The Role of Chemokines in Wound Healing


Abstract
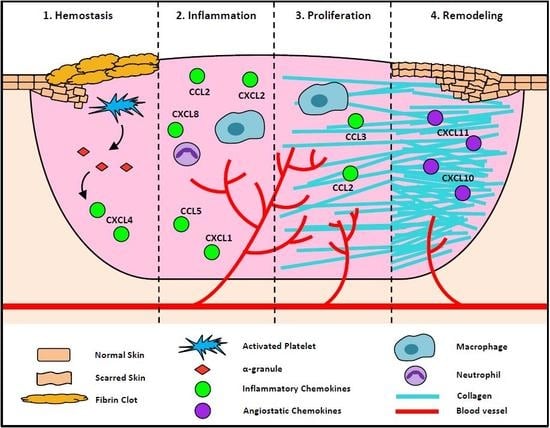
:
1. Introduction
2. The Roles of Chemokines at Each Stage of the Wound Healing Process
2.1. Chemokines in Hemostasis Stage of Wound Healing
2.2. Chemokines in the Inflammatory Stage of Wound Healing
2.3. Chemokines in the Proliferation Stage of Wound Healing
2.4. Chemokines in the Remodeling Stage of Wound Healing
3. Chemokines in Complex Wounds
3.1. Combat Wounds
3.2. Burns
3.3. Skin Grafts
4. Chemokines in Chronic Non-Healing Wounds
4.1. Diabetic Wounds
4.2. Ageing Wounds
5. The Therapeutic Targeting of Chemokines to Improve Wound Healing
5.1. Single Chemokine Targeting
5.2. Broad Spectrum Chemokine Targeting
6. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| BAOEC | Bovine aortic endothelial cell |
| bFGF | Basic fibroblast growth factor |
| ECM | Extra cellular matrix |
| EPC | Endothelial progenitor cells |
| HMEC-1 | Human microvascular endothelial cell |
| HUVEC | Human umbilical vein endothelial cell |
| IFN-γ | Interferon-γ |
| NF-κB | Nuclear factor κB |
| PDGF | Platelet derived growth factor |
| TGF-β | Transforming growth factor β |
| TNF-α | Tumor necrosis factor α |
| VEGF | Vascular endothelial growth factor |
References
- Tonnesen, M.G.; Feng, X.; Clark, R.A. Angiogenesis in wound healing. J. Investig. Dermatol. Symp. Proc. 2000, 5, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Salanga, C.L.; Handel, T.M. Chemokine and chemokine receptor structure and interactions: Implications for therapeutic strategies. Immunol. Cell Biol. 2015, 93, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Gleissner, C.A.; von Hundelshausen, P.; Ley, K. Platelet chemokines in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1920–1927. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, E.; Toksoy, A.; Goebeler, M.; Debus, S.; Brocker, E.B.; Gillitzer, R. Chemokines il-8, groalpha, mcp-1, ip-10, and mig are sequentially and differentially expressed during phase-specific infiltration of leukocyte subsets in human wound healing. Am. J. Pathol. 1998, 153, 1849–1860. [Google Scholar] [CrossRef]
- Kobayashi, Y. The role of chemokines in neutrophil biology. Front. Biosci. 2008, 13, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Goede, V.; Brogelli, L.; Ziche, M.; Augustin, H.G. Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int. J. Cancer 1999, 82, 765–770. [Google Scholar] [CrossRef] [Green Version]
- DiPietro, L.A.; Burdick, M.; Low, Q.E.; Kunkel, S.L.; Strieter, R.M. Mip-1alpha as a critical macrophage chemoattractant in murine wound repair. J. Clin. Investig. 1998, 101, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.C.; Whaley, D.; Kulasekeran, P.; Hancock, W.W.; Lu, B.; Bodnar, R.; Newsome, J.; Hebda, P.A.; Wells, A. Delayed and deficient dermal maturation in mice lacking the cxcr3 elr-negative cxc chemokine receptor. Am. J. Pathol. 2007, 171, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Zaja-Milatovic, S.; Richmond, A. Cxc chemokines and their receptors: A case for a significant biological role in cutaneous wound healing. Histol. Histopathol. 2008, 23, 1399–1407. [Google Scholar] [PubMed]
- Maione, T.E.; Gray, G.S.; Petro, J.; Hunt, A.J.; Donner, A.L.; Bauer, S.I.; Carson, H.F.; Sharpe, R.J. Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. Science 1990, 247, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Jouan, V.; Canron, X.; Alemany, M.; Caen, J.P.; Quentin, G.; Plouet, J.; Bikfalvi, A. Inhibition of in vitro angiogenesis by platelet factor-4–derived peptides and mechanism of action. Blood 1999, 94, 984–993. [Google Scholar] [PubMed]
- Wetzler, C.; Kämpfer, H.; Stallmeyer, B.; Pfeilschifter, J.; Frank, S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: Prolonged persistence of neutrophils and macrophages during the late phase of repair. J. Investig. Dermatol. 2000, 115, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Gillitzer, R.; Goebeler, M. Chemokines in cutaneous wound healing. J. Leukoc. Biol. 2001, 69, 513–521. [Google Scholar] [PubMed]
- Broughton, G., 2nd; Janis, J.E.; Attinger, C.E. The basic science of wound healing. Plast. Reconstr. Surg. 2006, 117, 12s–34s. [Google Scholar] [CrossRef] [PubMed]
- Abkowitz, J.L.; Robinson, A.E.; Kale, S.; Long, M.W.; Chen, J. Mobilization of hematopoietic stem cells during homeostasis and after cytokine exposure. Blood 2003, 102, 1249–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudek, A.Z.; Nesmelova, I.; Mayo, K.; Verfaillie, C.M.; Pitchford, S.; Slungaard, A. Platelet factor 4 promotes adhesion of hematopoietic progenitor cells and binds il-8: Novel mechanisms for modulation of hematopoiesis. Blood 2003, 101, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Hiti-Harper, J.; Wohl, H.; Harper, E. Platelet factor 4: An inhibitor of collagenase. Science 1978, 199, 991–992. [Google Scholar] [CrossRef] [PubMed]
- Gengrinovitch, S.; Greenberg, S.M.; Cohen, T.; Gitay-Goren, H.; Rockwell, P.; Maione, T.E.; Levi, B.Z.; Neufeld, G. Platelet factor-4 inhibits the mitogenic activity of vegf121 and vegf165 using several concurrent mechanisms. J. Biol. Chem. 1995, 270, 15059–15065. [Google Scholar] [CrossRef] [PubMed]
- Perollet, C.; Han, Z.C.; Savona, C.; Caen, J.P.; Bikfalvi, A. Platelet factor 4 modulates fibroblast growth factor 2 (fgf-2) activity and inhibits fgf-2 dimerization. Blood 1998, 91, 3289–3299. [Google Scholar] [PubMed]
- Bodnar, R.J.; Yates, C.C.; Wells, A. Ip-10 blocks vascular endothelial growth factor–induced endothelial cell motility and tube formation via inhibition of calpain. Circ. Res. 2006, 98. [Google Scholar] [CrossRef] [PubMed]
- Sarabi, A.; Kramp, B.K.; Drechsler, M.; Hackeng, T.M.; Soehnlein, O.; Weber, C.; Koenen, R.R.; Von Hundelshausen, P. Cxcl4l1 inhibits angiogenesis and induces undirected endothelial cell migration without affecting endothelial cell proliferation and monocyte recruitment. J. Thromb. Haemost. 2011, 9, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Struyf, S.; Burdick, M.D.; Proost, P.; Van Damme, J.; Strieter, R.M. Platelets release cxcl4l1, a nonallelic variant of the chemokine platelet factor-4/cxcl4 and potent inhibitor of angiogenesis. Circ. Res. 2004, 95, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Vandercappellen, J.; Liekens, S.; Bronckaers, A.; Noppen, S.; Ronsse, I.; Dillen, C.; Belleri, M.; Mitola, S.; Proost, P.; Presta, M.; et al. The cooh-terminal peptide of platelet factor-4 variant (cxcl4l1/pf-4var47-70) strongly inhibits angiogenesis and suppresses b16 melanoma growth in vivo. Mol. Cancer Res. 2010, 8, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Von Hundelshausen, P.; Koenen, R.R.; Sack, M.; Mause, S.F.; Adriaens, W.; Proudfoot, A.E.I.; Hackeng, T.M.; Weber, C. Heterophilic interactions of platelet factor 4 and rantes promote monocyte arrest on endothelium. Blood 2005, 105, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Lasagni, L.; Francalanci, M.; Annunziato, F.; Lazzeri, E.; Giannini, S.; Cosmi, L.; Sagrinati, C.; Mazzinghi, B.; Orlando, C.; Maggi, E.; et al. An alternatively spliced variant of cxcr3 mediates the inhibition of endothelial cell growth induced by ip-10, mig, and i-tac, and acts as functional receptor for platelet factor 4. J. Exp. Med. 2003, 197, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Petrai, I.; Rombouts, K.; Lasagni, L.; Annunziato, F.; Cosmi, L.; Romanelli, R.G.; Sagrinati, C.; Mazzinghi, B.; Pinzani, M.; Romagnani, S.; et al. Activation of p38mapk mediates the angiostatic effect of the chemokine receptor cxcr3-b. Int. J. Biochem. Cell Biol. 2008, 40, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Ebaid, H. Neutrophil depletion in the early inflammatory phase delayed cutaneous wound healing in older rats: Improvements due to the use of un-denatured camel whey protein. Diagn. Pathol. 2014, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Nedoszytko, B.; Sokołowska-Wojdyło, M.; Ruckemann-Dziurdzińska, K.; Roszkiewicz, J.; Nowicki, R.J. Chemokines and cytokines network in the pathogenesis of the inflammatory skin diseases: Atopic dermatitis, psoriasis and skin mastocytosis. Postepy Dermatol. Alergol. 2014, 31, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.R.; Seed, M.P.; Kircher, C.H.; Willoughby, D.A.; Winkler, J.D. The codependence of angiogenesis and chronic inflammation. FASEB J. 1997, 11, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Sunderkotter, C.; Steinbrink, K.; Goebeler, M.; Bhardwaj, R.; Sorg, C. Macrophages and angiogenesis. J. Leukoc. Biol. 1994, 55, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Gibran, N.S.; Ferguson, M.; Heimbach, D.M.; Isik, F.F. Monocyte chemoattractant protein-1 mrna expression in the human burn wound. J. Surg. Res. 1997, 70, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I.; Uguccioni, M. Modulation of chemokine responses: Synergy and cooperativity. Front. Immunol. 2016, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein hmgb1 by necrotic cells triggers inflammation. Nature 2002, 418, 191. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.-C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (hmg-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. Hmg-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Fiuza, C.; Bustin, M.; Talwar, S.; Tropea, M.; Gerstenberger, E.; Shelhamer, J.H.; Suffredini, A.F. Inflammation-promoting activity of hmgb1 on human microvascular endothelial cells. Blood 2003, 101, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, E.; Hain, A.; Vinci, M.; Carmona, G.; Bianchi, M.E.; Vajkoczy, P.; Zeiher, A.M.; Chavakis, T.; Dimmeler, S. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ. Res. 2007, 100, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Straino, S.; Di Carlo, A.; Mangoni, A.; De Mori, R.; Guerra, L.; Maurelli, R.; Panacchia, L.; Di Giacomo, F.; Palumbo, R.; Di Campli, C.; et al. High-mobility group box 1 protein in human and murine skin: Involvement in wound healing. J. Investig. Dermatol. 2008, 128, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Schiraldi, M.; Raucci, A.; Munoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. Hmgb1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with cxcl12 and signaling via cxcr4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Brancato, S.K.; Thomay, A.A.; Reichner, J.S.; Albina, J.E. The phenotype of murine wound macrophages. J. Leukoc. Biol. 2010, 87, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Devalaraja, R.M.; Nanney, L.B.; Du, J.; Qian, Q.; Yu, Y.; Devalaraja, M.N.; Richmond, A. Delayed wound healing in cxcr2 knockout mice. J. Investig. Dermatol. 2000, 115, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.C.; Whaley, D.; Hooda, S.; Hebda, P.A.; Bodnar, R.J.; Wells, A. Delayed reepithelialization and basement membrane regeneration after wounding in mice lacking cxcr3. Wound Repair Regen. 2009, 17, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Tortelli, F.; Pisano, M.; Briquez, P.S.; Martino, M.M.; Hubbell, J.A. Fibronectin binding modulates cxcl11 activity and facilitates wound healing. PLoS ONE 2013, 8, e79610. [Google Scholar] [CrossRef] [PubMed]
- Inokuma, D.; Abe, R.; Fujita, Y.; Sasaki, M.; Shibaki, A.; Nakamura, H.; McMillan, J.R.; Shimizu, T.; Shimizu, H. Ctack/ccl27 accelerates skin regeneration via accumulation of bone marrow-derived keratinocytes. Stem Cells 2006, 24, 2810–2816. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte–fibroblast interactions in wound healing. J. Investig. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Shen, Y.; Mohanasundaram, P.; Lindström, M.; Ivaska, J.; Ny, T.; Eriksson, J.E. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via tgf-β–slug signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E4320–E4327. [Google Scholar] [CrossRef] [PubMed]
- Hyman, K.M.; Seghezzi, G.; Pintucci, G.; Stellari, G.; Kim, J.H.; Grossi, E.A.; Galloway, A.C.; Mignatti, P. Transforming growth factor-beta1 induces apoptosis in vascular endothelial cells by activation of mitogen-activated protein kinase. Surgery 2002, 132, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.E.; Ballermann, B.J. Inhibition of capillary morphogenesis and associated apoptosis by dominant negative mutant transforming growth factor-beta receptors. J. Biol. Chem. 1995, 270, 21144–21150. [Google Scholar] [CrossRef] [PubMed]
- Gailit, J.; Welch, M.P.; Clark, R.A. Tgf-beta 1 stimulates expression of keratinocyte integrins during re-epithelialization of cutaneous wounds. J. Investig. Dermatol. 1994, 103, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.W.; Grobbelaar, A.O.; Rolfe, K.J. The role of the tgf-beta family in wound healing, burns and scarring: A review. Int. J. Burns Trauma 2012, 2, 18–28. [Google Scholar] [PubMed]
- Behm, B.; Babilas, P.; Landthaler, M.; Schreml, S. Cytokines, chemokines and growth factors in wound healing. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, M.; Jackson, C.J. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv. Wound Care 2015, 4, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Eckes, B.; Mauch, C.; Hartmann, K.; Krieg, T. Monocyte chemoattractant protein-1 enhances gene expression and synthesis of matrix metalloproteinase-1 in human fibroblasts by an autocrine il-1α loop. J. Immunol. 2000, 164, 6174–6179. [Google Scholar] [CrossRef] [PubMed]
- Johnatty, R.N.; Taub, D.D.; Reeder, S.P.; Turcovski-Corrales, S.M.; Cottam, D.W.; Stephenson, T.J.; Rees, R.C. Cytokine and chemokine regulation of prommp-9 and timp-1 production by human peripheral blood lymphocytes. J. Immunol. 1997, 158, 2327–2333. [Google Scholar] [PubMed]
- Wu, L.; Fan, J.; Matsumoto, S.-I.; Watanabe, T. Induction and regulation of matrix metalloproteinase-12 by cytokines and cd40 signaling in monocyte/macrophages. Biochem. Biophys. Res. Commun. 2000, 269, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, R.J.; Yates, C.C.; Rodgers, M.E.; Du, X.; Wells, A. Ip-10 induces dissociation of newly formed blood vessels. J. Cell Sci. 2009, 122, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Lisboa, F.A.; Forsberg, J.A.; Brown, T.S.; Gage, F.A.; Potter, B.K.; Elster, E.A. Bilateral lower-extremity amputation wounds are associated with distinct local and systemic cytokine response. Surgery 2013, 154, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, J.S.; Stojadinovic, A.; Gage, F.A.; Tadaki, D.K.; Perdue, P.W.; Forsberg, J.; Davis, T.A.; Dunne, J.R.; Denobile, J.W.; Brown, T.S.; et al. Inflammatory biomarkers in combat wound healing. Ann. Surg. 2009, 250, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.S.; Hawksworth, J.S.; Sheppard, F.R.; Tadaki, D.K.; Elster, E. Inflammatory response is associated with critical colonization in combat wounds. Surg. Infect. 2011, 12, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.K. Burn wound: How it differs from other wounds? Indian J. Plast. Surg. 2012, 45, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Valvis, S.M.; Waithman, J.; Wood, F.M.; Fear, M.W.; Fear, V.S. The immune response to skin trauma is dependent on the etiology of injury in a mouse model of burn and excision. J. Investig. Dermatol. 2015, 135, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Iocono, J.A.; Colleran, K.R.; Remick, D.G.; Gillespie, B.W.; Ehrlich, H.P.; Garner, W.L. Interleukin-8 levels and activity in delayed-healing human thermal wounds. Wound Repair Regen. 2000, 8, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Jolanda, P.; Gudula, K.; de Edith, B.; Esther, M.; Rik, S.; Sue, G. Comparison of Cytokine, Chemokine and Growth Factor Profiles in Burn Wounds, Chronic Wounds and Surgical Excision Wounds. 2012, pp. 71–84. Available online: https://pdfs.semanticscholar.org/d961/8cc306bdea13b4c6a39d2c7079bba9bd7f09.pdf (accessed on 17 October 2018).
- Stanojcic, M.; Chen, P.; Xiu, F.; Jeschke, M.G. Impaired immune response in elderly burn patients: New insights into the immune-senescence phenotype. Ann. Surg. 2016, 264, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Benichou, G.; Yamada, Y.; Yun, S.-H.; Lin, C.; Fray, M.; Tocco, G. Immune recognition and rejection of allogeneic skin grafts. Immunotherapy 2011, 3, 757–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syrbe, U.; Siveke, J.; Hamann, A. Th1/th2 subsets: Distinct differences in homing and chemokine receptor expression? Springer Semin. Immunopathol. 1999, 21, 263–285. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Novick, A.C.; Toma, H.; Fairchild, R.L. Induction of chemokine gene expression during allogeneic skin graft rejection1. Transplantation 1996, 61, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.M.; Park, J.O.; Kim, D.; Kim, J.Y.; Oh, K.H.; Park, C.G.; Oh, B.H.; Kim, S.; Ahn, C. Early up-regulation of cxc-chemokine expression is associated with strong cellular immune responses to murine skin xenografts. Xenotransplantation 2006, 13, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Kunstfeld, R.; Lechleitner, S.; Wolff, K.; Petzelbauer, P. Mcp-1 and mip-1α are most efficient in recruiting t cells into the skinin vivo. J. Investig. Dermatol. 1998, 111, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Blakytny, R.; Jude, E. The molecular biology of chronic wounds and delayed healing in diabetes. Diabet. Med. 2006, 23, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Bannon, P.; Wood, S.; Restivo, T.; Campbell, L.; Hardman, M.J.; Mace, K.A. Diabetes induces stable intrinsic changes to myeloid cells that contribute to chronic inflammation during wound healing in mice. Dis. Model Mech. 2013, 6, 1434–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through hif-1 induction of sdf-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-J.; Velazquez, O.C. Hyperoxia, endothelial progenitor cell mobilization, and diabetic wound healing. Antioxid. Redox Signal. 2008, 10, 1869–1882. [Google Scholar] [CrossRef] [PubMed]
- Balaji, S.; King, A.; Crombleholme, T.M.; Keswani, S.G. The role of endothelial progenitor cells in postnatal vasculogenesis: Implications for therapeutic neovascularization and wound healing. Adv. Wound Care 2013, 2, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.A.; Liu, Z.-J.; Xiao, M.; Chen, H.; Goldstein, L.J.; Buerk, D.G.; Nedeau, A.; Thom, S.R.; Velazquez, O.C. Diabetic impairments in no-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and sdf-1α. J. Clin. Investig. 2007, 117, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Restivo, T.E.; Mace, K.A.; Harken, A.H.; Young, D.M. Application of the chemokine cxcl12 expression plasmid restores wound healing to near normal in a diabetic mouse model. J. Trauma 2010, 69, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, D.M.; Xu, J.; Herdrich, B.J.; Radu, A.; Mitchell, M.E.; Liechty, K.W. Inhibition of stromal cell-derived factor-1alpha further impairs diabetic wound healing. J. Vasc. Surg. 2011, 53, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Bekeschus, S.; Schmidt, A.; Napp, M.; Kramer, A.; Kerner, W.; Woedtke, T.; Wende, K.; Hasse, S.; Masur, K. Distinct cytokine and chemokine patterns in chronic diabetic ulcers and acute wounds. Exp. Dermatol. 2017, 26, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Richmond, A. Chemokine regulation of neutrophil infiltration of skin wounds. Adv. Wound Care 2015, 4, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Schulz, G.; Stechmiller, J. Wound healing and nitric oxide production: Too little or too much may impair healing and cause chronic wounds. Int. J. Lower Extrem. Wounds 2006, 5, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Schwentker, A.; Vodovotz, Y.; Weller, R.; Billiar, T.R. Nitric oxide and wound repair: Role of cytokines? Nitric Oxide 2002, 7, 1–10. [Google Scholar] [CrossRef]
- Reed, M.J.; Corsa, A.; Pendergrass, W.; Penn, P.; Sage, E.H.; Abrass, I.B. Neovascularization in aged mice: Delayed angiogenesis is coincident with decreased levels of transforming growth factor beta1 and type i collagen. Am. J. Pathol. 1998, 152, 113–123. [Google Scholar] [PubMed]
- Gilchrest, B.A. In vitro assessment of keratinocyte aging. J. Investig. Dermatol. 1983, 81, 184s–189s. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Horan, M.A.; Ferguson, M.W. Aging is associated with reduced deposition of specific extracellular matrix components, an upregulation of angiogenesis, and an altered inflammatory response in a murine incisional wound healing model. J. Investig. Dermatol. 1997, 108, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Danon, D.; Kowatch, M.A.; Roth, G.S. Promotion of wound repair in old mice by local injection of macrophages. Proc. Natl. Acad. Sci. USA 1989, 86, 2018–2020. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, A.L.; Rendon, J.L.; Ramirez, L.; Choudhry, M.A.; Kovacs, E.J. Reduced neutrophil chemotaxis and infiltration contributes to delayed resolution of cutaneous wound infection with advanced age. J. Immunol. 2013, 4, 1746–1757. [Google Scholar] [CrossRef] [PubMed]
- Swift, M.E.; Kleinman, H.K.; DiPietro, L.A. Impaired wound repair and delayed angiogenesis in aged mice. Lab. Investig. 1999, 79, 1479–1487. [Google Scholar] [PubMed]
- Ashcroft, G.S.; Mills, S.J. Androgen receptor–mediated inhibition of cutaneous wound healing. J. Clin. Invest. 2002, 110, 615–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Xia, Y.P.; Roth, S.I.; Gruskin, E.; Mustoe, T.A. Transforming growth factor-beta1 fails to stimulate wound healing and impairs its signal transduction in an aged ischemic ulcer model: Importance of oxygen and age. Am. J. Pathol. 1999, 154, 301–309. [Google Scholar] [CrossRef]
- Shallo, H.; Plackett, T.P.; Heinrich, S.A.; Kovacs, E.J. Monocyte chemoattractant protein-1 (mcp-1) and macrophage infiltration into the skin after burn injury in aged mice. Burns 2003, 29, 641–647. [Google Scholar] [CrossRef]
- Loh, S.A.; Chang, E.I.; Galvez, M.G.; Thangarajah, H.; El-ftesi, S.; Vial, I.N.; Lin, D.A.; Gurtner, G.C. Sdf-1α expression during wound healing in the aged is hif dependent. Plast. Reconstr. Surg. 2009, 123, 65S–75S. [Google Scholar] [CrossRef] [PubMed]
- Low, Q.E.; Drugea, I.A.; Duffner, L.A.; Quinn, D.G.; Cook, D.N.; Rollins, B.J.; Kovacs, E.J.; DiPietro, L.A. Wound healing in mip-1alpha(-/-) and mcp-1(-/-) mice. Am. J. Pathol. 2001, 159, 457–463. [Google Scholar] [CrossRef]
- Wood, S.; Jayaraman, V.; Huelsmann, E.J.; Bonish, B.; Burgad, D.; Sivaramakrishnan, G.; Qin, S.; DiPietro, L.A.; Zloza, A.; Zhang, C.; et al. Pro-inflammatory chemokine ccl2 (mcp-1) promotes healing in diabetic wounds by restoring the macrophage response. PLoS ONE 2014, 9, e91574. [Google Scholar] [CrossRef] [PubMed]
- Kaesler, S.; Bugnon, P.; Gao, J.L.; Murphy, P.M.; Goppelt, A.; Werner, S. The chemokine receptor ccr1 is strongly up-regulated after skin injury but dispensable for wound healing. Wound Repair Regen. 2004, 12, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Berres, M.-L.; Koenen, R.R.; Rueland, A.; Zaldivar, M.M.; Heinrichs, D.; Sahin, H.; Schmitz, P.; Streetz, K.L.; Berg, T.; Gassler, N.; et al. Antagonism of the chemokine ccl5 ameliorates experimental liver fibrosis in mice. J. Clin. Investig. 2010, 120, 4129–4140. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yong, X.; Li, C.; Lu, M.; Liu, D.; Chen, L.; Hu, J.; Teng, M.; Zhang, D.; Fan, Y.; et al. Cxcl12/cxcr4 axis promotes mesenchymal stem cell mobilization to burn wounds and contributes to wound repair. J. Surg. Res. 2013, 183, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Abe, R.; Fujita, Y.; Ando, S.; Inokuma, D.; Shimizu, H. Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. J. Immunol. 2008, 180, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Guang, F.; Daifeng, H.; Jiake, C. Processing of cxcl12 impedes the recruitment of endothelial progenitor cells in diabetic wound healing. FEBS J. 2014, 281, 5054–5062. [Google Scholar]
- Nishimura, Y.; Ii, M.; Qin, G.; Hamada, H.; Asai, J.; Takenaka, H.; Sekiguchi, H.; Renault, M.A.; Jujo, K.; Katoh, N.; et al. Cxcr4 antagonist amd3100 accelerates impaired wound healing in diabetic mice. J. Investig. Dermatol. 2012, 132, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Vagesjo, E.; Ohnstedt, E.; Mortier, A.; Lofton, H.; Huss, F.; Proost, P.; Roos, S.; Phillipson, M. Accelerated wound healing in mice by on-site production and delivery of cxcl12 by transformed lactic acid bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 1895–1900. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Derider, M.; McCornack, M.A.; Jao, S.C.; Isern, N.; Ness, T.; Moyer, R.; LiWang, P.J. Solution structure of the complex between poxvirus-encoded cc chemokine inhibitor vcci and human mip-1beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13985–13990. [Google Scholar] [CrossRef] [PubMed]
- Ridiandries, A.; Bursill, C.; Tan, J. Broad-spectrum inhibition of the cc-chemokine class improves wound healing and wound angiogenesis. Int. J. Mol. Sci. 2017, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Reckless, J.; Tatalick, L.M.; Grainger, D.J. The pan-chemokine inhibitor nr58-3.14.3 abolishes tumour necrosis factor-α accumulation and leucocyte recruitment induced by lipopolysaccharide in vivo. Immunology 2001, 103, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Berkkanoglu, M.; Zhang, L.; Ulukus, M.; Cakmak, H.; Kayisli, U.A.; Kursun, S.; Arici, A. Inhibition of chemokines prevents intraperitoneal adhesions in mice. Hum. Reprod. 2005, 20, 3047–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Hemostasis | Inflammation | Proliferation | Remodelling | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CCL2 | + | [3] | CCL1 | + | [4,5] | CCL2 | +++ | [4,6,7] | CXCL10 | unknown | [8] |
| CCL3 | + | [3] | CCL2 | +++ | [4,5] | CCL3 | +++ | [4,6,7] | CXCL11 | unknown | [8] |
| CCL5 | + | [3] | CCL3 | ++ | [4,5] | CXCL1 | + | [9] | |||
| CXCL1 | + | [3] | CCL4 | + | [4] | CXCL2 | + | [9] | |||
| CXCL4 | +++ | [10,11] | CCL5 | +++ | [4,5] | CXCL3 | + | [9] | |||
| CXCL5 | + | [3] | CCL7 | + | [4] | CXCL5 | + | [9] | |||
| CXCL7 | + | [3] | CXCL1 | ++ | [3,5,12,13] | CXCL6 | + | [9] | |||
| CXCL8 | + | [3] | CXCL2 | ++ | [3,5,12,13] | CXCL7 | + | [9] | |||
| CXCL12 | + | [3] | CXCL5 | + | [13] | CXCL8 | + | [9] | |||
| CXCL7 | + | [13] | CXCL10 | + | [7] | ||||||
| CXCL8 | ++ | [3,4,5,12,13] | CXCL11 | + | [7] | ||||||
| CXCL12 | + | [13] | CXCL12 | + | [14,15] | ||||||
| Combat Wounds | Burn Wounds | Skin Grafts | Diabetic Wounds | Aged Wounds | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wound tissue | CCL2 | + (D) | [62] | CXCL8 | ++ | [66] | CCL2 | + (M) | [71,73] | CCL2 | ++ (M) | [12] | CXCL1 | − (M) | [90] |
| CCL3 | + (D) | [62] | CCL3 | + (M) | [71,73] | CXCL2 | ++ (M) | [12] | CXCL12 | − (M) | [95] | ||||
| CCL5 | − (D) | [62] | CCL4 | + (M) | [71] | CXCL12 | − (M) | [79,80] | |||||||
| CXCL10 | − (D) | [62] | CCL5 | + (M) | [71] | ||||||||||
| CXCL1 | + (M) | [71] | |||||||||||||
| CXCL9 | + (M) | [72] | |||||||||||||
| CXCL10 | + (M) | [71,72] | |||||||||||||
| Wound effluent | CCL2 | +, − (D) | [61] | CXCL8 | +++ | [67] | CCL5 | − | [82] | ||||||
| CCL3 | ++, + (B) | [61,63] | CXCL8 | + | [82] | ||||||||||
| CCL4 | ++, + (B) | [61] | CXCL10 | − | [82] | ||||||||||
| CCL11 | + | [61] | CXCL11 | − | [82] | ||||||||||
| CXCL10 | − (D) | [62] | |||||||||||||
| CXCL8 | ++, + (B) | [61,63] | |||||||||||||
| CXCL9 | − (D) | [61] | |||||||||||||
| Serum | CCL3 | ++ (D), + (B) | [61,62,63] | CCL2 | + (M) | [65,68] | |||||||||
| CCL4 | ++ (D), + (B) | [61,62] | CCL3 | + (M) | [65,68] | ||||||||||
| CCL5 | ++, ++ (D) | [61] | CCL7 | ++ | [68] | ||||||||||
| CXCL8 | ++, + (B) | [61,62,63] | CCL11 | + (M) | [65] | ||||||||||
| CXCL9 | ++, ++ (D) | [61] | CXCL1 | ++ (M) | [65] | ||||||||||
| CXCL10 | ++ (D), + (B) | [61,63] | CXCL10 | ++ | [68] | ||||||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ridiandries, A.; Tan, J.T.M.; Bursill, C.A. The Role of Chemokines in Wound Healing. Int. J. Mol. Sci. 2018, 19, 3217. https://doi.org/10.3390/ijms19103217
Ridiandries A, Tan JTM, Bursill CA. The Role of Chemokines in Wound Healing. International Journal of Molecular Sciences. 2018; 19(10):3217. https://doi.org/10.3390/ijms19103217
Chicago/Turabian StyleRidiandries, Anisyah, Joanne T. M. Tan, and Christina A. Bursill. 2018. "The Role of Chemokines in Wound Healing" International Journal of Molecular Sciences 19, no. 10: 3217. https://doi.org/10.3390/ijms19103217