the MHC molecule in the selection of the t cell repertoire. J. Immunol. 1991, 146, 1093–1098. [Google Scholar] [PubMed]
- Hammer, J.; Gallazzi, F.; Bono, E.; Karr, R.W.; Guenot, J.; Valsasnini, P.; Nagy, Z.A.; Sinigaglia, F. Peptide binding specificity of HLA-DR4 molecules: Correlation with rheumatoid arthritis association. J. Exp. Med. 1995, 181, 1847–1855. [Google Scholar] [CrossRef] [PubMed]
- Roudier, J. Association of MHC and rheumatoid arthritis. Association of RA with HLA-DR4: The role of repertoire selection. Arthritis Res. 2000, 2, 217–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wucherpfennig, K.W.; Strominger, J.L. Selective binding of self peptides to disease-associated major histocompatibility complex (MHC) molecules: A mechanism for MHC-linked susceptibility to human autoimmune diseases. J. Exp. Med. 1995, 181, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Southwood, S.; Sette, A.; Jevnikar, A.M.; Bell, D.A.; Cairns, E. Cutting edge: The conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class ii molecule. J. Immunol. 2003, 171, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Van Heemst, J.; Jansen, D.T.; Polydorides, S.; Moustakas, A.K.; Bax, M.; Feitsma, A.L.; Bontrop-Elferink, D.G.; Baarse, M.; van der Woude, D.; Wolbink, G.J.; et al. Crossreactivity to vinculin and microbes provides a molecular basis for HLA-based protection against rheumatoid arthritis. Nat. Commun. 2015, 6, 6681. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible mica. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Radaev, S.; Sun, P.D. Structure and function of natural killer cell surface receptors. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Cheng, A.; Pumpens, P.; Michalak, M.; Holoshitz, J. Identification of the rheumatoid arthritis shared epitope binding site on calreticulin. PLoS ONE 2010, 5, e11703. [Google Scholar] [CrossRef] [PubMed]
- Begovich, A.B.; Carlton, V.E.; Honigberg, L.A.; Schrodi, S.J.; Chokkalingam, A.P.; Alexander, H.C.; Ardlie, K.G.; Huang, Q.; Smith, A.M.; Spoerke, J.M.; et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 2004, 75, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Plenge, R.M.; Padyukov, L.; Remmers, E.F.; Purcell, S.; Lee, A.T.; Karlson, E.W.; Wolfe, F.; Kastner, D.L.; Alfredsson, L.; Altshuler, D.; et al. Replication of putative candidate-gene associations with rheumatoid arthritis in >4000 samples from north america and sweden: Association of susceptibility with PTPN22, CTLA4, and PADI4. Am. J. Hum. Genet. 2005, 77, 1044–1060. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Kallberg, H.; Bengtsson, C.; Grunewald, J.; Ronnelid, J.; Harris, H.E.; Ulfgren, A.K.; Rantapaa-Dahlqvist, S.; et al. A new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheumatol. 2006, 54, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Van der Helm-van Mil, A.H.; Verpoort, K.N.; le Cessie, S.; Huizinga, T.W.; de Vries, R.R.; Toes, R.E. The HLA-DRB1 shared epitope alleles differ in the interaction with smoking and predisposition to antibodies to cyclic citrullinated peptide. Arthritis Rheumatol. 2007, 56, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Wesoly, J.; van der Helm-van Mil, A.H.; Toes, R.E.; Chokkalingam, A.P.; Carlton, V.E.; Begovich, A.B.; Huizinga, T.W. Association of the PTPN22 C1858T single-nucleotide polymorphism with rheumatoid arthritis phenotypes in an inception cohort. Arthritis Rheumatol. 2005, 52, 2948–2950. [Google Scholar] [CrossRef] [PubMed]
- Plenge, R.M.; Seielstad, M.; Padyukov, L.; Lee, A.T.; Remmers, E.F.; Ding, B.; Liew, A.; Khalili, H.; Chandrasekaran, A.; Davies, L.R.; et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—A genomewide study. N. Engl. J. Med. 2007, 357, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Lorentzen, J.C.; Flornes, L.; Eklow, C.; Backdahl, L.; Ribbhammar, U.; Guo, J.P.; Smolnikova, M.; Dissen, E.; Seddighzadeh, M.; Brookes, A.J.; et al. Association of arthritis with a gene complex encoding C-type lectin-like receptors. Arthritis Rheumatol. 2007, 56, 2620–2632. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, S.; Padyukov, L.; Kurreeman, F.A.; Liljedahl, U.; Wiman, A.C.; Alfredsson, L.; Toes, R.; Ronnelid, J.; Klareskog, L.; Huizinga, T.W.; et al. Association of a haplotype in the promoter region of the interferon regulatory factor 5 gene with rheumatoid arthritis. Arthritis Rheumatol. 2007, 56, 2202–2210. [Google Scholar] [CrossRef] [PubMed]
- Mohan, V.K.; Ganesan, N.; Gopalakrishnan, R. Association of susceptible genetic markers and autoantibodies in rheumatoid arthritis. J. Genet. 2014, 93, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Di Giuseppe, D.; Discacciati, A.; Orsini, N.; Wolk, A. Cigarette smoking and risk of rheumatoid arthritis: A dose-response meta-analysis. Arthritis Res. Ther. 2014, 16, R61. [Google Scholar] [CrossRef] [PubMed]
- Rashid, T.; Ebringer, A. Rheumatoid arthritis is linked to proteus—The evidence. Clin. Rheumatol. 2007, 26, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Bengtsson, C.; Nordmark, B.; Lindblad, S.; Lundberg, I.; Klareskog, L.; Alfredsson, L.; EIRA Study Group. Quantification of the influence of cigarette smoking on rheumatoid arthritis: Results from a population based case-control study, using incident cases. Ann. Rheum. Dis. 2003, 62, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Bartold, P.M.; Marino, V.; Cantley, M.; Haynes, D.R. Effect of porphyromonas gingivalis-induced inflammation on the development of rheumatoid arthritis. J. Clin. Periodontol. 2010, 37, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Mikuls, T.R.; Payne, J.B.; Yu, F.; Thiele, G.M.; Reynolds, R.J.; Cannon, G.W.; Markt, J.; McGowan, D.; Kerr, G.S.; Redman, R.S.; et al. Periodontitis and porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; le Gall-David, S.; Bonnaure-Mallet, M.; Schaeverbeke, T.; Cantagrel, A.; Minet, J.; Gottenberg, J.E.; Chanson, P.; Ravaud, P.; Mariette, X. Association of anti-porphyromonas gingivalis antibody titers with nonsmoking status in early rheumatoid arthritis: Results from the prospective french cohort of patients with early rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 1729–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pablo, P.; Dietrich, T.; McAlindon, T.E. Association of periodontal disease and tooth loss with rheumatoid arthritis in the us population. J. Rheumatol. 2008, 35, 70–76. [Google Scholar] [PubMed]
- Klareskog, L.; Padyukov, L.; Lorentzen, J.; Alfredsson, L. Mechanisms of disease: Genetic susceptibility and environmental triggers in the development of rheumatoid arthritis. Nat. Clin. Pract. Rheumatol. 2006, 2, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, L.I.; Daoussis, D.; Liossis, S.N.; Bogdanos, D.P. The infectious basis of ACPA-positive rheumatoid arthritis. Front. Microbiol. 2017, 8, 1853. [Google Scholar] [CrossRef] [PubMed]
- Kharlamova, N.; Jiang, X.; Sherina, N.; Potempa, B.; Israelsson, L.; Quirke, A.M.; Eriksson, K.; Yucel-Lindberg, T.; Venables, P.J.; Potempa, J.; et al. Antibodies to porphyromonas gingivalis indicate interaction between oral infection, smoking, and risk genes in rheumatoid arthritis etiology. Arthritis Rheumatol. 2016, 68, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheumatol. 2010, 62, 2662–2672. [Google Scholar] [CrossRef] [PubMed]
- Harel-Meir, M.; Sherer, Y.; Shoenfeld, Y. Tobacco smoking and autoimmune rheumatic diseases. Nat. Clin. Pract. Rheumatol. 2007, 3, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Hoovestol, R.A.; Mikuls, T.R. Environmental exposures and rheumatoid arthritis risk. Curr. Rheumatol. Rep. 2011, 13, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Onozaki, K. Etiological and biological aspects of cigarette smoking in rheumatoid arthritis. Inflamm. Allergy Drug Targets 2009, 8, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Yang, S.M.; Kim, S.H.; Han, K.H.; Park, S.J.; Shin, J.I. Smoking and rheumatoid arthritis. Int. J. Mol. Sci. 2014, 15, 22279–22295. [Google Scholar] [CrossRef] [PubMed]
- Padyukov, L.; Silva, C.; Stolt, P.; Alfredsson, L.; Klareskog, L. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheumatol. 2004, 50, 3085–3092. [Google Scholar] [CrossRef] [PubMed]
- Linn-Rasker, S.P.; van der Helm-van Mil, A.H.; van Gaalen, F.A.; Kloppenburg, M.; de Vries, R.R.; le Cessie, S.; Breedveld, F.C.; Toes, R.E.; Huizinga, T.W. Smoking is a risk factor for anti-CCP antibodies only in rheumatoid arthritis patients who carry HLA-DRB1 shared epitope alleles. Ann. Rheum. Dis. 2006, 65, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.P.; Alfredsson, L.; Karlson, E.W. Environmental influences on risk for rheumatoid arthritis. Curr. Opin. Rheumatol. 2009, 21, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Draborg, A.; Izarzugaza, J.M.; Houen, G. How compelling are the data for Epstein-Barr virus being a trigger for systemic lupus and other autoimmune diseases? Curr. Opin. Rheumatol. 2016, 28, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr virus and systemic lupus erythematosus. Clin. Dev. Immunol. 2012, 2012, 370516. [Google Scholar] [CrossRef] [PubMed]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr virus in systemic autoimmune diseases. Clin. Dev. Immunol. 2013, 2013, 535738. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Mohl, B.S.; Chen, J.; Sathiyamoorthy, K.; Jardetzky, T.S.; Longnecker, R. Structural and mechanistic insights into the tropism of Epstein-Barr virus. Mol. Cell 2016, 39, 286–291. [Google Scholar]
- Sathiyamoorthy, K.; Chen, J.; Longnecker, R.; Jardetzky, T.S. The complexity in herpesvirus entry. Curr. Opin. Virol. 2017, 24, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Sathiyamoorthy, K.; Hu, Y.X.; Mohl, B.S.; Chen, J.; Longnecker, R.; Jardetzky, T.S. Structural basis for Epstein-Barr virus host cell tropism mediated by gp42 and gHgL entry glycoproteins. Nat. Commun. 2016, 7, 13557. [Google Scholar] [CrossRef] [PubMed]
- Sathiyamoorthy, K.; Jiang, J.; Hu, Y.X.; Rowe, C.L.; Mohl, B.S.; Chen, J.; Jiang, W.; Mellins, E.D.; Longnecker, R.; Zhou, Z.H.; et al. Assembly and architecture of the EBV B cell entry triggering complex. PLoS Pathog. 2014, 10, e1004309. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Rowe, M. Epstein barr virus entry; kissing and conjugation. Curr. Opin. Virol. 2014, 4, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Borza, C.M.; Hutt-Fletcher, L.M. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 2002, 8, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hutt-Fletcher, L.M. Epstein-Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J. Virol. 1998, 72, 158–163. [Google Scholar] [PubMed]
- Shaw, P.L.; Kirschner, A.N.; Jardetzky, T.S.; Longnecker, R. Characteristics of Epstein-Barr virus envelope protein gp42. Virus Genes 2010, 40, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Drickamer, K. C-type lectin-like domains. Curr. Opin. Struct. Biol. 1999, 9, 585–590. [Google Scholar] [CrossRef]
- Weis, W.I.; Taylor, M.E.; Drickamer, K. The C-type lectin superfamily in the immune system. Immunol. Rev. 1998, 163, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.L.; Omerovic, J.; Jardetzky, T.S.; Longnecker, R. Mutational analyses of Epstein-Barr virus glycoprotein 42 reveal functional domains not involved in receptor binding but required for membrane fusion. J. Virol. 2004, 78, 5946–5956. [Google Scholar] [CrossRef] [PubMed]
- Haan, K.M.; Kwok, W.W.; Longnecker, R.; Speck, P. Epstein-Barr virus entry utilizing HLA-DP or HLA-DQ as a coreceptor. J. Virol. 2000, 74, 2451–2454. [Google Scholar] [CrossRef] [PubMed]
- Janz, A.; Oezel, M.; Kurzeder, C.; Mautner, J.; Pich, D.; Kost, M.; Hammerschmidt, W.; Delecluse, H.J. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of additional viral ligands. J. Virol. 2000, 74, 10142–10152. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, A.N.; Sorem, J.; Longnecker, R.; Jardetzky, T.S. Structure of Epstein-Barr virus glycoprotein 42 suggests a mechanism for triggering receptor-activated virus entry. Structure 2009, 17, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Rowe, C.L.; Connolly, S.A.; Chen, J.; Jardetzky, T.S.; Longnecker, R. A soluble form of Epstein-Barr virus gH/gL inhibits EBV-induced membrane fusion and does not function in fusion. Virology 2013, 436, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Sorem, J.; Jardetzky, T.S.; Longnecker, R. Cleavage and secretion of Epstein-Barr virus glycoprotein 42 promote membrane fusion with B lymphocytes. J. Virol. 2009, 83, 6664–6672. [Google Scholar] [CrossRef] [PubMed]
- Spear, P.G.; Longnecker, R. Herpesvirus entry: An update. J. Virol. 2003, 77, 10179–10185. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, A.N.; Lowrey, A.S.; Longnecker, R.; Jardetzky, T.S. Binding-site interactions between Epstein-Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B-cell membrane fusion. J. Virol. 2007, 81, 9216–9229. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Marquardt, G.; Kirschner, A.N.; Longnecker, R.; Jardetzky, T.S. Mapping the n-terminal residues of Epstein-Barr virus gp42 that bind gH/gL by using fluorescence polarization and cell-based fusion assays. J. Virol. 2010, 84, 10375–10385. [Google Scholar] [CrossRef] [PubMed]
- Backovic, M.; Jardetzky, T.S.; Longnecker, R. Hydrophobic residues that form putative fusion loops of Epstein-Barr virus glycoprotein b are critical for fusion activity. J. Virol. 2007, 81, 9596–9600. [Google Scholar] [CrossRef] [PubMed]
- Mullen, M.M.; Haan, K.M.; Longnecker, R.; Jardetzky, T.S. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 2002, 9, 375–385. [Google Scholar] [CrossRef]
- McShane, M.P.; Mullen, M.M.; Haan, K.M.; Jardetzky, T.S.; Longnecker, R. Mutational analysis of the HLA class II interaction with Epstein-Barr virus glycoprotein 42. J. Virol. 2003, 77, 7655–7662. [Google Scholar] [CrossRef] [PubMed]
- Costenbader, K.H.; Karlson, E.W. Epstein-Barr virus and rheumatoid arthritis: Is there a link? Arthritis Res. Ther. 2006, 8, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erre, G.L.; Mameli, G.; Cossu, D.; Muzzeddu, B.; Piras, C.; Paccagnini, D.; Passiu, G.; Sechi, L.A. Increased Epstein-Barr virus DNA load and antibodies against EBNA1 and EA in sardinian patients with rheumatoid arthritis. Viral Immunol. 2015, 28, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.L.; Chibnik, L.B.; Karlson, E.W.; Costenbader, K.H. Epstein-Barr virus serologic abnormalities and risk of rheumatoid arthritis among women. Autoimmunity 2012, 45, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Toussirot, E.; Roudier, J. Pathophysiological links between rheumatoid arthritis and the Epstein-Barr virus: An update. Jt. Bone Spine 2007, 74, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Westergaard, M.W.; Draborg, A.H.; Troelsen, L.; Jacobsen, S.; Houen, G. Isotypes of Epstein-Barr virus antibodies in rheumatoid arthritis: Association with rheumatoid factors and citrulline-dependent antibodies. BioMed Res. Int. 2015, 2015, 472174. [Google Scholar] [CrossRef] [PubMed]
- Alspaugh, M.A.; Henle, G.; Lennette, E.T.; Henle, W. Elevated levels of antibodies to Epstein-Barr virus antigens in sera and synovial fluids of patients with rheumatoid arthritis. J. Clin. Investig. 1981, 67, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Billings, P.B.; Hoch, S.O.; White, P.J.; Carson, D.A.; Vaughan, J.H. Antibodies to the Epstein-Barr virus nuclear antigen and to rheumatoid arthritis nuclear antigen identify the same polypeptide. Proc. Natl. Acad. Sci. USA 1983, 80, 7104–7108. [Google Scholar] [CrossRef] [PubMed]
- Venables, P.J.; Pawlowski, T.; Mumford, P.A.; Brown, C.; Crawford, D.H.; Maini, R.N. Reaction of antibodies to rheumatoid arthritis nuclear antigen with a synthetic peptide corresponding to part of Epstein-Barr nuclear antigen 1. Ann. Rheum. Dis. 1988, 47, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Tosato, G.; Steinberg, A.D.; Blaese, R.M. Defective EBV-specific suppressor T-cell function in rheumatoid arthritis. N. Engl. J. Med. 1981, 305, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Tosato, G.; Steinberg, A.D.; Yarchoan, R.; Heilman, C.A.; Pike, S.E.; de Seau, V.; Blaese, R.M. Abnormally elevated frequency of Epstein-Barr virus-infected B cells in the blood of patients with rheumatoid arthritis. J. Clin. Investig. 1984, 73, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Balandraud, N.; Meynard, J.B.; Auger, I.; Sovran, H.; Mugnier, B.; Reviron, D.; Roudier, J.; Roudier, C. Epstein-Barr virus load in the peripheral blood of patients with rheumatoid arthritis: Accurate quantification using real-time polymerase chain reaction. Arthritis Rheumatol. 2003, 48, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Blaschke, S.; Schwarz, G.; Moneke, D.; Binder, L.; Muller, G.; Reuss-Borst, M. Epstein-Barr virus infection in peripheral blood mononuclear cells, synovial fluid cells, and synovial membranes of patients with rheumatoid arthritis. J. Rheumatol. 2000, 27, 866–873. [Google Scholar] [PubMed]
- Takeda, T.; Mizugaki, Y.; Matsubara, L.; Imai, S.; Koike, T.; Takada, K. Lytic Epstein-Barr virus infection in the synovial tissue of patients with rheumatoid arthritis. Arthritis Rheumatol. 2000, 43, 1218–1225. [Google Scholar] [CrossRef]
- Lotz, M.; Roudier, J. Epstein-Barr virus and rheumatoid arthritis: Cellular and molecular aspects. Rheumatol. Int. 1989, 9, 147–152. [Google Scholar] [PubMed]
- Roudier, J.; Petersen, J.; Rhodes, G.H.; Luka, J.; Carson, D.A. Susceptibility to rheumatoid arthritis maps to a T-cell epitope shared by the HLA-DW4 DR β-1 chain and the Epstein-Barr virus glycoprotein gp110. Proc. Natl. Acad. Sci. USA 1989, 86, 5104–5108. [Google Scholar] [CrossRef] [PubMed]
- Callan, M.F. Epstein-Barr virus, arthritis, and the development of lymphoma in arthritis patients. Curr. Opin. Rheumatol. 2004, 16, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Epstein-Barr virus infection. N. Engl. J. Med. 2000, 343, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Ball, R.J.; Avenell, A.; Aucott, L.; Hanlon, P.; Vickers, M.A. Systematic review and meta-analysis of the sero-epidemiological association between Epstein-Barr virus and rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherina, N.; Hreggvidsdottir, H.S.; Bengtsson, C.; Hansson, M.; Israelsson, L.; Alfredsson, L.; Lundberg, K. Low levels of antibodies against common viruses associate with anti-citrullinated protein antibody-positive rheumatoid arthritis; implications for disease aetiology. Arthritis Res. Ther. 2017, 19, 219. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, M.K.; Armitage, R.J.; Comeau, M.R.; Strockbine, L.; Farrah, T.; Macduff, B.; Ulrich, D.; Alderson, M.R.; Mullberg, J.; Cohen, J.I. The extracellular domain of the Epstein-Barr virus BZLF2 protein binds the HLA-DR β chain and inhibits antigen presentation. J. Virol. 1996, 70, 5557–5563. [Google Scholar] [PubMed]
- Li, Q.; Spriggs, M.K.; Kovats, S.; Turk, S.M.; Comeau, M.R.; Nepom, B.; Hutt-Fletcher, L.M. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 1997, 71, 4657–4662. [Google Scholar] [PubMed]
- Li, Q.; Turk, S.M.; Hutt-Fletcher, L.M. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of ebv and carries an epitope critical to infection of B cells but not of epithelial cells. J. Virol. 1995, 69, 3987–3994. [Google Scholar] [PubMed]
- Haan, K.M.; Longnecker, R. Coreceptor restriction within the HLA-DQ locus for Epstein-Barr virus infection. Proc. Natl. Acad. Sci. USA 2000, 97, 9252–9257. [Google Scholar] [CrossRef] [PubMed]
- Speck, P.; Haan, K.M.; Longnecker, R. Epstein-Barr virus entry into cells. Virology 2000, 277, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ou, D.; Mitchell, L.A.; Tingle, A.J. A new categorization of HLA DR alleles on a functional basis. Hum. Immunol. 1998, 59, 665–676. [Google Scholar] [CrossRef]
- Rosloniec, E.F.; Whittington, K.B.; Zaller, D.M.; Kang, A.H. HLA-DR1 (DRB1*0101) and DR4 (DRB1*0401) use the same anchor residues for binding an immunodominant peptide derived from human type II collagen. J. Immunol. 2002, 168, 253–259. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
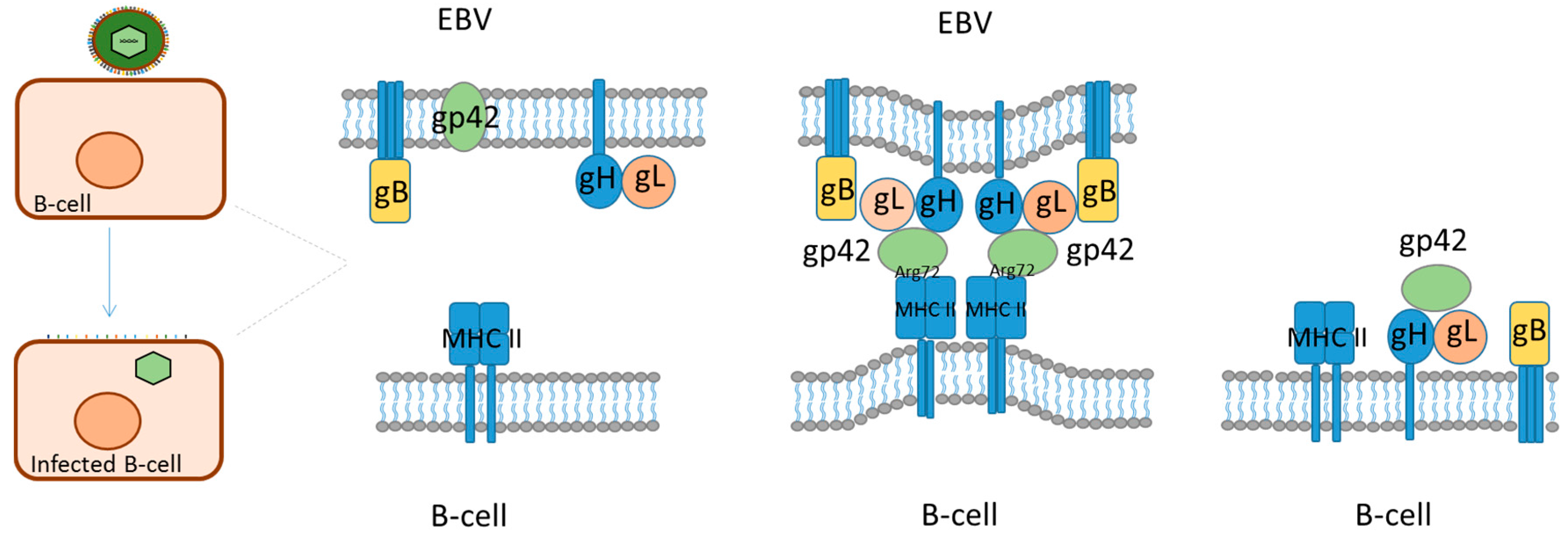{kind=link}
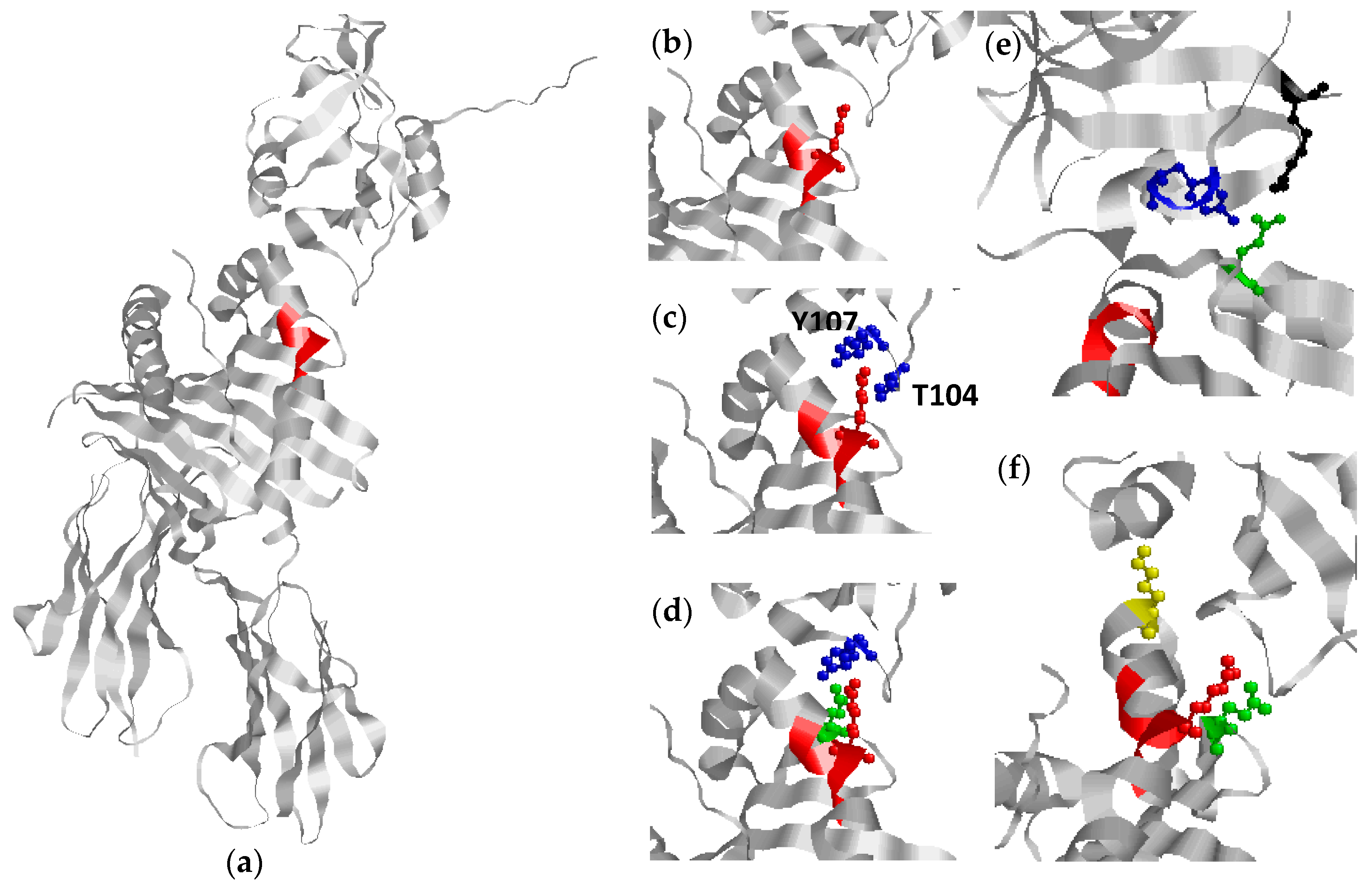{kind=link}
| Sequence | SE Motif | Alleles | Relative Genotype Risk * | References |
|---|---|---|---|---|
| QKRAA | + | *04:01, *04:09, *04:13, *04:16, *04:19, *04:21,*14:21 | 5.9 | [32] |
| DKRAA | - | *13:03 | 5.9 | [32] |
| QRRAA | + | *01:01, *01:02, *01:05, *04:04, *04:05, *04:08, *04:10, *04:19, *14:02, 14:06, *14:09, *14:13, *14:17, *14:20 | 3.3 | [31,32] |
| RRRAA | + | *10:01 | 3.3 | [32] |
| QRRAE | - | *04:03, *04:06, *04:07, *04:11, *04:17, *04:20 | 1 | [33] |
| RRRAE | - | *09:01, *14:01, *14:04, *14:05, *14:07, *14:08, *14:10, *14:11, *14:14, *14:18 | 1 | [33] |
| QARAA | - | *13:09, *15:01 | 1 | [33] |
| QKRGR | - | *03:01, *04:22, *11:07 | 1 | [33] |
| DRRGQ | - | *07:01 | 1 | [33] |
| DRRAL | - | *08:01 | 1 | [32] |
| DRRAA | - | *04:15, *08:05, *11:01, *11:04, *11:05, *11:06, *11:09, *11:10, *11:12, *11:15, *11:18, *11:19, *11:22, *12:01, *13:05, *13:06, *13:07, *13:11, *13:12, *13:14, *13:21, *13:25, *14:22, *16:01, *16:05 | 1 | [31,32] |
| DERAA | - | *01:03, *04:02, *11:02, *11:03, *11:16, *11:20, *11:21, *13:01, *13:02, *13:04, *13:08, *13:15, *13:17, *13:19, *13:22, *13:23, *14:16, *15:01 | 1 | [31,32] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trier, N.; Izarzugaza, J.; Chailyan, A.; Marcatili, P.; Houen, G. Human MHC-II with Shared Epitope Motifs Are Optimal Epstein-Barr Virus Glycoprotein 42 Ligands—Relation to Rheumatoid Arthritis. Int. J. Mol. Sci. 2018, 19, 317. https://doi.org/10.3390/ijms19010317
Trier N, Izarzugaza J, Chailyan A, Marcatili P, Houen G. Human MHC-II with Shared Epitope Motifs Are Optimal Epstein-Barr Virus Glycoprotein 42 Ligands—Relation to Rheumatoid Arthritis. International Journal of Molecular Sciences. 2018; 19(1):317. https://doi.org/10.3390/ijms19010317
Chicago/Turabian StyleTrier, Nicole, Jose Izarzugaza, Anna Chailyan, Paolo Marcatili, and Gunnar Houen. 2018. "Human MHC-II with Shared Epitope Motifs Are Optimal Epstein-Barr Virus Glycoprotein 42 Ligands—Relation to Rheumatoid Arthritis" International Journal of Molecular Sciences 19, no. 1: 317. https://doi.org/10.3390/ijms19010317
APA StyleTrier, N., Izarzugaza, J., Chailyan, A., Marcatili, P., & Houen, G. (2018). Human MHC-II with Shared Epitope Motifs Are Optimal Epstein-Barr Virus Glycoprotein 42 Ligands—Relation to Rheumatoid Arthritis. International Journal of Molecular Sciences, 19(1), 317. https://doi.org/10.3390/ijms19010317