Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases



Abstract
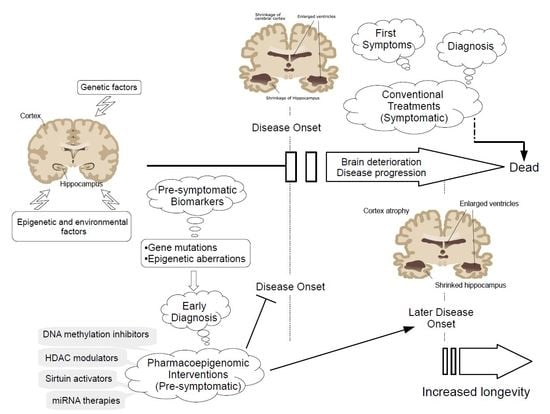
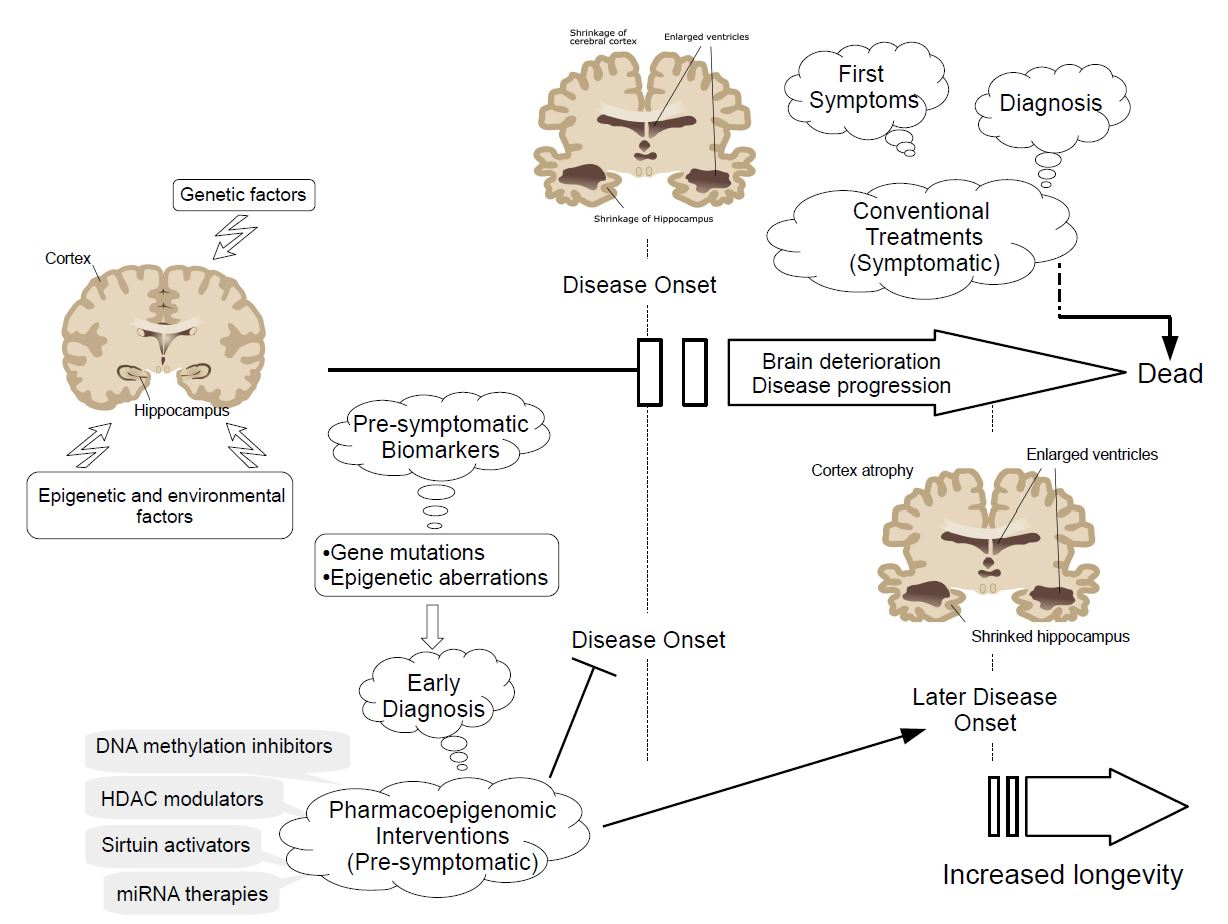
1. Introduction
2. Current Gene Targets and Pharmacological Treatments for Alzheimer’s and Parkinson’s Diseases
2.1. Alzheimer’s Disease (AD)
2.2. Parkinson’s Disease (PD)
3. Main Epigenetic Hallmarks of Neurodegeneration
3.1. DNA Methylation
3.1.1. Global DNA Methylation and Neurodegeneration
3.1.2. Gene Specific Methylation and Neurodegeneration
3.2. Histone Post-Translational Modifications Affecting Chromatin Remodeling
3.3. Non-Coding RNAs
3.4. Epigenetic Regulation of Telomeres
4. Current Epigenetic-Based Strategies Targeting Neurodegeneration
4.1. DNA Methylation Modulators
4.1.1. DNA Methylation Activators
4.1.2. DNA Methylation Inhibitors
4.2. Histone Deacetylase (HDAC) Modulators
4.2.1. Class I, II, and IV HDAC Inhibitors
4.2.2. Class III HDAC (SIRT) Inhibitors
4.2.3. Class III HDAC (SIRT) Activators
4.3. Histone Acetyltransferase (HAT) Modulators
4.4. Modulators of Histone Methylation
Histone Methyltransferase Inhibitors
4.5. Non-Coding RNAs
4.6. Other Potential Epigenetic Treatments
5. Neurodegeneration-Mediated DNA Methylation Patterns of Genes Involved in Drug Metabolism and Transport
6. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Botuyan, M.V.; Lee, J.; Ward, I.M.; Jim, J.E.; Thompson, J.R.; Chen, J.; Mer, G. Structural basis for the methylation statespecific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 2006, 127, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C. Epigenetic drug discovery for Alzheimer’s disease. Expert Opin. Drug Discov. 2014, 9, 1059–1086. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Pharmacogenomics in Alzheimer’s disease. Methods Mol. Biol. 2008, 448, 213–357. [Google Scholar] [PubMed]
- Cacabelos, R.; Fernández-Novoa, L.; Lombardi, V.; Kubota, Y.; Takeda, M. Molecular genetics of Alzheimer’s disease and aging. Methods Find. Exp. Clin. Pharmacol. 2005, 27 (Suppl. A), 1–573. [Google Scholar] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Feng, Y.; Jankovic, J.; Wu, Y.C. Epigenetic mechanisms in Parkinson’s disease. J. Neurol. Sci. 2015, 349, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ammal Kaidery, N.; Tarannum, S.; Thomas, B. Epigenetic landscape of Parkinson’s disease: Emerging role in disease mechanisms and therapeutic modalities. Neurotherapeutics 2013, 10, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F. Genetics and epigenetics of Parkinson’s disease. Sci. World J. 2012, 2012, 489830. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; McKnight, A.J.; Craig, D.; O’Neill, F. Epigenome-wide association study for Parkinson’s disease. Neuromol. Med. 2014, 16, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Dumaop, W.; Galasko, D.; Desplats, P. Distinctive patterns of DNA methylation associated with Parkinson disease: Identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 2013, 8, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goeder, M. Alpha-synuclein in Lewi bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Mallory, M.; Sundsmo, M.; Honer, W.; Hansen, L.; Masliah, E. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am. J. Pathol. 1998, 152, 367–372. [Google Scholar] [PubMed]
- Cacabelos, R.; Cacabelos, P.; Torrellas, C.; Tellado, I.; Carril, J.C. Pharmacogenomics of Alzheimer’s disease: Novel therapeutic strategies for drug development. Methods Mol. Biol. 2014, 1175, 323–556. [Google Scholar] [PubMed]
- Cacabelos, R. The path to personalized medicine in mental disorders. In The Handbook of Neuropsychiatric Biomarkers, Endophenotypes and Genes, 1st ed.; Ritsner, M., Ed.; Springer: Dordrecht, The Netherlands, 2011; Volume 4, pp. 3–63. ISBN 978-0-12-811060-7. [Google Scholar]
- Cacabelos, R. Pharmacogenetic basis for therapeutic optimization in Alzheimer’s disease. Mol. Diagn. Ther. 2007, 11, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Pharmacogenomics and therapeutic prospects in dementia. Eur. Arch. Psychiatry Clin. Neurosci. 2008, 258 (Suppl. A), 1–553. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Parkinson’s disease: From pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, J.T.; Tan, M.S.; Jiang, T.; Tan, L. Epigenetic mechanisms in Alzheimer’s disease: Implications for pathogenesis and therapy. Ageing Res. Rev. 2013, 12, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Epigenomic networking in drug development: From pathogenic mechanisms to pharmacogenomics. Drug Dev. Res. 2014, 75, 348–365. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Takae, H.; Miyake, K. Epigenetic mechanisms and therapeutic perspectives for neurodevelopmental disorders. Pharmaceuticals 2012, 5, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Teijido, O.; Cacabelos, R. Interrogating the epigenome to unveil the secrets of neurodegeneration: Promising epigenetic therapies. J. Genom. Med. Pharmacogenom. 2016, 1, 95–150. [Google Scholar]
- Takeda, M.; Martínez, R.; Kudo, T.; Tanaka, T.; Okochi, M.; Tagami, S.; Morihara, T.; Hashimoto, R.; Cacabelos, R. Apolipoprotein E and central nervous system disorders: Reviews of clinical findings. Psychiatry Clin. Neurosci. 2010, 64, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Fernández-Novoa, L.; Martínez-Bouza, R.; McKay, A.; Carril, J.C.; Lombardi, V.; Corzo, L.; Carrera, I.; Tellado, I.; Nebril, L.; et al. Future trends in the pharmacogenomics of brain disorders and dementia: Influence of APOE and CYP2D6 variants. Pharmaceuticals 2010, 3, 3040–3100. [Google Scholar] [CrossRef]
- Cacabelos, R.; Takeda, M. Pharmacogenomics, nutrigenomics and future therapeutics in Alzheimer’s disease. Drugs Future 2006, 31 (Suppl. B), 5–146. [Google Scholar]
- Cacabelos, R.; Martínez, R.; Fernández-Novoa, L.; Carril, J.C.; Lombardi, V.; Carrera, I.; Corzo, L.; Tellado, I.; Lescek, J.; McKay, A.; et al. Genomics of Dementia: APOE- and CYP2D6-Related Pharmacogenetics. Int. J. Alzheimer’s Dis. 2012, 2012, 518901. [Google Scholar]
- Cacabelos, R. World Guide for Drug Use and Pharmacogenomics, 1st ed.; EuroEspes Publishing: Corunna, Spain, 2012; pp. 1–3466. ISBN 978-84-940770-0-5. [Google Scholar]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dang, L.M.; Salvesen, G.S.; Pericak Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–80102. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, J.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Roses, A.D. Pharmacogenetics and drug development: The path to safer and more effective drugs. Nat. Rev. Genet. 2004, 5, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Kása, P.; Rakonczay, Z.; Gulya, K. The cholinergic system in Alzheimer’s disease. Prog. Neurobiol. 1977, 52, 511–535. [Google Scholar] [CrossRef]
- Qizilbash, N.; Birks, J.; López-Arrieta, J.; Lewington, S.; Szeto, S. Tacrine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2000, 2, CD000202. [Google Scholar] [CrossRef] [PubMed]
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Jörg Möbius, H. Memantine in moderater-to-severe Alzheimer’s disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.S.; Dagerman, K.S.; Higgins, J.P.; McShane, R. Lack of evidence for the efficacy of memantine in mild Alzheimer disease. Arch. Neurol. 2011, 68, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Yang, R.; Guo, J.C.; Ren, H.M.; Zha, X.L.; Chen, J.S.; Cai, D.F. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 2007, 18, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Cole, N.B.; Dieuliis, D.; Leo, P.; Mitchell, D.C.; Nussbaum, R.L. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp. Cell Res. 2008, 314, 2076–2089. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 2008, 65, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, C.; Zhu, Y.; Cai, Q.; Chan, P.; Uéda, K.; Yu, S.; Yang, H. Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: An immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res. 2008, 1244, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. α-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: The next (neurode) generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed]
- Elkon, H.; Don, J.; Melamed, E.; Ziv, I.; Shirvan, A.; Offen, D. Mutant and wild-type alpha-synuclein interact with mitochondrial cytochrome C oxidase. J. Mol. Neurosci. 2002, 18, 229–238. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. α-Synuclein Shows High Affinity Interaction with Voltage dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef] [PubMed]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Lardenoije, R.; Latrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.; Mastroeni, D.; Coleman, P.; Lemere, A.A.; Hof, P.R.; van den Hove, D.L.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef] [PubMed]
- Szulwach, K.E.; Jin, P. Integrating DNA methylation dynamics into a framework for understanding epigenetic codes. Bioessays 2014, 36, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Tsai, L.H. The potential of HDAC inhibitors as cognitive enhancers. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Mikaelsson, M.A.; Miller, C.A. The path to epigenetic treatment of memory disorders. Neurobiol. Learn. Mem. 2011, 96, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Van den Hove, D.L.; Kompotis, K.; Lardenoije, R.; Kenis, G.; Mill, J.; Steinbusch, H.W.; Lesch, K.P.; Fitzsimons, C.P.; De Strooper, B.; Rutten, B.P. Epigenetically regulated microRNAs in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H.; Edwards, J.R.; Boulard, M. Notes on the role of dynamic DNA methylation in mammalian development. Proc. Natl. Acad. Sci. USA 2015, 112, 6796–6809. [Google Scholar] [CrossRef] [PubMed]
- Gavery, M.R.; Roberts, S.B. Predominant intragenic methylation is associated with gene expression characteristics in a bivalve mollusc. PeerJ 2013, 1, e215. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Carafa, V.; Benedetti, R.; Altucci, L. Trials with epigenetic drugs: An update. Mol. Oncol. 2012, 6, 657–682. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Oyarzabal, J.; Lucas, M.P.; Franco, R.; García-Osta, A. Epigenetic drugs in Alzheimer’s disease. BioMol. Concepts 2013, 4, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Landgrave-Gómez, J.; Mercado-Gómez, O.; Guevara-Guzmán, R. Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 58. [Google Scholar] [PubMed]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Faltraco, F.; Lista, S.; Garaci, F.G.; Hampel, H. Epigenetic mechanisms in Alzheimer’s disease: State-of-the-art. Eur. J. Neurodegener. Dis. 2012, 1, 1–19. [Google Scholar]
- Lu, H.; Liu, X.; Deng, Y.; Qing, H. DNA methylation, a hand behind neurodegenerative diseases. Front. Aging Neurosci. 2013, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- Lovrečić, L.; Maver, A.; Zadel, M.; Peterlin, B. The Role of Epigenetics in Neurodegenerative Diseases. Chapter 14. 2013. Available online: http://dx.doi.org/10.5772/54744 (accessed on 2 September 2018).
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockeinstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef] [PubMed]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wüllner, U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef] [PubMed]
- O’Suilleabhain, P.E.; Sung, V.; Hernandez, C.; Lacritz, L.; Dewey, B.R.; Bottiglieri, T.; Diaz.Arrastia, R. Elevated plasma homocysteine level in patients with Parkinson disease. Arch. Neurol. 2004, 61, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Brattstrom, L. Plasma homocysteine and MTHFR C677T genotype in levodopa-treated patients with PD. Neurology 2001, 56, 281. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Ladenheim, B.; Cutler, R.G.; Kruman, I.I.; Cadet, J.L.; Mattson, M.P. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. J. Neurochem. 2002, 80, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Rutten, B.P.; Kenis, G.; Peerbooms, O.; Visser, P.J.; Verhey, F.; van Os, J.; Steinbusch, H.W.; van den Hove, D.L. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Prog. Neurobiol. 2010, 90, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Bottiglieri, T.; Godfrey, P.; Flynn, T.; Carney, M.W.; Toone, B.K.; Reynolds, E.H. Cerebrospinal fluid S-adenosyl-methionine in depression and dementia: Effects of treatment with parenteral and oral S-adenosyl-methionine. J. Neurol. Neurosurg. Psychiatry 1990, 53, 1096–1098. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.D.; Smith, D.D.; Kish, S.J. Brain S-adenosylmethionine levels are severely decreased in Alzheimer’s disease. J. Neurochem. 1996, 67, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Serot, J.M.; Christmann, D.; Dubost, T.; Bene, M.C.; Faure, G.C. CSF-folate levels are decreased in late-onset AD patients. J. Neural. Transm. 2001, 108, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F.; Tannorella, P.; Pezzini, I.; Migheli, F.; Ricci, G.; Caldarazzo lenco, E.; Piaceri, I.; Polini, A.; Nacmias, B.; Monzani, F.; et al. Folate, homocysteine, vitamin B12, and polymorphisms of genes participating in one-carbon metabolism in lateonset Alzheimer’s disease patients and healthy controls. Antioxid. Redox Signal. 2012, 17, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.C.; Lemos, R.; Ferreiro, E.; Martins, M.; de Mendonca, A.; Santana, I.; Outeiro, T.F.; Pereira, C.M. Epigenetic regulation of BACE1 in Alzheimer’s disease patients and in transgenic mice. Neuroscience 2012, 220, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Oelze, B.; Schumacher, A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent dysregulation of PS1 and BACE and beta-amyloid production. Mol. Cell. Neurosci. 2005, 28, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Ricceri, L.; D’Anselmi, F.; Coluccia, P.; Calamandrei, G.; Scarpa, S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice. Mol. Cell. Neurosci. 2008, 37, 731–746. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Pasqualato, A.; Fiorenza, M.T.; Cavallaro, R.A.; Scarpa, S. Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol. Aging 2011, 32, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Schadt, A.; Dillmann, U.; Kostopoulos, P.; Fassbender, K.; Herrmann, W. Methylation status and neurodegenerative markers in Parkinson disease. Clin. Chem. 2009, 55, 1852–1860. [Google Scholar] [CrossRef] [PubMed]
- Kalbe, E.; Kessler, J.; Calabrese, P.; Smith, R.; Passmore, A.P.; Brand, M.; Bullock, R. DemTect: A new, sensitive cognitive screening test to support the diagnosis of mild cognitive impairment and early dementia. Int. J. Geriatr. Psychiatry 2004, 19, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of Epigenetically Altered Genes in Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef] [PubMed]
- Paez-Colasante, X.; Figueroa-Romero, C.A.; Sakowski, S.; Goutman, S.A.; Feldman, E.L. Amyotrophic lateral sclerosis: Mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 2015, 11, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2010, 31, 16619–16636. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Yamagata, K.; Hong, K.; Wakayama, T.; Zhang, Y. A role for the elongator complex in zygotic paternal genome demethylation. Nature 2010, 463, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van der Berg, L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Genda, Y.; Ukitsu, M. Reduction with age in methylcytosine in the promoter region −224~−101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res. Mol. Brain Res. 1999, 70, 288–292. [Google Scholar] [CrossRef]
- Brohede, J.; Rinde, M.; Winblad, B.; Graff, C. A DNA methylation study of the amyloid precursor protein gene in several brain regions from patients with familial Alzheimer disease. J. Neurogenet. 2010, 24, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wu, X.; Ren, L.; Liu, G.; Li, L. Epigenetic mechanisms of amyloid-betaproduction in anisomycin-treated SH-SY5Y cells. Neuroscience 2011, 194, 272–281. [Google Scholar] [CrossRef] [PubMed]
- West, R.L.; Lee, J.M.; Maroun, L.E. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J. Mol. Neurosci. 1995, 6, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Barrachina, M.; Ferrer, I. DNA methylation of Alzheimer disease and tautopathy-related genes in postmortem brain. J. Neuropathol. Exp. Neurol. 2009, 68, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Nicolia, V.; Fuso, A.; Cavallaro, R.A.; Di Luzio, A.; Scarpa, S. B vitamin deficiency promotes tau phosphorylation through regulation of GSK3beta and PP2A. J. Alzheimer’s Dis. 2010, 19, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; van den Hove, D.L.; Kenis, G.; Keitel, S.; Hof, P.R.; van Os, J.; Steinbusch, H.W.; Schmitz, C.; Rutten, B.P. Age-related increase in levels of 5-hydroxymethylcytosine in mouse hippocampus is prevented by caloric restriction. Curr. Alzheimer Res. 2012, 9, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mut, J.V.; Aso, E.; Panayotis, N.; Lott, I.; Dierssen, M.; Rabano, A.; Urdinguio, R.G.; Fernandez, A.F.; Astudillo, A.; Martin-Subero, J.I.; et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 2013, 136, 3018–3027. [Google Scholar] [CrossRef] [PubMed]
- Caesar, I.; Gandy, S. Evidence that an APOE 4 ‘double whammy’ increases risk for Alzheimer’s disease. BMC Med. 2012, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Pihlstrøm, L.; Berge, V.; Rengmark, A.; Toft, M. Parkinson’s disease correlates with promoter methylation in the α-synuclein gene. Mov. Disord. 2015, 30, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Iwata, A. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS ONE 2010, 5, e15522. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, S.; Sothern, R.B.; Xu, S.; Chan, P. Expression of clock genes Per1 and Bmal1 in total leukocytes in health and Parkinson’s disease. Eur. J. Neurol. 2010, 17, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Hood, S.; Cassidy, P.; Cossette, M.P.; Weigl, Y.; Verwey, M.; Robisnson, B.; Stewart, J.; Amir, S. Endogenous dopamine regulates the rhythm of expression of the clock protein PER2 in the rat dorsal striatum via daily activation of D2 dopamine receptors. J. Neurosci. 2010, 30, 14046–14058. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Ding, H.; Zheng, Z.; Gu, Z.; Ma, J.; Chen, L.; Chan, P.; Cai, Y. Promoter methylation analysis of seven clock genes in Parkinson’s disease. Neurosci. Lett. 2012, 507, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Van Heesbeen, H.J.; Mesman, S.; Veenvliet, J.V.; Smidt, M.P. Epigenetic mechanisms in the development and maintenance of dopaminergic neurons. Development 2013, 140, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Farrer, I.A.; Cupples, L.A.; Kiely, D.K.; Conneally, P.M.; Myers, R.H. Inverse relationship between age at onset of Huntington disease and paternal age suggests involvement of genetic imprinting. Am. J. Hum. Genet. 1992, 50, 528–535. [Google Scholar] [PubMed]
- Behnkrappa, A.; Doerfler, W. Enzymatic amplification of synthetic oligodeoxyribonucleotides: Implication for triplet repeat expansions in the human genome. Hum. Mutat. 1994, 3, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A.; Mittelman, D.; Wilson, J.H. Genome-wide demethylation destabilizes CTG.CAG trinucleotide repeats in mammalian cells. Hum. Mol. Genet. 2004, 13, 2979–2989. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modification. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.L.; Casaccia, P. Epigenetic mechanisms in multiple sclerosis: Implications for pathogenesis and treatment. Lancet Neurol. 2013, 12, 195–206. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Konsoula, Z.; Barile, F.A. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J. Pharmacol. Toxicol. Methods 2012, 66, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Dai, X.L.; Huang, H.C.; Jiang, Z.F. Targeting HDACs: A promising therapy for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2011, 2011, 143269. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Ann. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Stilling, R.M.; Fischer, A. The role of histone acetylation in age-associated memory impairment and Alzheimer’s disease. Neurobiol. Learn. Mem. 2011, 96, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Schrag, M.; Crofton, A.; Trivedi, R.; Vinters, H.; Kirsch, W. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 2012, 12, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Francis, Y.I.; Fa, M.; Ashraf, H.; Zhang, H.; Staniszewski, A.; Latchman, D.S.; Arancio, O. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2009, 18, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lei, J.X.; Luo, C.; Lan, X.; Chi, L.; Deng, P.; Lei, S.; Ghribi, O.; Liu, Q.Y. Increased EID1 nuclear translocation impairs synaptic plasticity and memory function associated with pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2012, 45, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Maldonado, M.A.; Bokov, A.F.; Majumder, S.; Oddo, S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 22687–22692. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, R.; O’Brien, L.M.; Ahmad, S.T. Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson’s disease. Neuroscience 2013, 246, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Quintas, A.; de Solis, A.J.; Diez-Guerra, F.J.; Carrascosa, J.M.; Bogonez, E. Age-associated decrease of SIRT1 expression in rat hippocampus: Prevention by late onset caloric restriction. Exp. Gerontol. 2012, 47, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Sommer, M.; Poliak, N.; Upadhyay, S.; Ratovitski, E.; Nelkin, B.D.; Donehower, L.A.; Sidransky, D. DeltaNp63alpha overexpression induces downregulation of Sirt1 and an accelerated aging phenotype in the mouse. Cell Cycle 2006, 5, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Maier, B.; Bartke, A.; Scrable, H. Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell 2006, 5, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012, 483, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Balazs, R. Epigenetic mechanisms in Alzheimer’s disease. Degener. Neurol. Neuromuscul. Dis. 2014, 4, 85–102. [Google Scholar]
- Bannister, A.J.; Kouzarides, T. Reversing histone methylation. Nature 2005, 436, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Huyen, Y.; Zgheib, O.; Ditullio, R.A., Jr.; Gorgoulis, V.G.; Zacharatos, P.; Petty, T.J.; Sheston, E.A.; Mellert, H.S.; Stavridi, E.S.; Halazonetis, T.D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004, 432, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Lithner, C.U.; Lacor, P.N.; Zhao, W.Q.; Mustafiz, T.; Klein, W.L.; Sweatt, J.D.; Hernandez, C.M. Disruption of neocortical histone H3 homeostasis by soluble Abeta: Implications for Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A. Targeting histone-modifications in Alzheimer’s disease. What is the evidence that this is a promising therapeutic avenue? Neuropharmacology 2014, 80, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, T.; Li, J.; Yang, J.; Liu, H.; Zhang, X.J.; Le, W. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 2014, 21, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, A.P.; Lubin, F.D.; Hallett, P.J.; Vattem, P.; Ravenscroft, P.; Bezard, E.; Zhou, S.; Fox, S.H.; Brotchie, J.M.; Sweatt, J.D.; et al. Striatal histone modifications in models of levodopa-induced dyskinesia. J. Neurochem. 2008, 106, 486–494. [Google Scholar] [CrossRef] [PubMed]
- McCord, R.A.; Broccoli, D. Telomeric chromatin: Roles in aging, cancer and hereditary disease. Mutat. Res. 2008, 647, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.; Mitchell, J.R. Telomerase in the human organism. Oncogene 2002, 21, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Perrod, S.; Gasser, S.M. Long-range silencing and position effects at telomeres and centromeres: Parallels and differences. Cell. Mol. Life Sci. 2003, 60, 2303–2318. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. The epigenetic regulation of mammalian telomeres. Nat. Rev. Genet. 2007, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Benetti, R.; Garcia-Cao, M.; Blasco, M.A. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat. Genet. 2007, 39, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Jaco, I.; Fraga, M.F.; Chen, T.; Li, E.; Esteller, M.; Blasco, M.A. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat. Cell Biol. 2006, 8, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 2007, 318, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Luke, B.; Lingner, J. TERRA: Telomeric repeat-containing RNA. EMBO J. 2009, 28, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Schoeftner, S.; Blasco, M.A. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 2008, 10, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Nunbhakdi-Craig, V.; Sontag, J.M.; Diaz-Arrastia, R.; Ogris, E.; Dayal, S.; Lentz, S.R.; Arning, E.; Bottiglieri, T. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J. Neurosci. 2007, 27, 2751–2759. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Refsum, H.; Bottiglieri, T.; Fenech, M.; Hooshmand, B.; McCaddon, A.; Miller, J.W.; Rosenberg, I.H.; Obeid, R. Homocysteine and dementia: An international consensus statement. J. Alzheimer’s Dis. 2018, 62, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.L.; Hardy, T.M.; Tollefsbol, T.O. Medicinal chemistry of the epigenetic diet and caloric restriction. Curr. Med. Chem. 2013, 20, 4050–4059. [Google Scholar] [CrossRef] [PubMed]
- Dauncey, M.J. Genomic and epigenomic insights into nutrition and brain disorders. Nutrients 2013, 5, 887–914. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, H.; Jin, P. Epigenetics-Based Therapeutics for Neurodegenerative Disorders. Curr. Transl. Geriatr. Exp. Gerontol. Rep. 2012, 1, 229–236. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. US National Institutes of Health. Available online: http://clinicaltrials.gov/ (accessed on 1 August 2018).
- Aisen, P.S.; Schneider, L.S.; Sano, M.; Diaz-Arrastia, R.; van Dyck, C.H.; Weiner, M.F.; Bottiglieri, T.; Jin, S.; Stokes, K.T.; Thomas, R.G.; et al. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA 2008, 300, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Doshanjh, L.; Fishman, P.; Luo, Y.; Smyers, K.; Page, R.; Morrell, C.; et al. A Phase II Randomized Clinical Trial of a Nutritional Formulation for Cognition and Mood in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Page, R.; Morrell, C.; Shea, T.B. Maintenance of Cognitive Performance and Mood for Individuals with Alzheimer’s Disease Following Consumption of a Nutraceutical Formulation: A One-Year, Open-Label Study. J. Alzheimer’s Dis. 2016, 51, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Lökk, J. B-vitaminer kan prövas vid kognitiv svikt. Lakartidningen 2013, 110, 1528. [Google Scholar]
- Tsiachristas, A.; Smith, A.D. B-vitamins are potentially a cost-effective population health strategy to tackle dementia: Too good to be true? Alzheimers Dement. 2016, 2, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Whalley, L.J.; Duthie, S.J.; Collins, A.R.; Starr, J.M.; Deary, I.J.; Lemmon, H.; Duthie, A.C.; Murray, A.D.; Staff, R.T. Homocysteine, antioxidant micronutrients and late onset dementia. Eur. J. Nutr. 2014, 53, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Lötsch, J.; Schneider, G.; Reker, D.; Parnham, M.J.; Schneider, P.; Geisslinger, G.; Doehring, A. Common non-epigenetic drugs as epigenetic modulators. Trends Mol. Med. 2013, 19, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Garcia-Manero, G.; Giles, F.J.; Mannari, R.; Thomas, D.; Faderl, S.; Bayar, E.; Lyons, J.; Rosenfeld, C.S.; Cortes, J.; et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004, 103, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L.; Bouffard, D.Y.; Momparler, L.F.; Dionne, J.; Belanger, K.; Ayoub, J. Pilot phase I-II study on 5-aza-2’-deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anticancer Drugs 1997, 8, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J. Natural compounds may open new routes to treatment of amyloid diseases. Neurotherapeutics 2013, 10, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Smith, A.; Lin, X.; Yuan, F.; Copes, N.; Delic, V.; Tan, J.; Cao, C.; Shytle, R.D.; Bradshaw, P.C. Green tea epigallocatechin-3-gallate (EGCG) and other flavonoids reduce Alzheimer’s amyloid-induced mitochondrial dysfunction. J. Alzheimer’s Dis. 2011, 26, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, M.; Lu, F.; Luo, N.; He, Z.P.; Yang, H. Involvement of alpha7 nAChR signaling cascade in epigallocatechin gallate suppression of beta-amyloid-induced apoptotic cortical neuronal insults. Mol. Neurobiol. 2014, 49, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.; Püntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E. Perispinal etanercept for treatment of Alzheimer’s disease. Curr. Alzheimer Res. 2007, 4, 550–552. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sananbenesi, F.; Wang, X.; Dobbin, M.; Tsai, L.H. Recovery of learning and memory is associated with chromatin remodelling. Nature 2007, 447, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.K.; Schneider, J.S. Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1;2;3;6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 2011, 194, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.S.; Wang, C.C.; Bortner, C.D.; Peng, G.S.; Wu, X.; Pang, H.; Lu, R.B.; Gean, P.W.; Chuang, D.M.; Hong, J.S. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience 2007, 149, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.S.; Li, G.; Tzeng, N.S.; Chen, P.S.; Chuang, D.M.; Hsu, Y.D.; Yang, S.; Hong, J.S. Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: Role of microglia. Brain Res. Mol. Brain Res. 2005, 134, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Monti, B.; Gatta, V.; Piretti, F.; Raffaelli, S.S.; Virgili, M.; Contestabile, A. Valproic acid is neuroprotective in the rotenone rat model of Parkinson’s disease: Involvement of alpha-synuclein. Neurotox. Res. 2010, 17, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Marinova, Z.; Ren, M.; Wendland, J.R.; Leng, Y.; Liang, M.H.; Yasuda, S.; Leeds, P.; Chuang, D.M. Valproic acid induces functional heat-shock protein 70 via class I histone deacetylase inhibition in cortical neurons: A potential role of Sp1 acetylation. J. Neurochem. 2009, 111, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.S.; Peng, G.S.; Li, G.; Yang, S.; Wu, X.; Wang, C.C.; Wilson, B.; Lu, R.B.; Gean, P.W.; Cuang, D.M.; et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurothrophic factors from astrocytes. Mol. Psychiatry 2006, 11, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, P.S.; Dallas, S.; Wilson, B.; Block, M.L.; Wang, C.C.; Kinyamu, H.; Lu, N.; Gao, X.; Leng, Y.; et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int. J. Neuropsychopharmacol. 2008, 11, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.; Brazier, M.; Cerutti, I.; Pieri, F.; Tardivel, I.; Desmet, G.; Baillet, J.; Chany, C. Pharmacokinetic study of butyric acid administered in vivo as sodium and arginine butyrate salts. Clin. Chim. Acta 1989, 181, 255–263. [Google Scholar] [CrossRef]
- Egorin, M.J.; Yuan, Z.M.; Sentz, D.L.; Plaisance, K.; Eiseman, J.L. Plasma pharmacokinetics of butyrate after intravenous administration of sodium butyrate or oral administration of tributyrin or sodium butyrate to mice and rats. Cancer Chemother. Pharmacol. 1999, 43, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Kurschel, E.; Osieka, R.; Schmidt, C.G. Clinical pharmacology of sodium butyrate in patients with acute leukemia. Eur. J. Cancer Clin. Oncol. 1987, 23, 1283–1287. [Google Scholar] [CrossRef]
- Zhou, W.; Bercury, K.; Cummiskey, J.; Luong, N.; Lebin, J.; Freed, C.R. Phenylbutyrate-up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J. Biol. Chem. 2011, 286, 14941–14951. [Google Scholar] [CrossRef] [PubMed]
- Rane, P.; Shields, J.; Heffernan, M.; Guo, Y.; Akbarian, S.; King, J.A. The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD. Neuropharmacology 2012, 62, 2409–2412. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.G. Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J. Periodontol. 2008, 79, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Gardian, G.; Yang, L.; Cleren, C.; Calingasan, N.Y.; Klivenyi, P.; Beal, M.F. Neuroprotective effects of phenylbutyrate against MPTP neurotoxicity. Neuromol. Med. 2004, 5, 235–241. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.L.; Wang, S.S.; Yang, Y.Y.; Yuang, R.Y.; Chen, R.M.; Hu, C.J. The epigenetic effects of amyloid-beta(1-40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem. Biophys. Res. Commun. 2009, 378, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Rephaeli, A.; Zhuk, R.; Nudelman, A. Prodrugs of butyric acid from bench to bedside: Synthetic design, mechanisms of action, and clinical applications. Drug Dev. Res. 2000, 50, 379–391. [Google Scholar] [CrossRef]
- Tariot, P.N.; Schneider, L.S.; Cummings, J.; Thomas, R.G.; Raman, R.; Jakimovich, L.J.; Loy, R.; Bartocci, B.; Fleisher, A.; Ismail, M.S.; et al. Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch. Gen. Psychiatry 2011, 68, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D. Pharmacotherapy for Alzheimer’s disease: 2002. Clin. Neuropharmacol. 2003, 26, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [PubMed]
- Bollati, V.; Galimberti, D.; Pergoli, L.; Dalla Valle, E.; Barretta, F.; Cortini, F.; Scarpini, E.; Bertazzi, P.A.; Baccarelli, A. DNA methylation in repetitive elements and Alzheimer disease. Brain Behav. Immun. 2011, 25, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, K.D.; Connor, C.M.; Campan, M.; Long, T.; Weisenberg, D.J.; Biniszkiewicz, D.; Jaenisch, R.; Laird, P.W.; Akbarian, S. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS ONE 2007, 2, e895. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, J.; Hansmannel, F.; Gistelinck, M.; Mounier, A.; Van Cauwenberghe, C.; Kolen, K.V.; Geller, F.; Sottejeau, Y.; Harold, D.; Dourlen, P.; et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 2013, 18, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.K.; Schneider, J.S. Protection of dopaminergic cells from MPP+-mediated toxicity by histone deacetylase inhibition. Brain Res. 2010, 1354, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Dzitoyeva, S.; Manev, H. Effect of valproic acid on mitochondrial epigenetics. Eur. J. Pharmacol. 2012, 690, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schluesener, Y.H.J. Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinflammation and cerebral amyloidosis and improves behavior in a mouse model. J. Neuropathol. Exp. Neurol. 2013, 72, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.M.; Lee, T.; Yoon, H.; DiBattista, A.M.; Song, J.; Sohn, Y.; Moffat, E.I.; Turner, R.S.; Jung, M.; Kim, J.; et al. Mercaptoacetamide-based class II HDAC inhibitor lowers Aβ levels and improves learning and memory in a mouse model of Alzheimer’s disease. Exp. Neurol. 2013, 239, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, J.V.W. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.C.; Hong, R.; Schreiber, S.L. Structural basis for in-cell histone deacetylase paralog selectivity. J. Am. Chem. Soc. 2003, 125, 5586–5587. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.W.; Chang, P.T.; Hsin, L.W.; Chern, J.W. Quinazolin-4-one derivatives as selective histone deacetylase-6 inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2013, 56, 6775–6791. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.T.; Smith, B.C.; Jackson, M.D.; Denu, J.M. Coenzyme specificity of Sir2 protein deacetylases: Implications for physiological regulation. J. Biol. Chem. 2004, 279, 40122–40129. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Steffan, J.S.; Martinez-Coria, H.; Sun, X.; Schreiber, S.S.; Thompson, L.M.; LaFerla, F.M. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 2008, 28, 11500–11510. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattsson, M.P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wales, P.; Quinti, L.; Zuo, F.; Moniot, S.; Herisson, F.; Rauf, N.A.; Wang, H.; Silverman, R.B.; Ayata, C.; et al. The sirtuin-2 inhibitor AK7 is neuroprotective in models of Parkinson’s disease but not amyotrophic lateral sclerosis and cerebral ischemia. PLoS ONE 2015, 10, e0116919. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.; Quinti, L.; Kim, J.; Vollor, L.; Narayanan, K.L.; Edgerly, C.; Cipicchio, P.M.; Lauver, M.A.; Choi, S.H.; Silverman, R.B.; et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2012, 2, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Oláh, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef] [PubMed]
- Alcendor, R.R.; Kirshenbaum, L.A.; Imai, S.; Vatner, S.F.; Sadoshima, J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ. Res. 2004, 95, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased nuclear NAD biosynthesis and SirT1 activation prevent axonal degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Napper, A.D.; Hixon, J.; McDonagh, T.; Keavey, K.; Pons, J.F.; Barker, J.; Yau, W.T.; Amouzegh, P.; Flegg, A.; Hamelin, E.; et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 2005, 48, 8045–8054. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Fischer, F.; Nguyen, G.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, E2772–E2781. [Google Scholar] [CrossRef] [PubMed]
- Sussmuth, S.D.; Haider, S.; Landwehrmeyer, G.B.; Farmer, R.; Frost, C.; Tripepi, G.; Andersen, C.A.; Di Bacco, M.; Lamanna, C.; Diodato, E.; et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br. J. Clin. Pharmacol. 2015, 79, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef] [PubMed]
- Donmez, G.; Wang, D.; Cohen, D.E.; Guarente, L. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell 2010, 142, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Bordone, L.; Guarente, L. Calorie restriction, SIRT1 and metabolism: Understanding longevity. Nat. Rev. Mol. Cell Biol. 2005, 6, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Granzotto, A.; Zatta, P. Resveratrol acts not through anti-aggregative pathways but mainly via its scavenging properties against Abeta and Abeta-metal complexes toxicity. PLoS ONE 2011, 6, e21565. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Liang, N.; Zhu, D.; Gao, Q.; Peng, L.; Dong, H.; Yue, Q.; Liu, H.; Bao, L.; Zhang, J.; et al. Resveratrol inhibits beta-amyloid-induced neuronal apoptosis through regulation of SIRT1-ROCK1 signaling pathway. PLoS ONE 2013, 8, e59888. [Google Scholar]
- Zhao, Y.Q.; Jordan, I.K.; Lunyak, V.V. Epigenetics components of aging in the central nervous system. Neurotherapeutics 2013, 10, 647–663. [Google Scholar] [CrossRef] [PubMed]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Tuerner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Flores Guzmán, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Alcaín, F.J.; Villalba, J.M. Sirtuin activators. Expert Opin. Ther. Pat. 2009, 19, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.J.; Dragunow, M. High content analysis of histone acetylation in human cells and tissues. J. Neurosci. Methods 2010, 193, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Witkin, J.M.; Li, X. Curcumin, an active constituent of the ancient medicinal herb Curcuma longa L.: Some uses and the establishment and biological basis of medical efficacy. CNS Neurol. Disord. Drug Targets 2013, 12, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Sood, P.K.; Nahar, U.; Nehru, B. Curcumin attenuates aluminum-induced oxidative stress and mitocondrial dysfunction in rat brain. Neurotox. Res. 2011, 20, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, J.B.; Coradini, K.; Frozza, R.L.; Oliveira, C.M.; Meneghetti, A.B.; Bernardi, A.; Pires, E.S.; Beck, R.C.; Salbego, C.G. Free and nanoencapsulated curcumin suppress beta-amyloid –induced cognitive impairments in rats: Involvement of BDNF and Akt/GSK-3beta signaling pathway. Neurobiol. Learn. Mem. 2013, 106, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Gilani, A.H. Therapeutic potential of turmeric in Alzheimer’s disease: Curcumin or curcuminoids? Phytother. Res. 2013, 28, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, N.; Takahashi, Y.; Amakusa, Y.; Tanno, Y.; Tuji, Y.; Niwa, H.; Murakami, N.; Krishna, U.K. Effects of turmeric on Alzheimer’s disease with behavioral and psychological symptoms of dementia. Ayu 2012, 33, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Cheung, S.K.; Mok, V.C.; Lam, L.C.; Leung, V.P.; Hui, E.; Ng, C.C.; Chow, M.; Ho, P.C.; Lam, S.; et al. Curcumin effects on blood lipid profile in a 6-month human study. Pharmacol. Res. 2007, 56, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Lam, C.W.; Cheung, S.K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention Website. CDC Features: Alzheimer’s Disease. Available online: http://www.cdc.gov/Features/Alzheimers/ (accessed on 4 October 2018).
- Begum, A.N.; Jones, M.R.; Lim, G.P.; Morihara, T.; Kim, P.; Heath, D.D.; Rock, C.L.; Pruitt, M.A.; Yang, F.; Hudspeth, B.; et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2008, 326, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Peedicayil, J. Epigenetic drugs in cognitive disorders. Curr. Pharm. Des. 2014, 20, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Forneris, F.; Binda, C.; Vanoni, M.A.; Battaglioli, E.; Mattevi, A. Human histone demethylase LSD1 reads the histone code. J. Biol. Chem. 2005, 280, 41360–41365. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Schmidt, D.M.; McCafferty, D.G.; Shiekhattar, R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 2006, 13, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.M.; McCafferty, D.G. Trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–4416. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid- production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.R.; Wang, J.; Zhang, X.B.; Geng, Y.; Hu, Z.Y.; Rudd, J.A.; Ling, S.; Chen, W.; Han, S. The miR-124 regulates the expression of BACE1/beta-secretase correlated with cell death in Alzheimer’s disease. Toxicol. Lett. 2012, 209, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Li, L.; Huang, C.; Li, X.; Peng, Y.; Li, J. MicroRNA-323-3p with clinical potential in rheumatoid arthritis, Alzheimer’s disease and ectopic pregnancy. Expert Opin. Ther. Targets 2014, 18, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Sen, D. MicroRNAs in Parkinson’s disease. Exp. Brain Res. 2017, 235, 2359–2374. [Google Scholar] [CrossRef] [PubMed]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Mouradian, M.M.; Junn, E. Inhibition of miR-34b and miR-34c enhances α synuclein expression in Parkinson’s disease. FEBS Lett. 2015, 589, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Gillardon, F.; Mack, M.; Rist, W.; Schnack, C.; Lenter, M.; Hildebrandt, T.; Hengerer, B. MicroRNA and proteome expression profiling in early-symptomatic α-synuclein(A30P)-transgenic mice. Proteom. Clin. Appl. 2008, 2, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Wanet, A.; Tacheny, A.; Arnould, T.; Renard, P. miR-212/132 expression and functions: Within and beyond the neuronal compartment. Nucleic Acids Res. 2012, 40, 4742–4753. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Li, T.; Wang, Y.; Tang, Y.; Cui, H.; Tang, Y.; Zhang, X.; Chen, D.; Shen, N.; Le, W. miR-132 regulates the differentiation of dopamine neurons by directly targeting Nurr1 expression. J. Cell. Sci. 2012, 125, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Li, Y.; Wang, C.; Xu, F.; Wang, M.; Liu, Y. Serum miR-221 serves as a biomarker for Parkinson’s disease. Cell Biochem. Funct. 2016, 34, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, J.; Wu, M.; Hu, J.M. Protective role of microRNA-221 in Parkinson’s disease. Bratisl. Lek. Listy 2018, 119, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Sun, H. MiR-26a promotes neurite outgrowth by repressing PTEN expression. Mol. Med. Rep. 2013, 8, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.E.; Lee, P.R.; Chen, S.; Li, W.; Fields, R.D. MicroRNA regulation of homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 11650–11655. [Google Scholar] [CrossRef] [PubMed]
- Varela, M.A.; Roberts, T.C.; Wood, M.J. Epigenetics and ncRNAs in brain function and disease: Mechanisms and prospects for therapy. Neurotherapeutics 2013, 10, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Helin, K.; Dhanak, D. Chromatin proteins and modifications as drug targets. Nature 2013, 502, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, T.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C.; Teijido, O.; Carril, J.C. Pharmacogenetic considerations in the treatment of Alzheimer’s disease. Pharmacogenomics 2016, 17, 1041–1074. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C. Epigenetics of Aging and Alzheimer’s Disease: Implications for Pharmacogenomics and Drug Response. Int. J. Mol. Sci. 2015, 16, 30483–30543. [Google Scholar] [CrossRef] [PubMed]
- Riedl, A.G.; Watts, P.M.; Jenner, P.; Marsden, C.D. P450 enzymes and Parkinson’s disease: The story so far. Mov. Disord. 1998, 13, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Shahabi, H.N.; Westberg, L.; Melke, J.; Håkansson, A.; Belin, A.C.; Sydow, O.; Olson, L.; Holmberg, B.; Nissbrandt, H. Cytochrome P450 2E1 gene polymorphisms/haplotypes and Parkinson’s disease in a Swedish population. J. Neural. Transm. 2009, 116, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Kessova, I.; Cederbaum, A.I. CYP2E1: Biochemistry, toxicology, regulation and function in ethanol-induced liver injury. Curr. Mol. Med. 2003, 3, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Watts, P.M.; Riedl, A.G.; Douek, D.C.; Edwards, R.J.; Boobis, A.R.; Jenner, P.; Marsden, C.D. Co-localization of P450 enzymes in the rat substantia nigra with tyrosine hydroxylase. Neuroscience 1998, 86, 511–519. [Google Scholar] [CrossRef]
- Howard, L.A.; Miksys, S.; Hoffmann, E.; Mash, D.; Tyndale, R.F. Brain CYP2E1 is induced by nicotine and ethanol in rat and is higher in smokers and alcoholics. Br. J. Pharmacol. 2003, 138, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Shahabi, H.N.; Andersson, D.R.; Nissbrandt, H. Cytochrome P450 2E1 in the substantia nigra: Relevance for dopaminergic neurotransmission and free radical production. Synapse 2008, 62, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Kaut, O.; Schmitt, I.; Wüllner, U. Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics 2012, 13, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Roth, R. Human Body Index—Transcriptional Profiling; Gene Expression Omnibus (GEO) NCBI 2007, Series GSE7307; Neurocrine Biosciences, Inc.: San Diego, CA, USA, 2007. [Google Scholar]
- Lewandowski, N.M.; Ju, S.; Verbitsky, M.; Ross, B.; Geddie, M.L.; Rockenstein, E.; Adame, A.; Muhammad, A.; Vonsattel, J.P.; Ringe, D.; et al. Polyamine pathway contributes to the pathogenesis of Parkinson disease. Proc. Natl. Acad. Sci. USA 2010, 107, 16970–16975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; James, M.; Middleton, F.A.; Davis, R.L. Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 137B, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52–73. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chibnik, L.B.; Srivastava, G.P.; Pochet, N.; Yang, J.; Xu, J.; Kozubek, J.; Obholzer, N.; Leurgans, S.E.; Schneider, J.A.; et al. Association of Brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. 2015, 72, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, R.; van der Brug, M.; Vandrovcova, J.; Ding, J.; Sharma, S.; Renton, A.; Dillman, A.; Lees, A.; Cookson, M.R.; Bandopadhyay, R. Gene Expression Changes across Multiple Regions of the Parkinson’s Disease Brain; Gene Expression Omnibus (GEO) NCBI 2011, Series GSE28894; NIA, NIH: Bethesda, MD, USA, 2011. [Google Scholar]
- Moran, L.B.; Duke, D.C.; Deprez, M.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Whole genome expression profiling of the medial and lateral substantia nigra in Parkinson’s disease. Neurogenetics 2006, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Yoshino, Y.; Mori, T.; Yoshida, T.; Ozaki, Y.; Sao, T.; Mori, Y.; Ochi, S.; Iga, J.I.; Ueno, S.I. Gene Expression and Methylation Analysis of ABCA7 in Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; López-Muñoz, F. The ABCB1 transporter in Alzheimer’s disease. Clin. Exp. Pharmacol. 2014, 4, e128. [Google Scholar] [CrossRef]
- Abuznait, A.H.; Kaddoumi, A. Role of ABC transporters in the pathogenesis of Alzheimer’s disease. ACS Chem. Neurosci. 2012, 3, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Bauer, B.; Hartz, A.M. ABC transporters and the Alzheimer’s disease enigma. Front. Psychiatry 2012, 3, 54. [Google Scholar] [CrossRef] [PubMed]
- Qosa, H.; Abuznait, A.H.; Hill, R.A.; Kaddoumi, A. Enhanced brain amyloid-β clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 31, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Van Assema, D.M.; Lubberink, M.; Bauer, M.; van der Flier, W.M.; Schuit, R.C.; Windhorst, A.D.; Comans, E.F.; Hoetjes, N.J.; Tolboom, N.; Langer, O.; et al. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain 2012, 135, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, G.D.; Messier, A.A.; Miller, M.C.; Machan, J.T.; Majmudar, S.S.; Stopa, E.G.; Donahue, J.E.; Johanson, C.E. Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. J. Neuropathol. Exp. Neurol. 2010, 69, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.; Fitz, N.F.; Lefterov, I. The role of ATP-binding cassette transporter A1 in Alzheimer’s disease and neurodegeneration. Biochim. Biophys. Acta 2010, 1801, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Davis, W., Jr. The ATP-binding cassette transporter-2 (ABCA2) regulates esterification of plasma membrane cholesterol by modulation of sphingolipid metabolism. Biochim. Biophys. Acta 2014, 1841, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Fernández-Hernando, C. MicroRNAs: A connection between cholesterol metabolism and neurodegeneration. Neurobiol. Dis. 2014, 72 Pt A, 48–53. [Google Scholar] [CrossRef]
- Chan, S.L.; Kim, W.S.; Kwok, J.B.; Hill, A.F.; Cappai, R.; Rye, K.A.; Garner, B. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J. Neurochem. 2008, 106, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Li, H.; Ruberu, K.; Chan, S.; Elliott, D.A.; Low, J.K.; Cheng, D.; Karl, T.; Garner, B. Deletion of Abca7 increases cerebral amyloid-β accumulation in the J20 mouse model of Alzheimer’s disease. J. Neurosci. 2013, 33, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Duffy, D.L.; Zhu, G.; Liu, J.Z.; Macgregor, S.; McRae, A.F.; Wright, M.J.; Sturm, R.A.; Mackey, D.A.; Montgomery, G.W.; et al. GWAS findings for human iris patterns: Associations with variants in genes that influence normal neuronal pattern development. Am. J. Hum. Genet. 2011, 89, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Kajiho, H.; Saito, K.; Tsujita, K.; Kontani, K.; Araki, Y.; Kurosu, H.; Katada, T. RIN3: A novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J. Cell Sci. 2003, 116, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Lakhal, S.; Andaloussi, S.E.; O’Loughlin, A.J.; Li, J.; Wood, M.J.A. RNAi therapeutic delivery by exosomes. In RNA Interference from Biology to Therapeutics, 1st ed.; Howard, K.N., Ed.; Springer: London, UK, 2013; pp. 185–207. ISBN 978-1-4614-4743-6. [Google Scholar]
- Kuhn, D.E.; Nuovo, G.J.; Terry, A.V., Jr.; Martin, M.M.; Malana, G.E.; Sansom, S.E.; Pleister, A.P.; Beck, W.D.; Head, E.; Feldman, D.S.; et al. Chromosome 21-derived microRNAs provide an etiological basis for aberrant protein expression in human Down Syndrome brains. J. Biol. Chem. 2013, 288, 4228. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Mourdian, M.M. MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol. Ther. 2012, 133, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Fernández-Novoa, L.; Alejo, R.; Corzo, L.; Alcaraz, M.; Nebril, L.; Cacabelos, P.; Fraile, C.; Carrera, I.; Carril, J.C. E-PodoFavalin-15999 (Atremorine®)-induced dopamine response in Parkinson’s Disease: Pharmacogenetics-related effects. J. Genom. Med. Pharmacogenom. 2016, 1, 1–26. [Google Scholar]
- Cacabelos, R.; Fernández-Novoa, L.; Alejo, R.; Corzo, L.; Rodríguez, S.; Alcaraz, M.; Cacabelos, P.; Fraile, C.; Carrera, I.; Carril, J.C. E-PodoFavalin-15999 (Atremorine®)-induced neurotransmitter and hormonal response in Parkinson’s Disease. J. Exp. Res. Pharmacol. 2016, 1, 1–12. [Google Scholar]
- Cacabelos, R.; Carrera, I.; Fernández-Novoa, L.; Alejo, R.; Corzo, L.; Rodríguez, S.; Alcaraz, M.; Nebril, L.; Casas, A.; Fraile, C.; et al. Parkinson’s Disease: New solutions to old problems. EuroEspes J. 2017, 11, 74–96. [Google Scholar]
- Carrera, I.; Fernández-Novoa, L.; Sampedro, C.; Cacabelos, R. Neuroprotective effect of atremorine in an experimental model of Parkinson’s disease. Curr. Pharm. Des. 2017, 23, 2673–2684. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Bioactive Extract Obtained from Vicia Faba and Its Use in the Treatment and/or Prevention of Neurodegenerative Diseases. Eur. Pat. EP16382138, 29 March 2016. [Google Scholar]

{kind=link}
| Drug | Compound | Pharmacogenetics | Mechanisms of action | ClinicalTrials.gov ID |
|---|---|---|---|---|
 | Name: Vitamin B9; Folic acid; Folate; 59-30-3; Folacin; Pteroylglutamic acid IUPAC name: (2S)-2-[[4-[(2-amino-4-oxo-1H-pteridin-6-yl)methylamino]benzoyl]amino]pentanedioic acid Molecular formula: C19H19N7O6 Molecular Weight: 441.40 g/mol Category: SAMe methyl donors Targets: SAMe | Pathogenic genes: ADORA2A, AOX1, APOB, CDKN2A, COMPT Mechanistic genes: ALDH1A1, GSTA1, GSTP1, IL2, IL6, NAT2, SOD3, TNF, VGFA Metabolic genes: Substrate:ABCG2, MTHFR Inhibitor:ABCB1, ERCC2, MTHFR Inducer:CYP2C9 Transporter genes: ABCC1, ABCC2, ABCC3, SLC19A1, SLC22A8, SLC28A2, SLCOB1 Pleiotropic genes: PPAR, TNF, TP53, VCAM1 |
| NCT00056225-Phase III NCT01320527-Phase II NCT02457507-Phase IV |
 | Name: EGCG, (−)-epigallocatechin gallate, epigallocatechin 3-gallate, tea catechin, teavigo, catechin deriv., 989-51-5 IUPAC name: [(2R,3R)-5,7-dihydroxy-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-chromen-3-yl] 3,4,5-trihydroxybenzoate Molecular formula: C22H18O11 Molecular Weight: 458.37 g/mol Category: DNMT inhibitors Targets: DNMT1 | Pathogenic genes: APP, BACE1, CDX2, EGFR, FAS PIK3CA, ROS1 Mechanistic genes: APP, BACE1, BMP2, CDX2, CHRNA7, ECEs, EGFR, IRS1, PIK3CA, ROS1 Metabolic genes: Inhibitor:SOD Transporter genes: CD36, SLC5A1, SLC27A4, SLCO1B1, SLCO1B3 Pleiotropic genes: ACACA, CHRNA7, SCD |
| NTC00951834-Phases II, III |
 | Name: Quercetin; Sophoretin; Quercetol; Meletin; Xanthaurine; Quercitin; 3,3′,4′,5,7-Pentahydroxyflavone IUPAC name: 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxychromen-4-one Molecular formula: C15H10O7 Molecular Weight: 302.24 g/mol Category: DNMT inhibitors Targets: DNMT1 | Pathogenic genes: IL1R, NFkB, Ccl8, IKK, STAT3, CD4, CDK2, IL2 Mechanistic genes: MTND4, CDKN2A, PRDX4, DIO2, HSD17B1, MSH2, GSS, COMT, FOS, CRP, NR1I3, PON1 Metabolic genes: Substrate:UGT1A1, UGT1A3, GSTT1, CYP2J2, GSTK1, CYP2C8, CYP1A1, CYP1A2, CYP1B1, GSTA1, CYP19A1 Inhibitor:SULT1E1 Transporter genes: ABCB1, ABCG2 |
| NCT01716637-Phase I |
 | Name: Valproic acid, 2-propylpentanoic acid, depakene, depakine, ergenyl, dipropylacetic acid, mylproin, convulex, myproic acid IUPAC name: 2-propylpentanoic acid Molecular formula: C8H16O2 Molecular Weight: 144.21 g/mol Category: HDAC inhibitors Targets: Class I HDAC; Class II HDAC | Pathogenic genes: CREB1, IL6, LEP, SCN2A, TGFB1, TNF, TRNK Mechanistic genes: ABAT, CDK5, GSK3B, HDAC1, HDAC2, HDAC3, HDAC8, HDAC9, LEP, LEPR, SCNs, SMN2 Metabolic genes: Substrate:CYP2A6 (major), CYP2C9 (major), CYP4B1 (major), CYP1A1 (minor), CYP2B6 (minor), CYP2C19 (minor), CYP2E1 (minor), CYP3A4 (minor), CYP4F2 (minor), ABCB1 (minor), UGT1A4, UGT1A6, UGT1A8, UGT1A10, UGT2B7 Inhibitor: ABCB1, ACADSB, AKR1A1, CYP2C9 (strong), CYP2A6 (moderate), CYP2C19 (moderate), CYP3A4 (moderate), CYP2D6 (weak), HDAC1, HDAC2, HDAC3, HDAC8, HDAC9, UGT1A9, UGT2B1, UGT2B7 Inducer: ABCB1, AKR1C4, CASR, CYP2A6, CYP2B6, CYP3A4, CYP7A1, MAOA, NR1I2, SLC5A5, SLC6A2, SLC12A3, SLC22A16 Transporter genes: ABCB1, ABCC2, ABCG1, ABCG2, SCNs, SLC5A5, SLC6A2, SLC12A3, SLC22A16 Pleiotropic genes: ABL2, AGPAT2, ASL, ASS1, CDK4, CHRNA1, COL1A1, CPS1, CPT1A, DRD4, FMR1, FOS, HBB, HFE, HLA-A, HLA-B, ICAM1, IFNG, IL6, IL10, LEPR, NAGS, NR3C1, OTC, PTGES, STAT3, TGFB1, TNF, TP53 |
| NTC01729598-Phase I NTC00088387-Phase II NTC00071721-Phase III |
 | Name: sodium phenylbutyrate, buphenyl, 4-phenylbutiric acid, 4-phenylbutonoic acid, benzenebutanoic acid, benzenebutyric acid, butyric acid IUPAC name: sodium;4-phenylbutanoate Molecular formula: C10H11NaO2 Molecular Weight: 186.182909 g/mol Category: HDAC inhibitors Targets: Class I HDAC, Class IIa HDAC, Class IIb HDAC | Pathogenic genes: ARG1, ASS1, BCL2, CPS1, NAGS, OTC Mechanistic genes: BCL2, BDNF, EDN1, HDACs, HSPA8, ICAM1, NFKB2, NT3, VCAM1 Metabolic genes: Inhibitor:HDACs Inducer:ARG1, CFTR, CYP2B6, NFKB2 Transporter genes: CFTR Pleiotropic genes: ASL, BDNF, VCAM1 |
| NCT03533257-Phase II NCT02046434-Phase I |
 | Name: Nicotinamide, niacinamide, vitamin PP, aminicotin, nicotinic acid amide, amixicotyn, 3-pyridinecarboxamide, papulex, nicotylamide IUPAC name: pyridine-3-carboxamide Molecular formula: C6H6N2O Molecular Weight: 122.12 g/mol Category: SIRT inhibitors Targets: class III HDAC (SIRT1-7) | Pathogenic genes: IL6, IL8, PTGS2, TNF Mechanistic genes: ARTs, CAT, CLOCK, FOXO3, GPXs, IL6, IL8, PARP1, PTGS2, SIRT1, SOD1, TNF Metabolic genes: Inhibitor:CYP2D6, CYP3A4, CYP2E1, SIRT1-7 Pleiotropic genes: CAT, PARP1 |
| NTC00580931-Phases I, II NTC03061474-Phase II |
 | Name: Resveratrol, trans-resveratrol, 501-36-0, 3,4′,5-trihydroxystilbene, (E)-resveratrol, resvida IUPAC name: 5-[(E)-2-(4-hydroxyphenyl)ethenyl]benzene-1,3-diol Molecular formula: C14H12O3 Molecular Weight: 228.24 g/mol Category: SIRT inhibitors Targets: class III HDAC (SIRT1) | Pathogenic genes: BCL2, CAV1, ESR1, ESR2, GRIN2B, NOS3, PTGS2, TNFRSF10A, TNFRSF10B Mechanistic genes: APP, ATF3, BAX, BAK1, BBC3, BCL2, BCL2L1, BCL2L11, BIRC5, CASP3, CAV1, CFTR, ESR1, ESR2, GRIN1, GRIN2B, HTR3A, NFKB1, NOS3, PMAIP1, PTGS1, PTGS2, SIRT1, SIRT3, SIRT5, SRC, TNFRSF10A, TNFRSF10B, TRPs Metabolic genes: Substrate:CYP1A1, CYP1A2, CYP1B1, CYP2E1, GSTP1, PTGS1, PTGS2 Inhibitor:CYP1A1, CYP1B1, CYP2C9, CYP2D6, CYP3A4, NQO2 Inducer:CYP1A2, SIRT1 Transporter genes: ABCC1, ABCC2, ABCC3, ABCC4, ABCC8, ABCG1, ABCG2, CFTR, TRPs |
| NCT01504854-Phase II NCT00678431-Phase III NCT00743743-withdrown NCT02502253-Phase I |
 | Name: Curcumin, diferuloylmethane, turmeric yellow, turmeric, gelbwurz, kacha haldi, curcuma, haldar, souchet IUPAC name: (1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione Molecular formula: C21H20O6 Molecular Weight: 368.38 g/mol Category: HAT inhibitors Targets: HATs | Pathogenic genes: BACE1, CCND1, CDH1, GSK3B, IL1A, IL6, JUN, MSR1, PSEN1, PTGS2, SNCA, SREBF1, TNF Mechanistic genes: AKT1, PRKAs, BACE1, CCND1, CDH1, CDKs, CRM1, CTNNB1, EGF, GSK3B, HDACs, HIF1A, IL1A, IL6, JUN, MMPs, MSR1, NFKB1, NOS2, PDGFRs, PSEN1, PTGS2, SNCA, SOCS1, SOCS3, SREBF1, STAT3, TNF, VEGFA Metabolic genes: Inhibitor:CYP2C8, CYP2C9, EP300 Inducer:CYP2C8, CYP2C9, CYP2D6, CYP3A4 Transporter genes: ABCA1, SNCA Pleiotropic genes: CTNNB1, MSR1 |
| NTC00164749-Phases I, II NTC00099710-Phase II NTC01716637-Phase I NTC01811381-Recruting NTC02114372-Recruting |
 | Name: S-adenosylmethionine, ademetionine, AdoMet, donamet, methioninyladenylate, S-adenosyl-l-methionine, SAM-e IUPAC name: [(3S)-3-amino-3-carboxypropyl]-[[(2S,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl]-methylsulfanium Molecular formula: C15H23N6O5S+ Molecular Weight: 399.45 g/mol Category: HMT inhibitors Targets: HMTs | Pathogenic genes: AKT, ERK, GNMT, MAT1A, PSEN1 Mechanistic genes: AMD1, CAT, CBS, GCLC, GNMT, GSS, NOS2, ROS1, STAT1, TNF Metabolic genes: Substrate:COMT, GNMT, TPMT, SRM Inhibitor:ABCB1, CYP2E1, NOS2 Transporter genes: SLC25A26 Pleiotropic genes: CAT, TNF |
| NTC01320527-Phase II NTC00070941-Phases II, III |
| Category | Gene | Locus | OMIM | Pathology | Epigenetic changes |
|---|---|---|---|---|---|
| Phase I Drug Metabolizers | CYP2E1 | 10q26.3 | 124,040 | Parkinson’s Disease | Hypomethylated Up-regulated mRNA |
| Phase II Drug Metabolizers | GSTT1 | 22q11.23 | 600,436 | Parkinson’s Disease | Hypomethylated Upregulated mRNA |
| GSTTP1 | 22q11.23 | 600,436 | Parkinson’s Disease | Hypermethylated Downregulated mRNA | |
| GSTTP2 | 22q11.23 | 600,436 | Parkinson’s Disease | Hypermethylated Downregulated mRNA | |
| Phase III Drug Transporters | ABCA1 | 9q31.1 | 600,046 | Alzheimer’s Disease | Hypermethylated Downregulated mRNA |
| ABCB1 | 7q21.12 | 171,050 | Alzheimer’s Disease | Hypermethylated Downregulated mRNA | |
| ABCG2 | 4q22.1 | 603,756 | Parkinson’s Disease | Hypermethylated Downregulated mRNA | |
| ABCA3 | 16p13.3 | 601,615 | Parkinson’s Disease | Hypomethylated Upregulated mRNA | |
| ABCA7 | 19p13.3 | 605,414 | Alzheimer’s Disease | Hypomethylated Upregulated mRNA | |
| SLC12A5 | 20q13.12 | 606,726 | Parkinson’s Disease | Hypomethylated Upregulated mRNA | |
| SLC24A4 | 14q32.12 | 609,840 | Alzheimer’s Disease | Hypomethylated Upregulated mRNA | |
| SLC25A24 | 1p13.3 | 608,744 | Parkinson’s Disease | Hypomethylated Upregulated mRNA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teijido, O.; Cacabelos, R. Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases. Int. J. Mol. Sci. 2018, 19, 3199. https://doi.org/10.3390/ijms19103199
Teijido O, Cacabelos R. Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases. International Journal of Molecular Sciences. 2018; 19(10):3199. https://doi.org/10.3390/ijms19103199
Chicago/Turabian StyleTeijido, Oscar, and Ramón Cacabelos. 2018. "Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases" International Journal of Molecular Sciences 19, no. 10: 3199. https://doi.org/10.3390/ijms19103199
APA StyleTeijido, O., & Cacabelos, R. (2018). Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases. International Journal of Molecular Sciences, 19(10), 3199. https://doi.org/10.3390/ijms19103199