Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases


Abstract

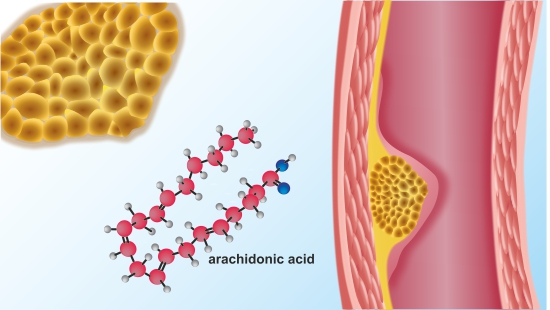{kind=link}
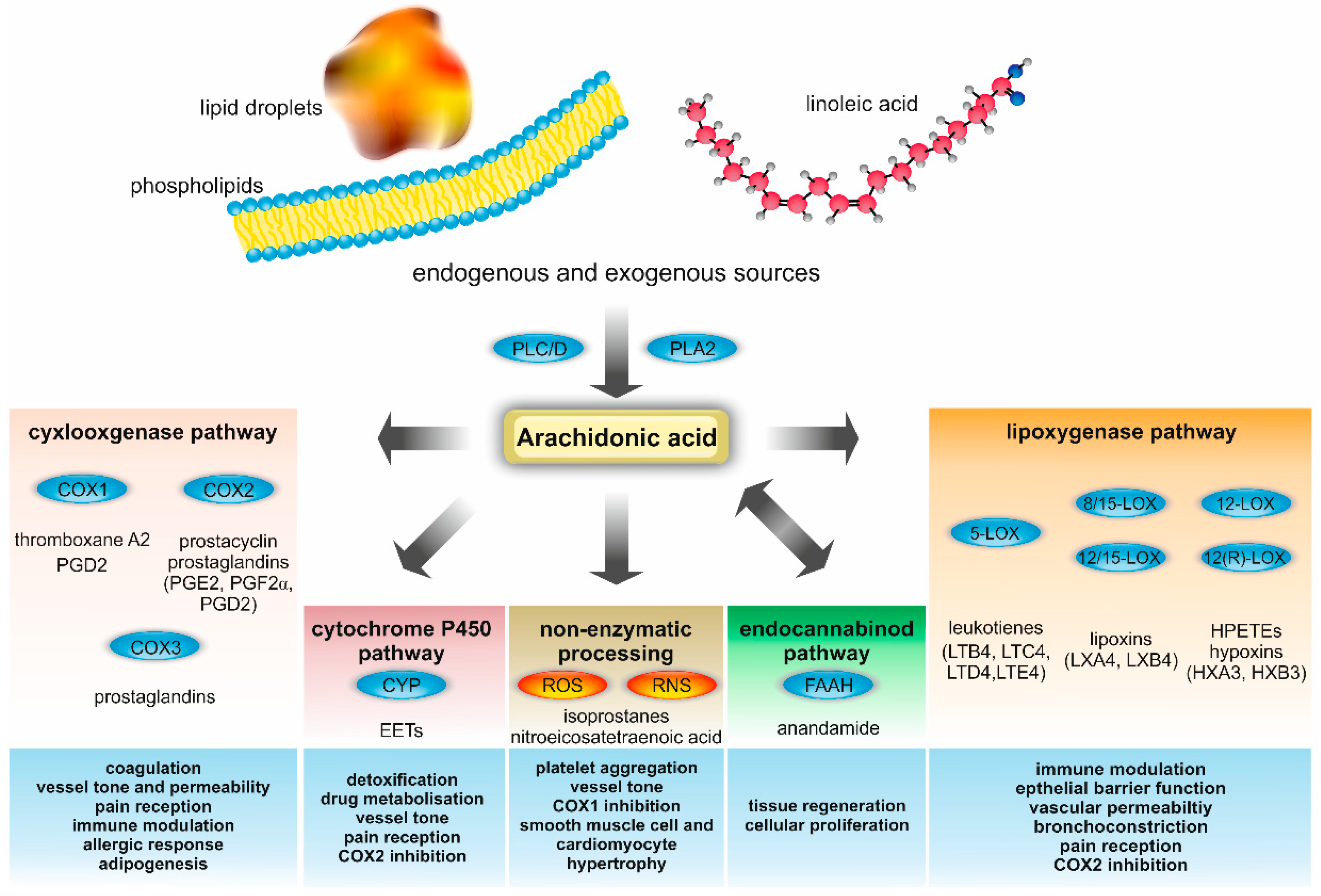{kind=link}
1. Introduction
2. AA Metabolism
3. AA Metabolism in Human Health
4. Alterations of Immunity and Wound Healing in Obesity and Metabolic Disease
5. AA Metabolism in the Pathogenesis of Diabetes Mellitus
6. AA Metabolites in Hepatic Steatosis and Steatohepatitis
7. AA Metabolome in Atherosclerosis and Cardiovascular Disease
8. Conclusions and Perspectives
9. Methodology
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| %E | Percent of energy |
| AA | Arachidonic acid |
| ACSL1 | Acyl-CoA synthetase 1 |
| ALOX; LOX | Lipoxygenase |
| apoE | Apolipoprotein E |
| BMI | Body mass index |
| CB1 | Cannabinoid type 1 receptor |
| COX | Cyclooxygenase |
| CYP450 | Cytochrome P450 |
| CysLTs | Cysteinyl leukotrienes (LTC4, LTD4, LTE4 and LTF4) |
| CYSLTR | Cysteinyl leukotriene receptor |
| DAG | Diacylglycerol |
| DHA | Docosahexaenoic acid |
| DM | Diabetes mellitus |
| EET | Epoxyeicosatrienoic acid |
| EPA | Eicosapentaenoic acid |
| FAAH | Fatty acid amide hydrolase |
| FFA | Free fatty acid |
| HETE | Hydroxyeicosatetraenoic acid |
| HPETE | Hydroperoxyeicosatetraenoic acid |
| HHT | 12-Hydroxyheptadecatrienoic acid |
| LDL | Low density lipoprotein |
| LDL-C | Low density lipoprotein cholesterol |
| LPS | Lipopolysaccharide |
| LT | Leukotriene |
| LTB4 | Leukotriene B4 |
| LTB4R | Leukotriene B4 receptor |
| LXA4 | Lipoxin A4 |
| MyD88 | Myeloid differentiation primary response 88 |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| NLRP3-ASC | NACHT, LRR and PYD domains-containing protein 3/apoptosis-associated Speck-like protein containing a CARD |
| NLR | Nod-like receptor |
| oxLDL-C | Oxidized low density lipoprotein cholesterol |
| P2RY12 | Purinergic receptor P2Y12 |
| PAF | Platelet-activating factor |
| PG | Prostaglandin |
| PGI2 | Prostacyclin |
| PLA2 | Phospholipase A2 |
| PLC | Phospholipase C |
| PLD | Phospholipase D |
| PMN | Polymorphonuclear leukocyte |
| PPAR | Peroxisome proliferator activated receptor gamma |
| PUFA | Polyunsaturated omega fatty acid |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| sEH | Soluble epoxide hydratase |
| SMC | Smooth muscle cell |
| SPM | Specialized pro-resolving mediators |
| TG | Triglyceride |
| TH | T helper cell |
| TLR | Toll-like receptor |
| TNFα | Tumor necrosis factor alpha |
| TXA2 | Thromboxane A2 |
| VLDL | Very low-density lipoprotein |
| vWF | Von Willebrand factor |
| WHO | World Health Organization |
References
- Haffner, S.M. The metabolic syndrome: Inflammation, diabetes mellitus, and cardiovascular disease. Am. J. Cardiol. 2006, 97, 3A–11A. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report from the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Flegal, K.M.; Carroll, M.D.; Kit, B.K.; Ogden, C.L. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA 2012, 307, 491–497. [Google Scholar] [CrossRef] [PubMed]
- WHO. Obesity and Overweight: Key Facts. 2018. Available online: http://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 22 October 2018).
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed]
- Vanni, E.; Marengo, A.; Mezzabotta, L.; Bugianesi, E. Systemic Complications of Nonalcoholic Fatty Liver Disease: When the Liver Is Not an Innocent Bystander. Semin. Liver Dis. 2015, 35, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Harker, L. Hyperlipidemia and atherosclerosis. Science 1976, 193, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, S.; Newburgh, L.H. The Relation between Atherosclerosis and Ingested Cholesterol in the Rabbit. J. Exp. Med. 1926, 43, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Walker, W.J. Relationship of adiposity to serum cholesterol and lipoprotein levels and their modification by dietary means. Ann. Intern. Med. 1953, 39, 705–716. [Google Scholar] [PubMed]
- Ross, R. Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Gilroy, D.W. Lipid Mediators in Inflammation. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Inflammation. Signalling the fat controller. Nature 1996, 384, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information: Pubchem. 5,8,11,14-Eicosatetraenoic Acid. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5312542#section=Top (accessed on 22 October 2018).
- Rich, M.R. Conformational analysis of arachidonic and related fatty acids using molecular dynamics simulations. Biochim. Biophys. Acta 1993, 1178, 87–96. [Google Scholar] [CrossRef]
- Weller, P.F. Leukocyte Lipid Bodies—Structure and Function as “Eicosasomes”. Trans. Am. Clin. Climatol. Assoc. 2016, 127, 328–340. [Google Scholar] [PubMed]
- McArthur, M.J.; Atshaves, B.P.; Frolov, A.; Foxworth, W.D.; Kier, A.B.; Schroeder, F. Cellular uptake and intracellular trafficking of long chain fatty acids. J. Lipid Res. 1999, 40, 1371–1383. [Google Scholar] [PubMed]
- Taber, L.; Chiu, C.H.; Whelan, J. Assessment of the arachidonic acid content in foods commonly consumed in the American diet. Lipids 1998, 33, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.J.; Johnson, L.; O’Dea, K.; Holman, R.T. Diets rich in lean beef increase arachidonic acid and long-chain omega 3 polyunsaturated fatty acid levels in plasma phospholipids. Lipids 1994, 29, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ng, A.; Mann, N.J.; Sinclair, A.J. Contribution of meat fat to dietary arachidonic acid. Lipids 1998, 33, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, H.; Kang, S.; Park, W.J. Fatty Acid Desaturases, Polyunsaturated Fatty Acid Regulation, and Biotechnological Advances. Nutrients 2016, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Investig. 2001, 107, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Yamamoto, K.; Taketomi, Y.; Sato, H.; Shimo, K.; Kobayashi, T.; Ishikawa, Y.; Ishii, T.; Nakanishi, H.; Ikeda, K.; et al. Lymphoid tissue phospholipase A2 group IID resolves contact hypersensitivity by driving antiinflammatory lipid mediators. J. Exp. Med. 2013, 210, 1217–1234. [Google Scholar] [CrossRef] [PubMed]
- Kano, M. Control of synaptic function by endocannabinoid-mediated retrograde signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2014, 90, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Akiba, S.; Sato, T.; Fujii, T. Contribution of phospholipases A2 and D to arachidonic acid liberation and prostaglandin D2 formation with increase in intracellular Ca2+ concentration in rat peritoneal mast cells. FEBS J. 1994, 219, 401–406. [Google Scholar] [CrossRef]
- Ahn, K.; McKinney, M.K.; Cravatt, B.F. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 2008, 108, 1687–1707. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, P.; Samuelsson, B. On the organization and mechanism of prostaglandin synthetase. J. Biol. Chem. 1973, 248, 5673–5678. [Google Scholar] [PubMed]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.H.; Bokoch, G.M.; Carpenter, C.L.; Janmey, P.A.; Taylor, L.A.; Toker, A.; Stossel, T.P. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell 1995, 82, 643–653. [Google Scholar] [CrossRef]
- Kabashima, K.; Murata, T.; Tanaka, H.; Matsuoka, T.; Sakata, D.; Yoshida, N.; Katagiri, K.; Kinashi, T.; Tanaka, T.; Miyasaka, M.; et al. Thromboxane A2 modulates interaction of dendritic cells and T cells and regulates acquired immunity. Nat. Immunol. 2003, 4, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Cogolludo, A.; Moreno, L.; Bosca, L.; Tamargo, J.; Perez-Vizcaino, F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: Role of protein kinase Czeta. Circ. Res. 2003, 93, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Wu, T.J.; Wu, K.K.; Sturino, C.; Metters, K.; Gottesdiener, K.; Wright, S.D.; Wang, Z.; O’Neill, G.; Lai, E.; et al. Antagonism of the prostaglandin D2 receptor 1 suppresses nicotinic acid-induced vasodilation in mice and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 6682–6687. [Google Scholar] [CrossRef] [PubMed]
- Taketomi, Y.; Ueno, N.; Kojima, T.; Sato, H.; Murase, R.; Yamamoto, K.; Tanaka, S.; Sakanaka, M.; Nakamura, M.; Nishito, Y.; et al. Mast cell maturation is driven via a group III phospholipase A2-prostaglandin D2-DP1 receptor paracrine axis. Nat. Immunol. 2013, 14, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Basu, S. Novel cyclooxygenase-catalyzed bioactive prostaglandin F2alpha from physiology to new principles in inflammation. Med. Res. Rev. 2007, 27, 435–468. [Google Scholar] [CrossRef] [PubMed]
- Brock, T.G.; McNish, R.W.; Peters-Golden, M. Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J. Biol. Chem. 1999, 274, 11660–11666. [Google Scholar] [CrossRef] [PubMed]
- Stockley, P. Female multiple mating behaviour, early reproductive failure and litter size variation in mammals. Proc. Biol. Sci. 2003, 270, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R.; Chang, S.H.; Tolle, K.C.; Dillon, R.; Young, L.J.; Cardiff, R.D.; Newman, R.A.; Yang, P.; Thaler, H.T.; Muller, W.J.; et al. HER2/neu-induced mammary tumorigenesis and angiogenesis are reduced in cyclooxygenase-2 knockout mice. Cancer Res. 2005, 65, 10113–10119. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Tieu, K.; Choi, D.K.; Wu, D.C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef] [PubMed]
- Minami, T.; Nakano, H.; Kobayashi, T.; Sugimoto, Y.; Ushikubi, F.; Ichikawa, A.; Narumiya, S.; Ito, S. Characterization of EP receptor subtypes responsible for prostaglandin E2-induced pain responses by use of EP1 and EP3 receptor knockout mice. Br. J. Pharmacol. 2001, 133, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Shinomiya, S.; Naraba, H.; Ueno, A.; Utsunomiya, I.; Maruyama, T.; Ohuchida, S.; Ushikubi, F.; Yuki, K.; Narumiya, S.; Sugimoto, Y.; et al. Regulation of TNFalpha and interleukin-10 production by prostaglandins I(2) and E(2): Studies with prostaglandin receptor-deficient mice and prostaglandin E-receptor subtype-selective synthetic agonists. Biochem. Pharmacol. 2001, 61, 1153–1160. [Google Scholar] [CrossRef]
- Murata, T.; Ushikubi, F.; Matsuoka, T.; Hirata, M.; Yamasaki, A.; Sugimoto, Y.; Ichikawa, A.; Aze, Y.; Tanaka, T.; Yoshida, N.; et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature 1997, 388, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.J.; Turitto, V.T. Prostacyclin (prostaglandin I2, PGI2) inhibits platelet adhesion and thrombus formation on subendothelium. Blood 1979, 53, 244–250. [Google Scholar] [PubMed]
- Chandrasekharan, N.V.; Dai, H.; Roos, K.L.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression. Proc. Natl. Acad. Sci. USA 2002, 99, 13926–13931. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.S.; Rokach, J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim. Biophys. Acta 2015, 1851, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, B. Leukotrienes: Mediators of immediate hypersensitivity reactions and inflammation. Science 1983, 220, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Lammermann, T.; Afonso, P.V.; Angermann, B.R.; Wang, J.M.; Kastenmuller, W.; Parent, C.A.; Germain, R.N. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 2013, 498, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.A.; Cunningham, F.M.; Ford-Hutchinson, A.W.; Smith, M.J. Leukotriene B4: A mediator of vascular permeability. Br. J. Pharmacol. 1981, 72, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, Y.; Okuno, T.; Saeki, K.; Uozaki, H.; Okada, S.; Misaka, T.; Sato, T.; Toh, H.; Fukayama, M.; Takeda, N.; et al. Protective role of the leukotriene B4 receptor BLT2 in murine inflammatory colitis. FASEB J. 2010, 24, 4678–4690. [Google Scholar] [CrossRef] [PubMed]
- Moos, M.P.; Mewburn, J.D.; Kan, F.W.; Ishii, S.; Abe, M.; Sakimura, K.; Noguchi, K.; Shimizu, T.; Funk, C.D. Cysteinyl leukotriene 2 receptor-mediated vascular permeability via transendothelial vesicle transport. FASEB J. 2008, 22, 4352–4362. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R.; Boeglin, W.E.; Chang, M.S. Discovery of a second 15S-lipoxygenase in humans. Proc. Natl. Acad. Sci. USA 1997, 94, 6148–6152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Ran, Y. Role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in hypoxia-induced pulmonary hypertension. J. Physiol. Sci. 2012, 62, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Cianci, E.; Simiele, F.; Recchiuti, A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur. J. Pharmacol. 2015, 760, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Feltenmark, S.; Gautam, N.; Brunnstrom, A.; Griffiths, W.; Backman, L.; Edenius, C.; Lindbom, L.; Bjorkholm, M.; Claesson, H.E. Eoxins are proinflammatory arachidonic acid metabolites produced via the 15-lipoxygenase-1 pathway in human eosinophils and mast cells. Proc. Natl. Acad. Sci. USA 2008, 105, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.W.; Cho, H.; Kwak, J.; Lee, S.Y.; Kang, C.J.; Jung, J.; Cho, S.; Min, K.H.; Suh, Y.G.; Kim, D.; et al. Direct activation of capsaicin receptors by products of lipoxygenases: Endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. USA 2000, 97, 6155–6160. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.T.; Welch, J.S.; Ricote, M.; Binder, C.J.; Willson, T.M.; Kelly, C.; Witztum, J.L.; Funk, C.D.; Conrad, D.; Glass, C.K. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 1999, 400, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Chavis, C.; Vachier, I.; Godard, P.; Bousquet, J.; Chanez, P. Lipoxins and other arachidonate derived mediators in bronchial asthma. Thorax 2000, 55, S38–S41. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids 2005, 73, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Ng, V.Y.; Huang, Y.; Reddy, L.M.; Falck, J.R.; Lin, E.T.; Kroetz, D.L. Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor alpha. Drug Metab. Dispos. 2007, 35, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.; Schmelzer, K.; Lee, T.S.; Fang, X.; Zhu, Y.; Spector, A.A.; Gill, S.; Morisseau, C.; Hammock, B.D.; et al. The antiinflammatory effect of laminar flow: The role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA 2005, 102, 16747–16752. [Google Scholar] [CrossRef] [PubMed]
- Inceoglu, B.; Schmelzer, K.R.; Morisseau, C.; Jinks, S.L.; Hammock, B.D. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs). Prostaglandins Other Lipid Mediat. 2007, 82, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Muroya, Y.; Roman, R.J. Cytochrome P450 eicosanoids in hypertension and renal disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Tsuboi, K.; Uyama, T. Metabolism of endocannabinoids and related N-acylethanolamines: Canonical and alternative pathways. FEBS J. 2013, 280, 1874–1894. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Deutsch, D.G. Unique pathway for anandamide synthesis and liver regeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 6339–6340. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, B.; Cinar, R.; Yin, S.; Liu, J.; Tam, J.; Godlewski, G.; Harvey-White, J.; Mordi, I.; Cravatt, B.F.; Lotersztajn, S.; et al. Hyperactivation of anandamide synthesis and regulation of cell-cycle progression via cannabinoid type 1 (CB1) receptors in the regenerating liver. Proc. Natl. Acad. Sci. USA 2011, 108, 6323–6328. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J., 2nd. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef] [PubMed]
- Balazy, M.; Poff, C.D. Biological nitration of arachidonic acid. Curr. Vasc. Pharmacol. 2004, 2, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Fam, S.S.; Morrow, J.D. The isoprostanes: Unique products of arachidonic acid oxidation-a review. Curr. Med. Chem. 2003, 10, 1723–1740. [Google Scholar] [CrossRef] [PubMed]
- Kunapuli, P.; Lawson, J.A.; Rokach, J.A.; Meinkoth, J.L.; FitzGerald, G.A. Prostaglandin F2alpha (PGF2alpha) and the isoprostane, 8, 12-iso-isoprostane F2alpha-III, induce cardiomyocyte hypertrophy. Differential activation of downstream signaling pathways. J. Biol. Chem. 1998, 273, 22442–22452. [Google Scholar] [CrossRef] [PubMed]
- Trostchansky, A.; Bonilla, L.; Thomas, C.P.; O’Donnell, V.B.; Marnett, L.J.; Radi, R.; Rubbo, H. Nitroarachidonic acid, a novel peroxidase inhibitor of prostaglandin endoperoxide H synthases 1 and 2. J. Biol. Chem. 2011, 286, 12891–12900. [Google Scholar] [CrossRef] [PubMed]
- Tallima, H.; El Ridi, R. Arachidonic acid: Physiological roles and potential health benefits—A review. J. Adv. Res. 2018, 11, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.X.; Leaf, A. Prevention of fatal cardiac arrhythmias by polyunsaturated fatty acids. Am. J. Clin. Nutr. 2000, 71, 202S–207S. [Google Scholar] [CrossRef] [PubMed]
- Kawanabe, A.; Okamura, Y. Effects of unsaturated fatty acids on the kinetics of voltage-gated proton channels heterologously expressed in cultured cells. J. Physiol. 2016, 594, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.M.; Thomas, S.; Banting, G.; Chappell, J.B. The arachidonate-activatable, NADPH oxidase-associated H+ channel is contained within the multi-membrane-spanning N-terminal region of gp91-phox. Biochem. J. 1997, 325, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Mitjavila, M.T.; Moreno, J.J. The effects of polyphenols on oxidative stress and the arachidonic acid cascade. Implications for the prevention/treatment of high prevalence diseases. Biochem. Pharmacol. 2012, 84, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.; Matabosch, X.; Llebaria, A.; Balboa, M.A.; Balsinde, J. Blockade of arachidonic acid incorporation into phospholipids induces apoptosis in U937 promonocytic cells. J. Lipid Res. 2006, 47, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Gamma-linolenic acid, arachidonic acid, and eicosapentaenoic acid as potential anticancer drugs. Nutrition 1990, 6, 429–434. [Google Scholar] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Abdelmagid, S.A.; Clarke, S.E.; Nielsen, D.E.; Badawi, A.; El-Sohemy, A.; Mutch, D.M.; Ma, D.W. Comprehensive profiling of plasma fatty acid concentrations in young healthy Canadian adults. PLoS ONE 2015, 10, e0116195. [Google Scholar] [CrossRef] [PubMed]
- Pompeia, C.; Lima, T.; Curi, R. Arachidonic acid cytotoxicity: Can arachidonic acid be a physiological mediator of cell death? Cell Biochem. Funct. 2003, 21, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M. Impact of maternal diet on human milk composition and neurological development of infants. Am. J. Clin. Nutr. 2014, 99, 734S–741S. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Folsom, A.R.; Eckfeldt, J.H. Plasma fatty acid composition and incidence of coronary heart disease in middle aged adults: The Atherosclerosis Risk in Communities (ARIC) Study. Nutr. Metab. Cardiovasc. Dis. 2003, 13, 256–266. [Google Scholar] [CrossRef]
- Salonen, J.T.; Salonen, R.; Penttila, I.; Herranen, J.; Jauhiainen, M.; Kantola, M.; Lappetelainen, R.; Maenpaa, P.H.; Alfthan, G.; Puska, P. Serum fatty acids, apolipoproteins, selenium and vitamin antioxidants and the risk of death from coronary artery disease. Am. J. Cardiol. 1985, 56, 226–231. [Google Scholar] [CrossRef]
- Miettinen, T.A.; Naukkarinen, V.; Huttunen, J.K.; Mattila, S.; Kumlin, T. Fatty-acid composition of serum lipids predicts myocardial infarction. Br. Med. J. (Clin Res. Ed.) 1982, 285, 993–996. [Google Scholar] [CrossRef]
- Goetzl, E.J.; An, S.; Smith, W.L. Specificity of expression and effects of eicosanoid mediators in normal physiology and human diseases. FASEB J. 1995, 9, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Folco, G.; Murphy, R.C. Eicosanoid transcellular biosynthesis: From cell-cell interactions to in vivo tissue responses. Pharmacol. Rev. 2006, 58, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Norris, P.C.; Reichart, D.; Dumlao, D.S.; Glass, C.K.; Dennis, E.A. Specificity of eicosanoid production depends on the TLR-4-stimulated macrophage phenotype. J. Leukoc. Biol. 2011, 90, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Astarita, G.; Kendall, A.C.; Dennis, E.A.; Nicolaou, A. Targeted lipidomic strategies for oxygenated metabolites of polyunsaturated fatty acids. Biochim. Biophys. Acta 2015, 1851, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Fredman, G.; Backhed, F.; Oh, S.F.; Vickery, T.; Schmidt, B.A.; Serhan, C.N. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012, 484, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhang, M.J.; Hellmann, J.; Kosuri, M.; Bhatnagar, A.; Spite, M. Proresolution therapy for the treatment of delayed healing of diabetic wounds. Diabetes 2013, 62, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Spite, M.; Claria, J.; Serhan, C.N. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 2014, 19, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Gilroy, D.W. Eicosanoids and the endogenous control of acute inflammatory resolution. Int. J. Biochem. Cell Biol. 2010, 42, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Drazen, J.M. Antiinflammatory potential of lipoxygenase-derived eicosanoids: A molecular switch at 5 and 15 positions? J. Clin. Investig. 1997, 99, 1147–1148. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell. 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.P.; Eckman, K.; Lee, Y.K.; Abdelmegeed, M.A.; Esterle, A.; Chilian, W.M.; Chiang, J.Y.; Song, B.J. Eicosanoids in metabolic syndrome. Adv. Pharmacol. 2013, 66, 157–266. [Google Scholar] [PubMed]
- Back, M.; Hansson, G.K. Leukotriene receptors in atherosclerosis. Ann. Med. 2006, 38, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Kirkby, N.S. Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. Br. J. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Glass, C.K. Anti-inflammatory therapy in chronic disease: Challenges and opportunities. Science 2013, 339, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Cyrus, T.; Witztum, J.L.; Rader, D.J.; Tangirala, R.; Fazio, S.; Linton, M.F.; Funk, C.D. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J. Clin. Investig. 1999, 103, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Della Bona, R.; Cardillo, M.T.; Leo, M.; Biasillo, G.; Gustapane, M.; Trotta, F.; Biasucci, L.M. Polymorphonuclear neutrophils and instability of the atherosclerotic plaque: A causative role? Inflamm. Res. 2013, 62, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Filep, J.G.; Zouki, C.; Petasis, N.A.; Hachicha, M.; Serhan, C.N. Anti-inflammatory actions of lipoxin A(4) stable analogs are demonstrable in human whole blood: Modulation of leukocyte adhesion molecules and inhibition of neutrophil-endothelial interactions. Blood 1999, 94, 4132–4142. [Google Scholar] [PubMed]
- Merched, A.J.; Ko, K.; Gotlinger, K.H.; Serhan, C.N.; Chan, L. Atherosclerosis: Evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 2008, 22, 3595–3606. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Nakahira, K.; Dalli, J.; Siempos, I.I.; Norris, P.C.; Colas, R.A.; Moon, J.S.; Shinohara, M.; Hisata, S.; Howrylak, J.A.; et al. NLRP3 Inflammasome Deficiency Protects against Microbial Sepsis via Increased Lipoxin B4 Synthesis. Am. J. Respir. Crit. Care Med. 2017, 196, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Rocha, V.Z.; Libby, P. Obesity, inflammation, and atherosclerosis. Nat. Rev. Cardiol. 2009, 6, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Bornfeldt, K.E.; Tabas, I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell metab. 2011, 14, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization of the United Nations (FAO). Fats and Fatty Acids in Human nutrition—Report of An Expert Consultation. 2010. Available online: http://www.who.int/nutrition/publications/nutrientrequirements/fatsandfattyacids_humannutrition/en/ (accessed on 22 October 2018).
- Djousse, L.; Akinkuolie, A.O.; Wu, J.H.; Ding, E.L.; Gaziano, J.M. Fish consumption, omega-3 fatty acids and risk of heart failure: A meta-analysis. Clin. Nutr. 2012, 31, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, A.S.; Brown, T.J.; Brainard, J.S.; Biswas, P.; Thorpe, G.C.; Moore, H.J.; Deane, K.H.; AlAbdulghafoor, F.K.; Summerbell, C.D.; Worthington, H.V.; et al. Omega-3 fatty acids for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2018, 7, CD003177. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.S.; Mozaffarian, D.; Rimm, E.; Kris-Etherton, P.; Rudel, L.L.; Appel, L.J.; Engler, M.M.; Engler, M.B.; Sacks, F. Omega-6 fatty acids and risk for cardiovascular disease: A science advisory from the American Heart Association Nutrition Subcommittee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Cardiovascular Nursing; and Council on Epidemiology and Prevention. Circulation 2009, 119, 902–907. [Google Scholar] [PubMed]
- Aung, T.; Halsey, J.; Kromhout, D.; Gerstein, H.C.; Marchioli, R.; Tavazzi, L.; Geleijnse, J.M.; Rauch, B.; Ness, A.; Galan, P.; et al. Associations of Omega-3 Fatty Acid Supplement Use With Cardiovascular Disease Risks: Meta-analysis of 10 Trials Involving 77917 Individuals. JAMA cardiol. 2018, 3, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Vanden Heuvel, J.P. Cardiovascular disease-related genes and regulation by diet. Curr. Atheroscler. Rep. 2009, 11, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Vanden Heuvel, J.P. Nutrigenomics and nutrigenetics of omega3 polyunsaturated fatty acids. Prog. Mol. Biol. Transl. Sci. 2012, 108, 75–112. [Google Scholar] [PubMed]
- Nguyen, M.T.; Favelyukis, S.; Nguyen, A.K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef] [PubMed]
- Turinsky, J.; O’Sullivan, D.M.; Bayly, B.P. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J. Biol. Chem. 1990, 265, 16880–16885. [Google Scholar] [PubMed]
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef] [PubMed]
- Kennerly, D.A.; Sullivan, T.J.; Sylwester, P.; Parker, C.W. Diacylglycerol metabolism in mast cells: A potential role in membrane fusion and arachidonic acid release. J. Exp. Med. 1979, 150, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, M.; Suraamornkul, S.; Romanelli, A.; Cline, G.W.; Mandarino, L.J.; Shulman, G.I.; DeFronzo, R.A. Effect of a sustained reduction in plasma free fatty acid concentration on intramuscular long-chain fatty Acyl-CoAs and insulin action in type 2 diabetic patients. Diabetes 2005, 54, 3148–3153. [Google Scholar] [CrossRef] [PubMed]
- Pickens, C.A.; Sordillo, L.M.; Zhang, C.; Fenton, J.I. Obesity is positively associated with arachidonic acid-derived 5- and 11-hydroxyeicosatetraenoic acid (HETE). Metabolism 2017, 70, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, J.; Zhang, M.J.; Tang, Y.; Rane, M.; Bhatnagar, A.; Spite, M. Increased saturated fatty acids in obesity alter resolution of inflammation in part by stimulating prostaglandin production. J. Immunol. 2013, 191, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Borgeson, E.; Johnson, A.M.; Lee, Y.S.; Till, A.; Syed, G.H.; Ali-Shah, S.T.; Guiry, P.J.; Dalli, J.; Colas, R.A.; Serhan, C.N.; et al. Lipoxin A4 Attenuates Obesity-Induced Adipose Inflammation and Associated Liver and Kidney Disease. Cell Metab. 2015, 22, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Kompoti, M. Obesity and infection. Lancet 2006, 6, 438–446. [Google Scholar] [CrossRef]
- O’Brien, B.A.; Huang, Y.; Geng, X.; Dutz, J.P.; Finegood, D.T. Phagocytosis of apoptotic cells by macrophages from NOD mice is reduced. Diabetes 2002, 51, 2481–2488. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sun, Y.; Liang, C.P.; Thorp, E.B.; Han, S.; Jehle, A.W.; Saraswathi, V.; Pridgen, B.; Kanter, J.E.; Li, R.; et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ. Res. 2009, 105, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Biswas, S.; Shang, Y.; Collard, E.; Azad, A.; Kauh, C.; Bhasker, V.; Gordillo, G.M.; Sen, C.K.; Roy, S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS ONE 2010, 5, e9539. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, K.A.; Morris, J.L.; Feterl, M.L.; Govan, B.L.; Ketheesan, N. Altered macrophage function is associated with severe Burkholderia pseudomallei infection in a murine model of type 2 diabetes. Microbes Infect. 2011, 13, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Canetti, C.; Serezani, C.H.; Atrasz, R.G.; White, E.S.; Aronoff, D.M.; Peters-Golden, M. Activation of phosphatase and tensin homolog on chromosome 10 mediates the inhibition of FcgammaR phagocytosis by prostaglandin E2 in alveolar macrophages. J. Immunol. 2007, 179, 8350–8356. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, Y.; Matsumura, K.; Ballou, L.R.; Morham, S.G.; Blatteis, C.M. The febrile response to lipopolysaccharide is blocked in cyclooxygenase-2(−/−), but not in cyclooxygenase-1(−/−) mice. Brain res. 1999, 825, 86–94. [Google Scholar] [CrossRef]
- Collin, M.; Rossi, A.; Cuzzocrea, S.; Patel, N.S.; Di Paola, R.; Hadley, J.; Collino, M.; Sautebin, L.; Thiemermann, C. Reduction of the multiple organ injury and dysfunction caused by endotoxemia in 5-lipoxygenase knockout mice and by the 5-lipoxygenase inhibitor zileuton. J. Leukoc. Biol. 2004, 76, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Jeffcoate, W.J.; Harding, K.G. Diabetic foot ulcers. Lancet 2003, 361, 1545–1551. [Google Scholar] [CrossRef]
- Wetzler, C.; Kampfer, H.; Stallmeyer, B.; Pfeilschifter, J.; Frank, S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: Prolonged persistence of neutrophils and macrophages during the late phase of repair. J. Investig. Dermatol. 2000, 115, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Berry, E.; Liu, Y.; Chen, L.; Guo, A.M. Eicosanoids: Emerging contributors in stem cell-mediated wound healing. Prostaglandins Other Lipid Mediat. 2017, 132, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.L.; Wang, S.; Dejani, N.N.; Klopfenstein, N.; Winfree, S.; Filgueiras, L.; McCarthy, B.P.; Territo, P.R.; Serezani, C.H. Excessive localized leukotriene B4 levels dictate poor skin host defense in diabetic mice. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Reis, M.B.; Pereira, P.A.T.; Caetano, G.F.; Leite, M.N.; Galvao, A.F.; Paula-Silva, F.W.G.; Frade, M.A.C.; Faccioli, L.H. Lipoxin A4 encapsulated in PLGA microparticles accelerates wound healing of skin ulcers. PLoS ONE 2017, 12, e0182381. [Google Scholar] [CrossRef] [PubMed]
- Dejani, N.N.; Brandt, S.L.; Pineros, A.; Glosson-Byers, N.L.; Wang, S.; Son, Y.M.; Medeiros, A.I.; Serezani, C.H. Topical Prostaglandin E Analog Restores Defective Dendritic Cell-Mediated Th17 Host Defense against Methicillin-Resistant Staphylococcus Aureus in the Skin of Diabetic Mice. Diabetes 2016, 65, 3718–3729. [Google Scholar] [CrossRef] [PubMed]
- Gyurko, R.; Siqueira, C.C.; Caldon, N.; Gao, L.; Kantarci, A.; Van Dyke, T.E. Chronic hyperglycemia predisposes to exaggerated inflammatory response and leukocyte dysfunction in Akita mice. J. Immunol. 2006, 177, 7250–7256. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Kanter, J.E.; Kramer, F.; Barnhart, S.; Averill, M.M.; Vivekanandan-Giri, A.; Vickery, T.; Li, L.O.; Becker, L.; Yuan, W.; Chait, A.; et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc. Natl. Acad. Sci. USA 2012, 109, E715–E724. [Google Scholar] [CrossRef] [PubMed]
- Abrass, C.K.; Hori, M. Alterations in Fc receptor function of macrophages from streptozotocin-induced diabetic rats. J. Immunol. 1984, 133, 1307–1312. [Google Scholar] [PubMed]
- Luo, P.; Wang, M.H. Eicosanoids, beta-cell function, and diabetes. Prostaglandins Other Lipid Mediat. 2011, 95, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yan, S.; Xiao, B.; Zuo, S.; Zhang, Q.; Chen, G.; Yu, Y.; Chen, D.; Liu, Q.; Liu, Y.; et al. Prostaglandin F2alpha Facilitates Hepatic Glucose Production Through CaMKIIgamma/p38/FOXO1 Signaling Pathway in Fasting and Obesity. Diabetes 2018, 67, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Bleich, D.; Chen, S.; Zipser, B.; Sun, D.; Funk, C.D.; Nadler, J.L. Resistance to type 1 diabetes induction in 12-lipoxygenase knockout mice. J. Clin. Investig. 1999, 103, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Dobrian, A.D.; Morris, M.A.; Taylor-Fishwick, D.A.; Nadler, J.L. Lipids and immunoinflammatory pathways of beta cell destruction. Diabetologia 2016, 59, 673–678. [Google Scholar] [CrossRef] [PubMed]
- McDuffie, M.; Maybee, N.A.; Keller, S.R.; Stevens, B.K.; Garmey, J.C.; Morris, M.A.; Kropf, E.; Rival, C.; Ma, K.; Carter, J.D.; et al. Nonobese diabetic (NOD) mice congenic for a targeted deletion of 12/15-lipoxygenase are protected from autoimmune diabetes. Diabetes 2008, 57, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Zafiriou, M.P.; Zelarayan, L.C.; Noack, C.; Renger, A.; Nigam, S.; Siafaka-Kapadai, A. Hepoxilin A(3) protects beta-cells from apoptosis in contrast to its precursor, 12-hydroperoxyeicosatetraenoic acid. Biochim. Biophys. Acta 2011, 1811, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Wollam, J.; Ofrecio, J.M.; Bandyopadhyay, G.; El Ouarrat, D.; Lee, Y.S.; Oh, D.Y.; Li, P.; Osborn, O.; Olefsky, J.M. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J. Clin. Investig. 2017, 127, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Tunaru, S.; Bonnavion, R.; Brandenburger, I.; Preussner, J.; Thomas, D.; Scholich, K.; Offermanns, S. 20-HETE promotes glucose-stimulated insulin secretion in an autocrine manner through FFAR1. Nat. Commun. 2018, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Gangadhariah, M.H.; Dieckmann, B.W.; Lantier, L.; Kang, L.; Wasserman, D.H.; Chiusa, M.; Caskey, C.F.; Dickerson, J.; Luo, P.; Gamboa, J.L.; et al. Cytochrome P450 epoxygenase-derived epoxyeicosatrienoic acids contribute to insulin sensitivity in mice and in humans. Diabetologia 2017, 60, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Falck, J.R.; Manthati, V.L.; Jat, J.L.; Campbell, W.B. 20-Iodo-14,15-epoxyeicosa-8(Z)-enoyl-3-azidophenylsulfonamide: Photoaffinity labeling of a 14,15-epoxyeicosatrienoic acid receptor. Biochemistry 2011, 50, 3840–3848. [Google Scholar] [CrossRef] [PubMed]
- Gundala, N.K.V.; Naidu, V.G.M.; Das, U.N. Amelioration of streptozotocin-induced type 2 diabetes mellitus in Wistar rats by arachidonic acid. Biochem. Biophys. Res. Commun. 2018, 496, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Borgeson, E.; McGillicuddy, F.C.; Harford, K.A.; Corrigan, N.; Higgins, D.F.; Maderna, P.; Roche, H.M.; Godson, C. Lipoxin A4 attenuates adipose inflammation. FASEB J. 2012, 26, 4287–4294. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Arachidonic acid and lipoxin A4 as possible endogenous anti-diabetic molecules. Prostaglandins Leukot. Essent. Fatty Acids 2013, 88, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Is There a Role for Bioactive Lipids in the Pathobiology of Diabetes Mellitus? Front. Endocrinol. (Lausanne) 2017, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.Y.; Marklund, M.; Imamura, F.; Tintle, N.; Ardisson Korat, A.V.; de Goede, J.; Zhou, X.; Yang, W.S.; de Oliveira Otto, M.C.; Kroger, J.; et al. Omega-6 fatty acid biomarkers and incident type 2 diabetes: Pooled analysis of individual-level data for 39 740 adults from 20 prospective cohort studies. Lancet Diabetes Endocrinol. 2017, 5, 965–974. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Zoller, H.; Tilg, H. Nonalcoholic fatty liver disease and hepatocellular carcinoma. Metabolism 2016, 65, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H. Adipocytokines in nonalcoholic fatty liver disease: Key players regulating steatosis, inflammation and fibrosis. Curr. Pharm. Des. 2010, 16, 1893–1895. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Bozza, P.T.; Bakker-Abreu, I.; Navarro-Xavier, R.A.; Bandeira-Melo, C. Lipid body function in eicosanoid synthesis: An update. Prostaglandins Leukot. Essent. Fatty Acids 2011, 85, 205–213. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Adolph, T.E.; Grander, C.; Grabherr, F.; Tilg, H. Adipokines and Non-Alcoholic Fatty Liver Disease: Multiple Interactions. Int. J. Mol. Sci. 2017, 18. [Google Scholar]
- Puri, P.; Wiest, M.M.; Cheung, O.; Mirshahi, F.; Sargeant, C.; Min, H.K.; Contos, M.J.; Sterling, R.K.; Fuchs, M.; Zhou, H.; et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Horrillo, R.; Gonzalez-Periz, A.; Martinez-Clemente, M.; Lopez-Parra, M.; Ferre, N.; Titos, E.; Moran-Salvador, E.; Deulofeu, R.; Arroyo, V.; Claria, J. 5-lipoxygenase activating protein signals adipose tissue inflammation and lipid dysfunction in experimental obesity. J. Immunol. 2010, 184, 3978–3987. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Clemente, M.; Ferre, N.; Gonzalez-Periz, A.; Lopez-Parra, M.; Horrillo, R.; Titos, E.; Moran-Salvador, E.; Miquel, R.; Arroyo, V.; Funk, C.D.; et al. 5-lipoxygenase deficiency reduces hepatic inflammation and tumor necrosis factor alpha-induced hepatocyte damage in hyperlipidemia-prone ApoE-null mice. Hepatology 2010, 51, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Spite, M.; Hellmann, J.; Tang, Y.; Mathis, S.P.; Kosuri, M.; Bhatnagar, A.; Jala, V.R.; Haribabu, B. Deficiency of the leukotriene B4 receptor, BLT-1, protects against systemic insulin resistance in diet-induced obesity. J. Immunol. 2011, 187, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Clemente, M.; Ferre, N.; Titos, E.; Horrillo, R.; Gonzalez-Periz, A.; Moran-Salvador, E.; Lopez-Vicario, C.; Miquel, R.; Arroyo, V.; Funk, C.D.; et al. Disruption of the 12/15-lipoxygenase gene (Alox15) protects hyperlipidemic mice from nonalcoholic fatty liver disease. Hepatology 2010, 52, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, N.; Ikejima, K.; Yamashina, S.; Enomoto, A.; Nishiura, T.; Nishimura, T.; Brenner, D.A.; Schemmer, P.; Bradford, B.U.; Rivera, C.A.; et al. Kupffer cell-derived prostaglandin E(2) is involved in alcohol-induced fat accumulation in rat liver. Am. J. Physiol. 2000, 279, G100–G106. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.Y.; Dannenberg, A.J.; Sung, J.J.; Subbaramaiah, K.; Du, B.; Olinga, P.; Friedman, S.L. Prostaglandin E2 inhibits transforming growth factor beta 1-mediated induction of collagen alpha 1(I) in hepatic stellate cells. J. Hepatol. 2004, 41, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, S.; Kishina, M.; Koda, M.; Yamamoto, Y.; Tanaka, K.; Harada, Y.; Yoshida, A.; Hisatome, I. Nimesulide, a cyclooxygenase-2 selective inhibitor, suppresses obesity-related non-alcoholic fatty liver disease and hepatic insulin resistance through the regulation of peroxisome proliferator-activated receptor gamma. Int. J. Mol. Med. 2016, 38, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Theken, K.N.; Deng, Y.; Schuck, R.N.; Oni-Orisan, A.; Miller, T.M.; Kannon, M.A.; Poloyac, S.M.; Lee, C.R. Enalapril reverses high-fat diet-induced alterations in cytochrome P450-mediated eicosanoid metabolism. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E500–E509. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Yoo, S.H.; Jang, S.; Gonzalez, F.J.; Song, B.J. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J. Hepatol. 2012, 57, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Rankinen, T.; Zuberi, A.; Chagnon, Y.C.; Weisnagel, S.J.; Argyropoulos, G.; Walts, B.; Perusse, L.; Bouchard, C. The human obesity gene map: The 2005 update. Obesity (Silver Spring) 2006, 14, 529–644. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Azar, S.; Nemirovski, A.; Webb, M.; Halpern, Z.; Shibolet, O.; Tam, J. Serum levels of endocannabinoids are independently associated with nonalcoholic fatty liver disease. Obesity (Silver Spring) 2017, 25, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Periz, A.; Horrillo, R.; Ferre, N.; Gronert, K.; Dong, B.; Moran-Salvador, E.; Titos, E.; Martinez-Clemente, M.; Lopez-Parra, M.; Arroyo, V.; et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: A role for resolvins and protectins. FASEB J. 2009, 23, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Vicario, C.; Gonzalez-Periz, A.; Rius, B.; Moran-Salvador, E.; Garcia-Alonso, V.; Lozano, J.J.; Bataller, R.; Cofan, M.; Kang, J.X.; Arroyo, V.; et al. Molecular interplay between Delta5/Delta6 desaturases and long-chain fatty acids in the pathogenesis of non-alcoholic steatohepatitis. Gut 2014, 63, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Fracanzani, A.L.; Fargion, S.; Valenti, L. Iron in fatty liver and in the metabolic syndrome: A promising therapeutic target. J. Hepatol. 2011, 55, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Aigner, E.; Theurl, I.; Theurl, M.; Lederer, D.; Haufe, H.; Dietze, O.; Strasser, M.; Datz, C.; Weiss, G. Pathways underlying iron accumulation in human nonalcoholic fatty liver disease. Am. J. Clin. Nutr. 2008, 87, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D. Thematic review series: The pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: Part II: The early evidence linking hypercholesterolemia to coronary disease in humans. J. Lipid Res. 2005, 46, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D. Thematic review series: The pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: Part I. J. Lipid Res. 2004, 45, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Osaka, M.; Aikawa, M.; Uematsu, S.; Akira, S.; Libby, P.; Shimokado, K.; Sacks, F.M.; Yoshida, M. Toll-like receptor 2 mediates apolipoprotein CIII-induced monocyte activation. Circ. Res. 2008, 103, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Witztum, J.L.; Steinberg, D. Role of oxidized low density lipoprotein in atherogenesis. J. Clin. Investig. 1991, 88, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Engler, R.L.; Schmid-Schonbein, G.W.; Pavelec, R.S. Leukocyte capillary plugging in myocardial ischemia and reperfusion in the dog. Am. J. Pathol. 1983, 111, 98–111. [Google Scholar] [PubMed]
- Jolly, S.R.; Kane, W.J.; Hook, B.G.; Abrams, G.D.; Kunkel, S.L.; Lucchesi, B.R. Reduction of myocardial infarct size by neutrophil depletion: Effect of duration of occlusion. Am. Heart J. 1986, 112, 682–690. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013, 339, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 2007, 117, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Yoshida, N.; Shinke, T.; Otake, H.; Kuroda, M.; Sakaguchi, K.; Hirota, Y.; Toba, T.; Takahashi, H.; Terashita, D.; et al. Impact of CD14(++)CD16(+) monocytes on coronary plaque vulnerability assessed by optical coherence tomography in coronary artery disease patients. Atherosclerosis 2018, 269, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Rogacev, K.S.; Cremers, B.; Zawada, A.M.; Seiler, S.; Binder, N.; Ege, P.; Grosse-Dunker, G.; Heisel, I.; Hornof, F.; Jeken, J.; et al. CD14++CD16+ monocytes independently predict cardiovascular events: A cohort study of 951 patients referred for elective coronary angiography. J. Am. Coll. Cardiol. 2012, 60, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Demetz, E.; Schroll, A.; Auer, K.; Heim, C.; Patsch, J.R.; Eller, P.; Theurl, M.; Theurl, I.; Theurl, M.; Seifert, M.; et al. The arachidonic acid metabolome serves as a conserved regulator of cholesterol metabolism. Cell Metab. 2014, 20, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Ge, S.; Liu, D.; Xu, G.; Zhang, R.; Yin, Q.; Zhu, W.; Chen, J.; Liu, X. Atorvastatin reduces plaque vulnerability in an atherosclerotic rabbit model by altering the 5-lipoxygenase pathway. Cardiology 2010, 115, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Zernecke, A.; Libby, P. The multifaceted contributions of leukocyte subsets to atherosclerosis: Lessons from mouse models. Nat. Rev. Immunol. 2008, 8, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Bot, I.; Djalali-Talab, Y.; Shagdarsuren, E.; Bidzhekov, K.; Meiler, S.; Krohn, R.; Schober, A.; Sperandio, M.; Soehnlein, O.; et al. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ. Res. 2008, 102, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.S.; et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, T.; Runge, S.; Chatterjee, A.; Chen, M.; Mottola, G.; Fitzgerald, J.M.; Serhan, C.N.; Conte, M.S. D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J. 2013, 27, 2220–2232. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.J.; Spite, M.; Owens, C.D.; Lancero, H.; Kroemer, A.H.; Pande, R.; Creager, M.A.; Serhan, C.N.; Conte, M.S. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am. J. Pathol. 2010, 177, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
- Hachicha, M.; Pouliot, M.; Petasis, N.A.; Serhan, C.N. Lipoxin (LX)A4 and aspirin-triggered 15-epi-LXA4 inhibit tumor necrosis factor 1alpha-initiated neutrophil responses and trafficking: Regulators of a cytokine-chemokine axis. J. Exp. Med. 1999, 189, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Sodin-Semrl, S.; Taddeo, B.; Tseng, D.; Varga, J.; Fiore, S. Lipoxin A4 inhibits IL-1 beta-induced IL-6, IL-8, and matrix metalloproteinase-3 production in human synovial fibroblasts and enhances synthesis of tissue inhibitors of metalloproteinases. J. Immunol. 2000, 164, 2660–2666. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.H.; Laguna-Fernandez, A.; Arnardottir, H.; Wheelock, C.E.; Perretti, M.; Hansson, G.K.; Back, M. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E(−/−) mice. Br. J. Pharmacol. 2017, 174, 4043–4054. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Moos, M.P.; Grabner, R.; Pedrono, F.; Fan, J.; Kaiser, B.; John, N.; Schmidt, S.; Spanbroek, R.; Lotzer, K.; et al. The 5-lipoxygenase pathway promotes pathogenesis of hyperlipidemia-dependent aortic aneurysm. Nat. Med. 2004, 10, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Brink, C.; Dahlen, S.E.; Drazen, J.; Evans, J.F.; Hay, D.W.; Nicosia, S.; Serhan, C.N.; Shimizu, T.; Yokomizo, T. International Union of Pharmacology XXXVII. Nomenclature for leukotriene and lipoxin receptors. Pharmacol. Rev. 2003, 55, 195–227. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, A.; Kanaoka, Y.; Xing, W.; Austen, K.F. Functional recognition of a distinct receptor preferential for leukotriene E4 in mice lacking the cysteinyl leukotriene 1 and 2 receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 16695–16700. [Google Scholar] [CrossRef] [PubMed]
- Paruchuri, S.; Jiang, Y.; Feng, C.; Francis, S.A.; Plutzky, J.; Boyce, J.A. Leukotriene E4 activates peroxisome proliferator-activated receptor gamma and induces prostaglandin D2 generation by human mast cells. J. Biol. Chem. 2008, 283, 16477–16487. [Google Scholar] [CrossRef] [PubMed]
- Paruchuri, S.; Tashimo, H.; Feng, C.; Maekawa, A.; Xing, W.; Jiang, Y.; Kanaoka, Y.; Conley, P.; Boyce, J.A. Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J. Exp. Med. 2009, 206, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, G. Leukotrienes and the cardiovascular system. Prostaglandins 1984, 27, 781–802. [Google Scholar] [CrossRef]
- Folco, G.; Rossoni, G.; Buccellati, C.; Berti, F.; Maclouf, J.; Sala, A. Leukotrienes in cardiovascular diseases. Am. J. Respir. Crit. Care Med. 2000, 161, 1121. [Google Scholar] [CrossRef] [PubMed]
- Walch, L.; Norel, X.; Gascard, J.P.; Brink, C. Functional studies of leukotriene receptors in vascular tissues. Am. J. Respir. Crit. Care Med. 2000, 161, 1071. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Leukotrienes C4 and D4 stimulate human endothelial cells to synthesize platelet-activating factor and bind neutrophils. Proc. Natl. Acad. Sci. USA 1986, 83, 2204–2208. [Google Scholar] [CrossRef] [PubMed]
- Datta, Y.H.; Romano, M.; Jacobson, B.C.; Golan, D.E.; Serhan, C.N.; Ewenstein, B.M. Peptido-leukotrienes are potent agonists of von Willebrand factor secretion and P-selectin surface expression in human umbilical vein endothelial cells. Circulation 1995, 92, 3304–3311. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Mazzone, A.; Giannessi, D.; Sicari, R.; Pelosi, W.; Lazzerini, G.; Azzara, A.; Forder, R.; Carey, F.; Caruso, D.; et al. Leukotriene B4 production in human atherosclerotic plaques. Biomed. Biochim. Acta 1988, 47, 1821. [Google Scholar]
- Spanbroek, R.; Grabner, R.; Lotzer, K.; Hildner, M.; Urbach, A.; Ruhling, K.; Moos, M.P.; Kaiser, B.; Cohnert, T.U.; Wahlers, T.; et al. Expanding expression of the 5-lipoxygenase pathway within the arterial wall during human atherogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Cipollone, F.; Mezzetti, A.; Fazia, M.L.; Cuccurullo, C.; Iezzi, A.; Ucchino, S.; Spigonardo, F.; Bucci, M.; Cuccurullo, F.; Prescott, S.M.; et al. Association between 5-lipoxygenase expression and plaque instability in humans. Arter. Thromb. Vasc. Biol. 2005, 25, 1665–1670. [Google Scholar] [CrossRef] [PubMed]
- Ketelhuth, D.F.; Hermansson, A.; Hlawaty, H.; Letourneur, D.; Yan, Z.Q.; Back, M. The leukotriene B4 receptor (BLT) antagonist BIIL284 decreases atherosclerosis in ApoE−/− mice. Prostaglandins Other Lipid Mediat. 2015, 121, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hall, S.R.; Moos, M.P.; Cao, R.Y.; Ishii, S.; Ogunyankin, K.O.; Melo, L.G.; Funk, C.D. Endothelial cysteinyl leukotriene 2 receptor expression mediates myocardial ischemia-reperfusion injury. Am. J. Pathol. 2008, 172, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Hui, Y.; Cheng, Y.; Smalera, I.; Jian, W.; Goldhahn, L.; Fitzgerald, G.A.; Funk, C.D. Directed vascular expression of human cysteinyl leukotriene 2 receptor modulates endothelial permeability and systemic blood pressure. Circulation 2004, 110, 3360–3366. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Gabrielsen, A.; Agardh, H.E.; Wan, M.; Wetterholm, A.; Wong, C.H.; Hedin, U.; Swedenborg, J.; Hansson, G.K.; Samuelsson, B.; et al. Expression of 5-lipoxygenase and leukotriene A4 hydrolase in human atherosclerotic lesions correlates with symptoms of plaque instability. Proc. Natl. Acad. Sci. USA 2006, 103, 8161–8166. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, A.; Manolescu, A.; Thorleifsson, G.; Gretarsdottir, S.; Jonsdottir, H.; Thorsteinsdottir, U.; Samani, N.J.; Gudmundsson, G.; Grant, S.F.; Thorgeirsson, G.; et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat. Genet. 2004, 36, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, A.; Manolescu, A.; Helgason, A.; Thorleifsson, G.; Thorsteinsdottir, U.; Gudbjartsson, D.F.; Gretarsdottir, S.; Magnusson, K.P.; Gudmundsson, G.; Hicks, A.; et al. A variant of the gene encoding leukotriene A4 hydrolase confers ethnicity-specific risk of myocardial infarction. Nat. Genet. 2006, 38, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Mehrabian, M.; Allayee, H.; Wong, J.; Shi, W.; Wang, X.P.; Shaposhnik, Z.; Funk, C.D.; Lusis, A.J. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ. Res. 2002, 91, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Jawien, J.; Gajda, M.; Rudling, M.; Mateuszuk, L.; Olszanecki, R.; Guzik, T.J.; Cichocki, T.; Chlopicki, S.; Korbut, R. Inhibition of five lipoxygenase activating protein (FLAP) by MK-886 decreases atherosclerosis in apoE/LDLR-double knockout mice. Eur. J. Clin. Investig. 2006, 36, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Jawien, J.; Gajda, M.; Olszanecki, R.; Korbut, R. BAY x 1005 attenuates atherosclerosis in apoE/LDLR—Double knockout mice. J. Physiol. Pharmacol. 2007, 58, 583–588. [Google Scholar] [PubMed]
- Mawhin, M.A.; Tilly, P.; Zirka, G.; Charles, A.L.; Slimani, F.; Vonesch, J.L.; Michel, J.B.; Back, M.; Norel, X.; Fabre, J.E. Neutrophils recruited by leukotriene B4 induce features of plaque destabilization during endotoxemia. Cardiovasc. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Moos, M.P.; Funk, C.D. Endothelial cysteinyl leukotriene 2 receptor expression and myocardial ischemia/reperfusion injury. Trends Cardiovasc. Med. 2008, 18, 268–273. [Google Scholar] [CrossRef] [PubMed]
- De Hoog, V.C.; Bovens, S.M.; de Jager, S.C.; van Middelaar, B.J.; van Duijvenvoorde, A.; Doevendans, P.A.; Pasterkamp, G.; de Kleijn, D.P.; Timmers, L. BLT1 antagonist LSN2792613 reduces infarct size in a mouse model of myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2015, 108, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Becher, U.M.; Ghanem, A.; Tiyerili, V.; Furst, D.O.; Nickenig, G.; Mueller, C.F. Inhibition of leukotriene C4 action reduces oxidative stress and apoptosis in cardiomyocytes and impedes remodeling after myocardial injury. J. Mol. Cell. Cardiol. 2011, 50, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Nobili, E.; Salvado, M.D.; Folkersen, L.; Castiglioni, L.; Kastrup, J.; Wetterholm, A.; Tremoli, E.; Hansson, G.K.; Sironi, L.; Haeggstrom, J.Z.; et al. Cysteinyl leukotriene signaling aggravates myocardial hypoxia in experimental atherosclerotic heart disease. PLoS ONE 2012, 7, e41786. [Google Scholar] [CrossRef] [PubMed]
- Allayee, H.; Hartiala, J.; Lee, W.; Mehrabian, M.; Irvin, C.G.; Conti, D.V.; Lima, J.J. The effect of montelukast and low-dose theophylline on cardiovascular disease risk factors in asthmatics. Chest 2007, 132, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, E.; Yin, L.; Back, M. Nationwide cohort study of the leukotriene receptor antagonist montelukast and incident or recurrent cardiovascular disease. J. Allergy Clin. Immunol. 2012, 129, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, M.; Rovati, G.E.; Cavanillas, A.B. The leukotriene receptor antagonist montelukast and its possible role in the cardiovascular field. Eur. J. Clin. Pharmacol. 2017, 73, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New. Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.A.S.; Dias, A.T.; Porto, M.L.; Brun, B.F.; Gava, A.L.; Meyrelles, S.S.; Gil-Longo, J.; Campos-Toimil, M.; Pereira, T.M.C.; Vasquez, E.C. Sildenafil (Viagra®) Prevents Cox-1/ TXA2 Pathway-Mediated Vascular Hypercontractility in ApoE−/− Mice. Cell. Physiol. Biochem. 2017, 44, 1796–1809. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Cook, N.R.; Gaziano, J.M.; Price, J.F.; Belch, J.F.F.; Roncaglioni, M.C.; Morimoto, T.; Mehta, Z. Effects of aspirin on risks of vascular events and cancer according to bodyweight and dose: Analysis of individual patient data from randomised trials. Lancet 2018, 392, 387–399. [Google Scholar] [CrossRef]
- MacDonald, T.M.; Hawkey, C.J.; Ford, I.; McMurray, J.J.V.; Scheiman, J.M.; Hallas, J.; Findlay, E.; Grobbee, D.E.; Hobbs, F.D.R.; Ralston, S.H.; et al. Randomized trial of switching from prescribed non-selective non-steroidal anti-inflammatory drugs to prescribed celecoxib: The Standard care vs. Celecoxib Outcome Trial (SCOT). Eur. Heart J. 2017, 38, 1843–1850. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Yeomans, N.D.; Solomon, D.H.; Luscher, T.F.; Libby, P.; Husni, M.E.; Graham, D.Y.; Borer, J.S.; Wisniewski, L.M.; Wolski, K.E.; et al. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N. Engl. J. Med. 2016, 375, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Celotti, F.; Durand, T. The metabolic effects of inhibitors of 5-lipoxygenase and of cyclooxygenase 1 and 2 are an advancement in the efficacy and safety of anti-inflammatory therapy. Prostaglandins Other Lipid Mediat. 2003, 71, 147–162. [Google Scholar] [CrossRef]
- Imig, J.D. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am. J. Physiol. Renal. Physiol. 2005, 289, F496–F503. [Google Scholar] [CrossRef] [PubMed]
- Neckar, J.; Kopkan, L.; Huskova, Z.; Kolar, F.; Papousek, F.; Kramer, H.J.; Hwang, S.H.; Hammock, B.D.; Imig, J.D.; Maly, J.; et al. Inhibition of soluble epoxide hydrolase by cis-4-[4-(3-adamantan-1-ylureido)cyclohexyl-oxy]benzoic acid exhibits antihypertensive and cardioprotective actions in transgenic rats with angiotensin II-dependent hypertension. Clin. Sci. (Lond.) 2012, 122, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Duflot, T.; Roche, C.; Lamoureux, F.; Guerrot, D.; Bellien, J. Design and discovery of soluble epoxide hydrolase inhibitors for the treatment of cardiovascular diseases. Expert Opin. Drug Discov. 2014, 9, 229–243. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 3285. https://doi.org/10.3390/ijms19113285
Sonnweber T, Pizzini A, Nairz M, Weiss G, Tancevski I. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. International Journal of Molecular Sciences. 2018; 19(11):3285. https://doi.org/10.3390/ijms19113285
Chicago/Turabian StyleSonnweber, Thomas, Alex Pizzini, Manfred Nairz, Günter Weiss, and Ivan Tancevski. 2018. "Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases" International Journal of Molecular Sciences 19, no. 11: 3285. https://doi.org/10.3390/ijms19113285
APA StyleSonnweber, T., Pizzini, A., Nairz, M., Weiss, G., & Tancevski, I. (2018). Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. International Journal of Molecular Sciences, 19(11), 3285. https://doi.org/10.3390/ijms19113285