Stable Isotope Labeling Highlights Enhanced Fatty Acid and Lipid Metabolism in Human Acute Myeloid Leukemia

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Lipid Metabolism is Dysregulated in IDH1 Mutant Cells
2.2. Lipidomic Experiments Confirmed Changes in Fatty Acid Reorganization in IDH1 R132H AML Cells
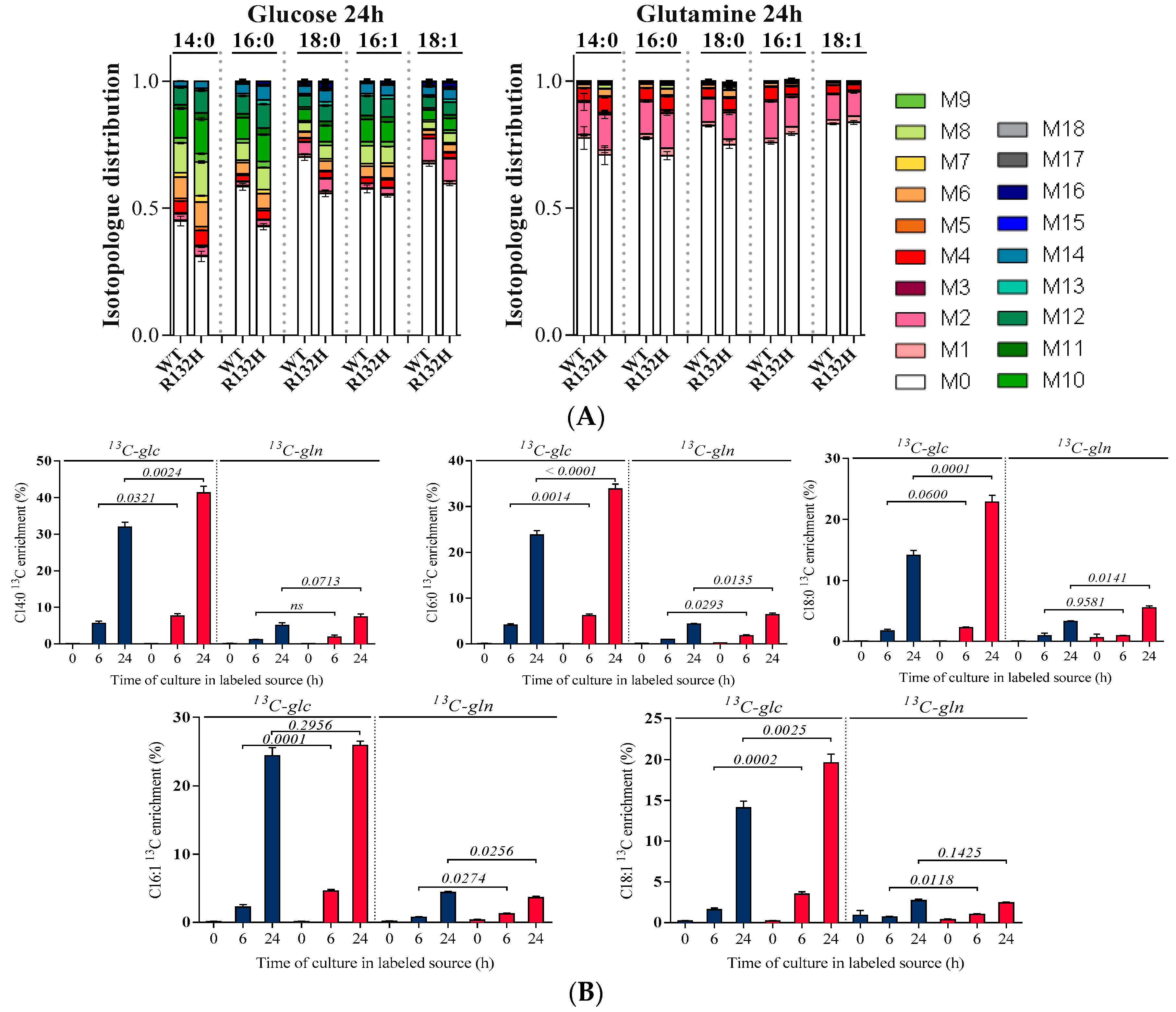
2.3. Isotopic Measurements of FAs Revealed Enhanced Lipid Anabolic Fluxes in IDH1 Mutant AML Cells
2.4. Lipogenesis is Regulated by 2-HG Production in IDH1 Mutant Cells
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Cell Culture
3.3. Proteomics
3.3.1. Protein Preparation
3.3.2. Analysis, Identification and Quantification of Proteins
3.3.3. Data Exploration and Mining
3.4. Lipidomic Analysis
3.4.1. Preparation of Total Lipid Extracts
3.4.2. Phospholipid and Sphingolipid Relative Quantification
3.4.3. Sphingoid Bases
3.4.4. Neutral Lipid Relative Quantification
3.4.5. Total FA Profiling
3.5. FA Isotopic Labeling Profiling
3.5.1. Sample Preparation
3.5.2. GC-MS Analysis
3.5.3. Data Processing
3.6. Immunoblotting of Total Proteins
3.7. ChIP Assays
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Acetic acid |
| ACN | Acetonitrile HPLC-grade |
| AML | Accute Myeloide Leukemia |
| AraC | Cytarabine |
| AU | Arbitrary Unit |
| B&D | Bligh and Dyer |
| BF3-MeOH | Boron trifluoride-methanol solution 14% |
| Cer | Ceramides |
| CH2Cl2 | Dichloromethane HPLC-grade |
| ESI | Electrospray ionization |
| FA | Fatty acids |
| FAMEs | Fatty acid methyl esters |
| FAO | Fatty acids oxidation |
| FID | Flame Ionization Detector |
| GC | Gas chromatography |
| GO | Gene Ontology |
| H2O | Ultrapure water |
| HPLC | High performance liquid chromatography |
| IDH | Isocitrate dehydrogenase |
| 2-HG | 2-hydroxyglatarate |
| KOH | Acetic acid potassium hydroxide |
| LC-MS/MS | Liquid chromatography coupled to detector MS/MS |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| m/z | Mass-to-charge ratio |
| MeOH | Methanol HPLC-grade |
| MS | Mass spectrometry |
| PC | Phosphatidylcholine |
| PE | Phosphatidylethanolamine |
| PI | Phosphatidylinositol |
| PL | Glycerophospholipids |
| PS | Phosphatidylserine |
| PTFB-Br | Pentafluorobenzyl-bromide |
| PUFAs | Polyunsaturated fatty acids |
| RLCs | Resistant leukemic cells |
| S/N | Signal to noise ratio |
| Sa | Sphinganine |
| SIM | Selected ion monitoring |
| SM | Sphingomyelins |
| So | Sphingosine |
| SRM | Selected-reaction monitoring |
| MRM | Multiple-reaction monitoring |
| MS | Mass SPectrometry |
| UPLC | Ultra performance liquid chromatography |
| WT | Wild Type |
References
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Model Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. (Lond.) 2018, 38, 27. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Holthuis, J.C.; Menon, A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Lee, M.Y.; Park, S.W.; Moon, J.S.; Koh, Y.K.; Ahn, Y.H.; Park, B.W.; Kim, K.S. Up-regulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J. Biol. Chem. 2007, 282, 26122–26131. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Wilson, R.K. Cancer genome sequencing: A review. Hum. Mol. Genet. 2009, 18, R163–R168. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrozek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A cancer and leukemia group b study. J. Clin. Oncol. 2010, 28, 2348–2355. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Kronke, J.; Bullinger, L.; Spath, D.; Kayser, S.; Zucknick, M.; Gotze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.C.; Chou, S.C.; Liu, C.Y.; Chen, C.Y.; Hou, H.A.; Kuo, Y.Y.; Lee, M.C.; Ko, B.S.; Tang, J.L.; Yao, M.; et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 2011, 118, 3803–3810. [Google Scholar] [CrossRef] [PubMed]
- Boissel, N.; Nibourel, O.; Renneville, A.; Gardin, C.; Reman, O.; Contentin, N.; Bordessoule, D.; Pautas, C.; de Revel, T.; Quesnel, B.; et al. Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid leukemia: A study by the acute leukemia french association group. J. Clin. Oncol. 2010, 28, 3717–3723. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Elkashef, S.M.; Lin, A.P.; Myers, J.; Sill, H.; Jiang, D.; Dahia, P.L.M.; Aguiar, R.C.T. IDH mutation, competitive inhibition of FTO, and RNA methylation. Cancer Cell 2017, 31, 619–620. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zou, T.; Eberhart, C.G.; Villalobos, M.A.V.; Heo, H.Y.; Zhang, Y.; Wang, Y.; Wang, X.; Yu, H.; Du, Y.; et al. Predicting IDH mutation status in grade II gliomas using amide proton transfer-weighted (APTW) MRI. Magn. Reson. Med. 2017, 78, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M. Expanding the reach of cancer metabolomics. Cancer Prev. Res. (Phila.) 2012, 5, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; DeBerardinis, R.J. Genetically-defined metabolic reprogramming in cancer. Trends Endocrinol. Metab. 2012, 23, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Burla, B.; Arita, M.; Arita, M.; Bendt, A.K.; Cazenave-Gassiot, A.; Dennis, E.A.; Ekroos, K.; Han, X.; Ikeda, K.; Liebisch, G.; et al. MS-based lipidomics of human blood plasma: A community-initiated position paper to develop accepted guidelines. J. Lipid Res. 2018, 59, 2001–2017. [Google Scholar] [CrossRef] [PubMed]
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallee, J.C.; Touboul, D.; Bertrand-Michel, J.; et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7, 46658. [Google Scholar] [CrossRef] [PubMed]
- Lillington, J.M.; Trafford, D.J.; Makin, H.L. A rapid and simple method for the esterification of fatty acids and steroid carboxylic acids prior to gas-liquid chromatography. Clin. Chim. Acta 1981, 111, 91–98. [Google Scholar] [CrossRef]
- Barrans, A.; Jaspard, B.; Barbaras, R.; Chap, H.; Perret, B.; Collet, X. Pre-beta HDL: Structure and metabolism. Biochim. Biophys. Acta 1996, 1300, 73–85. [Google Scholar] [CrossRef]
- Daniels, V.W.; Smans, K.; Royaux, I.; Chypre, M.; Swinnen, J.V.; Zaidi, N. Cancer cells differentially activate and thrive on de novo lipid synthesis pathways in a low-lipid environment. PLoS ONE 2014, 9, e106913. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [PubMed]
- German, N.J.; Yoon, H.; Yusuf, R.Z.; Murphy, J.P.; Finley, L.W.; Laurent, G.; Haas, W.; Satterstrom, F.K.; Guarnerio, J.; Zaganjor, E.; et al. PHD3 loss in cancer enables metabolic reliance on fatty acid oxidation via deactivation of ACC2. Mol. Cell 2016, 63, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.J.; Duvvuri, S.; Clise-Dwyer, K.; Watt, J.C.; Mak, D.; Kantarjian, H.; Yang, D.; Ruvolo, V.; Borthakur, G. Activation of p53 signaling by MI-63 induces apoptosis in acute myeloid leukemia cells. Leuk. Lymphoma 2010, 51, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Saulnier-Blache, J.S.; Feigerlova, E.; Halimi, J.M.; Gourdy, P.; Roussel, R.; Guerci, B.; Dupuy, A.; Bertrand-Michel, J.; Bascands, J.L.; Hadjadj, S.; et al. Urinary lysophopholipids are increased in diabetic patients with nephropathy. J. Diabetes Complicat. 2017, 31, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Chaumeil, M.M.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Metabolic reprogramming in mutant IDH1 glioma cells. PLoS ONE 2015, 10, e0118781. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [PubMed]
- Viswanath, P.; Radoul, M.; Izquierdo-Garcia, J.L.; Luchman, H.A.; Gregory Cairncross, J.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Mutant IDH1 gliomas downregulate phosphocholine and phosphoethanolamine synthesis in a 2-hydroxyglutarate-dependent manner. Cancer Metab. 2018, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Sikora, J.; Dworski, S.; Jones, E.E.; Kamani, M.A.; Micsenyi, M.C.; Sawada, T.; Le Faouder, P.; Bertrand-Michel, J.; Dupuy, A.; Dunn, C.K.; et al. Acid ceramidase deficiency in mice results in a broad range of central nervous system abnormalities. Am. J. Pathol. 2017, 187, 864–883. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Don, A.S.; Lim, X.Y.; Couttas, T.A. Re-configuration of sphingolipid metabolism by oncogenic transformation. Biomolecules 2014, 4, 315–353. [Google Scholar] [CrossRef] [PubMed]
- Fielding, B. Tracing the fate of dietary fatty acids: Metabolic studies of postprandial lipaemia in human subjects. Proc. Nutr. Soc. 2011, 70, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Persson, X.M.; Blachnio-Zabielska, A.U.; Jensen, M.D. Rapid measurement of plasma free fatty acid concentration and isotopic enrichment using lc/ms. J. Lipid Res. 2010, 51, 2761–2765. [Google Scholar] [CrossRef] [PubMed]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting (13)c metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Argus, J.P.; Yu, A.K.; Wang, E.S.; Williams, K.J.; Bensinger, S.J. An optimized method for measuring fatty acids and cholesterol in stable isotope-labeled cells. J. Lipid Res. 2017, 58, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; van Dijk, T.H.; Tietge, U.J.; Boer, T.; Havinga, R.; Stellaard, F.; Groen, A.K.; Kuipers, F.; Reijngoud, D.J. High fat feeding induces hepatic fatty acid elongation in mice. PLoS ONE 2009, 4, e6066. [Google Scholar] [CrossRef] [PubMed]
- Millard, P.; Letisse, F.; Sokol, S.; Portais, J.C. Isocor: Correcting ms data in isotope labeling experiments. Bioinformatics 2012, 28, 1294–1296. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of ag-120 (ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Ricoult, S.J.; Dibble, C.C.; Asara, J.M.; Manning, B.D. Sterol regulatory element binding protein regulates the expression and metabolic functions of wild-type and oncogenic IDH1. Mol. Cell Biol. 2016, 36, 2384–2395. [Google Scholar] [CrossRef] [PubMed]
- Boutzen, H.; Saland, E.; Larrue, C.; de Toni, F.; Gales, L.; Castelli, F.A.; Cathebas, M.; Zaghdoudi, S.; Stuani, L.; Kaoma, T.; et al. Isocitrate dehydrogenase 1 mutations prime the all-trans retinoic acid myeloid differentiation pathway in acute myeloid leukemia. J. Exp. Med. 2016, 213, 483–497. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stuani, L.; Riols, F.; Millard, P.; Sabatier, M.; Batut, A.; Saland, E.; Viars, F.; Tonini, L.; Zaghdoudi, S.; Linares, L.K.; et al. Stable Isotope Labeling Highlights Enhanced Fatty Acid and Lipid Metabolism in Human Acute Myeloid Leukemia. Int. J. Mol. Sci. 2018, 19, 3325. https://doi.org/10.3390/ijms19113325
Stuani L, Riols F, Millard P, Sabatier M, Batut A, Saland E, Viars F, Tonini L, Zaghdoudi S, Linares LK, et al. Stable Isotope Labeling Highlights Enhanced Fatty Acid and Lipid Metabolism in Human Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2018; 19(11):3325. https://doi.org/10.3390/ijms19113325
Chicago/Turabian StyleStuani, Lucille, Fabien Riols, Pierre Millard, Marie Sabatier, Aurélie Batut, Estelle Saland, Fanny Viars, Laure Tonini, Sonia Zaghdoudi, Laetitia K. Linares, and et al. 2018. "Stable Isotope Labeling Highlights Enhanced Fatty Acid and Lipid Metabolism in Human Acute Myeloid Leukemia" International Journal of Molecular Sciences 19, no. 11: 3325. https://doi.org/10.3390/ijms19113325
APA StyleStuani, L., Riols, F., Millard, P., Sabatier, M., Batut, A., Saland, E., Viars, F., Tonini, L., Zaghdoudi, S., Linares, L. K., Portais, J.-C., Sarry, J.-E., & Bertrand-Michel, J. (2018). Stable Isotope Labeling Highlights Enhanced Fatty Acid and Lipid Metabolism in Human Acute Myeloid Leukemia. International Journal of Molecular Sciences, 19(11), 3325. https://doi.org/10.3390/ijms19113325