Abstract
Increases in the prevalence of obesity, insulin resistance, and metabolic syndrome has led to the increase of atrial fibrillation (AF) cases in the developed world. These AF risk factors are associated with mitochondrial dysfunction, previously modelled using peroxisome proliferator activated receptor-γ (PPARγ) coactivator-1 (Pgc-1)-deficient murine cardiac models. We explored gene and protein expression profiles of selected molecular targets related to electrophysiological function in murine Pgc-1α−/− atria. qPCR analysis surveyed genes related to Na+-K+-ATPase, K+ conductance, hyperpolarisation-activated cyclic nucleotide-gated (Hcn), Na+ channels, Ca2+ channels, and indicators for adrenergic and cholinergic receptor modulation. Western blot analysis for molecular targets specific to conduction velocity (Nav1.5 channel and gap junctions) was performed. Transcription profiles revealed downregulation of molecules related to Na+-K+-ATPase transport, Hcn-dependent pacemaker function, Na+ channel-dependent action potential activation and propagation, Ca2+ current generation, calsequestrin-2 dependent Ca2+ homeostasis, and adrenergic α1D dependent protection from hypertrophic change. Nav1.5 channel protein expression but not gap junction expression was reduced in Pgc-1α−/− atria compared to WT. Nav1.5 reduction reflects corresponding reduction in its gene expression profile. These changes, as well as the underlying Pgc-1α−/− alteration, suggest potential pharmacological targets directed towards either upstream PGC-1 signalling mechanisms or downstream ion channel changes.
1. Introduction
Cardiac arrhythmias follow breakdown of the ordered action potential (AP) excitation and propagation triggering physiological activity in successive myocardial regions that thereby generates the heartbeat. This, in turn, involves ordered sequences of ion channel activation and inactivation causing AP initiation and recovery. The most common arrhythmia, atrial fibrillation (AF), affects 1–3% of the population in the developed world. Chronic AF increases risks of morbidity, often in the form of stroke and all-cause mortality. Model-based estimates predict substantially increased AF incidences and prevalence in coming decades, resulting in ~9 and ~18 million cases in the United States and Europe, respectively, by 2060 [1].
Both age and acquired metabolic disorders constitute major risk factors for AF. AF thus affects 0.1%, 4%, and 20% of individuals aged <55, 60–70, and >80 years, respectively. Factors such as physical inactivity, obesity, diabetes mellitus and metabolic syndrome, whose occurrence and effects are themselves age-dependent, may explain ~60% of current upward trends in AF incidence [1]. Observational and experimental evidence in turn associate both ageing and metabolic disorder with mitochondrial dysfunction and impaired oxidative capacity [2]. Age-associated mitochondrial DNA damage and compromised respiratory chain function have been demonstrated in a range of human and other mammalian species [3]. Increased mitochondrial dysfunction and defective electron transport chain complex 1 occurs in obese mice on high fat diets [4]. Mitochondrial dysfunction, whether acute or chronic, is known in turn to promote cardiac arrhythmogenesis [5]. The latter is the typical result of altered expression or properties in ion channels underlying electrophysiological activity, or fibrotic or cardiomyopathic change influencing cardiomyocyte or cardiac tissue structure. The latter structural changes have also been reported in experimental diabetes [6] and metabolic syndrome [7].
Mitochondrial function is strongly influenced by members of the peroxisome proliferator activated receptor-γ (PPARγ) coactivator-1 transcriptional coactivator (Pgc-1) family. PPARγ coactivators (PGCs) are abundant in oxidative tissues, including cardiac and skeletal muscle, and brown adipose tissue [8]. Modifications in these have been used to examine pathological changes associated with energetic deficiencies. Their expression is impaired in obesity, insulin resistance, type 2 diabetes mellitus (T2DM), and first-degree relatives of diabetic patients [9]. Increasing gene expression related to fatty acid β-oxidation, the tricarboxylic acid cycle and the electron transport chain [10] allows for PGC to regulate mitochondrial biogenesis, mass and function [11]. This, in turn, influences cellular metabolism, and its link to external stimuli driving cellular energy demands [8]. They, thus, constitute potential therapeutic targets. For instance, the PPAR-α agonist fenofibrate, inhibited atrial metabolic remodelling in AF [12,13]. Specific PPARγ ligands such as thiazolidinediones (TZDs) are used in clinical practice to improve insulin sensitivity T2DM [14]. Improvement of cardiac function and reduction in myocardial fibrosis by TZDs have also been previously reported [14]. Finally, rosiglitazone reduces atrial interstitial fibrosis and AF promotion in diabetic rabbits via modulating oxidative stress and inflammation [15]. Of major Pgc-1α, Pgc-1β, and Prc (Pgc-1-related) groups of coactivators [11], Pgc-1α appears to have a more dynamic function: Pgc-1α, but not Pgc-1β, expression is upregulated by physiological stimuli such as fasting, exercise and cold temperatures, thus adapting tissues to high energy demand as opposed to basal mitochondrial function [11].
Experimental models for mitochondrial disorders directed at associated pro-arrhythmic electrophysiological changes are thus central to understanding of their relationships to development of pro-arrhythmic phenotypes. The present experiments utilise mice with a Pgc-1α−/− genotype to recapitulate features of energetic deficiency. The widespread use of murine models for cardiac electrophysiology research stems from the ability to genetically modify mice to generate targeted disruption modelling human diseases, and thus avoiding the need to use potentially nonspecific pharmacological models [16]. In terms of translatability to human cardiac electrophysiology, murine hearts show similar rapid Na+ current (INa) mediated depolarisation phases and transmural AP conduction velocities [17,18,19]. Thus, although murine hearts show anatomical differences, have higher pacing rates and shorter APs compared to human hearts, they have been established to substantially recapitulate human clinical arrhythmic phenotypes [20,21]. Previous studies have successfully validated both Pgc-1α−/− and Pgc-1β−/− murine models in the study of mitochondrial dysfunction and arrhythmogenesis [22,23,24,25,26,27,28].
Thus, a higher incidence of extra-systolic provoked atrial arrhythmias, manifesting as atrial tachycardia and ectopic activity, were observed in Pgc-1β−/− atria [27]. This was attributed to a reduced rate of AP depolarisation (dV/dt), prolonged AP latencies representing reduced AP conduction and an increased level of myocardial fibrosis [26,27]. Furthermore, at a cellular level, extensive loose patch clamp studies implicated reduced Na+ currents but unchanged K+ currents as a factor for the previously determined slowed conduction velocity in Pgc-1β−/− atria [28]. At a systems level, electrocardiographic (ECG) studies in these mice have shown irregular PR, PP and RR intervals with differing P wave morphologies as well as reduced atrioventricular node (AVN) function following adrenergic stimulation [23,24]. Interestingly, murine studies of Pgc-1α−/− overexpression have shown changes in Ca2+ signalling, electrophysiology, and contractile properties [25].
Whilst previous murine electrophysiological studies have shown slowed myocardial conduction velocity as an arrhythmogenic substrate in conditions of mitochondrial dysfunction, the underlying molecular changes remains unexplored. Therefore, the present study firstly uses Pgc-1α−/− atria in an exploration by quantitative PCR of the gene transcriptional background for changes in genes strategic for the electrophysiological and, therefore, potential arrhythmic phenotypes that may offer possible novel pharmacological targets [29]. Genes were selected and grouped according to the physiological processes underlying excitable activity [16], adapting an approach first applied to rat as opposed to genetically modified mouse hearts [30] comprising: (1) energetically-dependent Na+-K+-ATPase mediated membrane transport processes generating the ionic gradients driving excitable activity; (2) ion, particularly background K+, channels mediating the resting potential; (3) voltage-dependent processes generating both cardiac automaticity and Na+ current mediated AP activation; (4) Ca2+ homeostatic changes involving both Ca2+ channel mediated entry and the subsequent alterations in intracellular Ca2+ homeostasis between cellular compartments; (5) electrophysiological recovery from such activity through voltage-dependent K+ channel activation; (6) cellular capacity for autonomic modulation through both adrenergic and cholinergic receptor mediated activation of intracellular signalling; (7) a range of cellular and tissue changes ultimately impacting such AP generation and propagation, including inflammatory and tissue fibrotic changes reported in experimental diabetes on earlier occasions. Secondly, this study uses western blots to determine the protein expression levels of Nav1.5, Cx40 and Cx43, all critical for determining myocardial conduction velocity.
2. Results
Table 1, Table 2, Table 3, Table 4, Table 5, Table 6, Table 7 and Table 8 summarise results of investigations for independent effects of the Pgc-1α−/− genotype on expression of selected murine atrial genes. As indicated in the Methods, samples were obtained by isolating RNA from murine atrial tissue and performing qPCR using ThermoFisher custom Taqman array cards pre-probed with the 60 different genes of potential interest for cardiac function. The samples were divided into two groups, WT (n = 3) and Pgc-1α−/− (n = 3). The genetic, fold change, differences between WT and Pgc-1α−/− atria were calculated using the previously established ΔΔCt method [31]. The statistical results summarised here arise from applications of (a) paired student t-tests to assess for differences in expression of functional gene groups between WT and Pgc-1α−/− atria and (b) unpaired student t-tests to assess for differences in expression of individual genes between WT and Pgc-1α−/− atria. The genes explored were subgrouped by function bearing on their mediation of, or maintenance of cellular capacity for electrophysiological function. We stratified the resulting p values into the p < 0.001, p < 0.01, p < 0.05, and p < 0.10 levels.

Table 1.
The mean gene-fold changes underlying Na+-K+ ATPase activity in Pgc-1α−/− compared to WT atria.
Table 2.
The mean gene-fold changes underlying channels relating to the resting membrane potential in Pgc-1α−/− compared to WT atria.
Table 3.
The mean gene fold changes underlying channels relating to the initiation of excitable activity in Pgc-1α−/− compared to WT atria.
Table 4.
The mean gene fold changes underlying Ca2+ homeostasis in Pgc-1α−/− compared to WT atria.
Table 5.
The mean gene fold changes underlying channels relating to repolarisation in Pgc-1α−/− compared to WT atria.
Table 6.
The mean gene fold changes underlying adrenergic and cholinergic receptors in Pgc-1α−/− compared to WT atria.
Table 7.
The mean gene fold changes underlying the cAMP pathway in Pgc-1α−/− compared to WT atria.
Table 8.
Other mean gene fold changes in Pgc-1α−/− compared to WT atria.
2.1. Genes Encoding Transport Proteins Mediating Na+/K+ ATPase Activity
Electrophysiological activity first requires establishment and maintenance of transmembrane ionic gradients that drive its underlying ion fluxes. The gradients are established by energetically dependent membrane transport processes, of which the most fundamental is Na+-K+ ATPase activity. The resulting Na+ and K+ transmembrane gradients support cellular osmoregulation, Na+-coupled organic and inorganic solute transport and electrical excitability [32]. We accordingly tested transcription activity for the Na+-K+ ATPase catalytic α1 subunit and α2 peptide, and accessory β1 subunit. These proteins are encoded by Atpa1, Atpa2 and Atpb1, respectively. Table 1 summarises mean gene fold changes underlying Na+-K+ ATPase activity in Pgc-1α−/− compared to WT atria. It indicates a significant decrease in the level of overall gene expression within the gene group evaluated in Pgc-1α−/− compared to WT (p = 0.01). This appeared to arise from decreased expression of Atp1a1, Atp1a2 and Atp1b1 in Pgc-1α−/− compared to WT at the p = 0.063, 0.062, and 0.028 levels, respectively, compatible with diminished capacities for ATP-driven Na+ and K+ transmembrane transport.
2.2. Genes Encoding Ion Channel Molecules Mediating Ion Permeabilities Underlying Cardiomyocyte Resting Potential
The ionic, particularly K+, gradients arising from such metabolically dependent transport, sustain a resting potential baseline dependent upon the K+ permeabilities of inward rectifier and ATP-sensitive K+ and Cl− channels. First, of genes encoding Kir2.1 (Kcnj2), Kir2.2 (Kcnj12), and Kir2.3 (Kcnj4) mediating the IK1 K+ current, Kir2.1 and Kir2.2 predominate in human and rabbit ventricle with Kir2.3 having less prominent roles in mammalian heart [33]. We surveyed expression of Kcnj12, known to encode the ATP-sensitive inward rectifier K+ channel 12, Kir2.2. Secondly, KATP channels couple resting potentials to metabolic state through their regulation by intracellular nucleotides [34,35]. This would be relevant to situations with an expected deficiency in intracellular ATP as in the Pgc-1α−/− mutant. KATP channels each comprise four regulatory and four pore-forming subunits. The regulatory subunits are formed by ATP-binding cassette (ABC) transporter subunits of the ATP-sensitive K+ channels, members 8 and 9, which also form sulfonylurea receptors, SUR1 and 2 encoded by Abcc8 and Abcc9, respectively. The inwardly rectifying pore-forming K+ channel Kir6.1 and Kir6.2 subunits are encoded by Kcnj8 and Kcnj11, respectively. Cardiac Kir6.2/SUR2A KATP channels may be critical for ischaemic pre-conditioning. Table 2 accordingly surveys Abcc8 and Abcc9, and Kcnj8 and Kcnj11, respectively.
Thirdly, the G-protein regulated potassium inwardly-rectifying Kir3.1 channel mediating IKACh (subfamily J, member 3), encoded by Kcnj3, may influence background potentials and exert bradycardic effects in the sinus node [36]. Table 2 also includes Kcnj5, encoding the G protein-activated inward rectifier potassium channel 4, GIRK4, whose mutations can cause abnormal aldosterone secretion and hypertension. Finally, we tabulated Clcn3 encoding the chloride voltage-gated channel 3, important for ischemic preconditioning-induced second-window protection against myocardial infarction [37]. Table 2 summarises our analysis of mean gene-fold changes underlying channels relating to the resting membrane potential in the Pgc-1α−/− compared to WT atria. The functional group made up of the K+ conductance indicated little change in expression levels as a whole and low levels of significance (p = 0.66). At the level of individual genes, there were no significant changes in the expression of individual genes between WT and Pgc-1α−/− to a significance level of p < 0.05, with expression levels of only the regulatory Abcc8 and Abcc9 subunits of the KATP channel showing increases with p = 0.078 and 0.071, respectively, consistent with little change in the molecular basis for the baseline resting potential.
2.3. Genes Encoding Ion Channels Related to Automaticity and Initiation and Propagation of Excitable Activity
Resting potentials provide the background level from which inward currents drive depolarisation events in pacemaker tissue. Normal pacemaking depends on hyperpolarisation-activated cyclic nucleotide-gated (HCN) channels that drive repetitive action potential activation through generating pacemaker current Ih in the sinoatrial node. The HCN family comprises four members that all occur in the heart (HCN1-4) at different expression levels between cardiac regions and species. HCN4 accounts for ~80% of Ih. Of the remaining fractions, HCN1 and HCN2 dominate in rabbits, and in mice and humans, respectively [38].
Table 3 summarises mean gene-fold changes underlying channels relating to pacemaker activity and AP initiation in Pgc-1α−/− compared to WT atria. It demonstrates decreases in the level of gene expression at the functional group level including Hcn1, Hcn3 and Hcn4 in Pgc-1α−/− compared to WT to a p < 0.01 level. At the individual gene level, there was a decreased Hcn1 and Hcn4 expression in Pgc-1α−/− compared to WT to significance levels of p = 0.022 and 0.10, respectively. Pacemaker depolarisation in turn initiates Na+ current dependent regenerative processes depolarising the membrane potential thereby resulting in the AP upstroke. The major cardiac Na+ channel isoform is Nav1.5 [39], but contributions arise from other isoforms in specialized cardiac tissues such as the sino-atrial node [40]. Table 3 demonstrates a marked decrease in cardiac Nav1.5 expression to a significance level of p = 0.012, strongly suggestive of a potentially pro-arrhythmic compromise in both excitability and its propagation. There was also a downregulation in the atypical Nav2.1 encoded by Scn7a.
2.4. Genes Encoding Ion Channel Molecules Related to Surface Membrane Ca2+ Current
AP depolarisation results in surface membrane Ca2+ channel activation responsible for both the plateau phase of the cardiac AP and alterations in Ca2+ channel homeostasis. The latter arise from release of intracellularly stored sarcoplasmic reticular (SR) Ca2+ and both its re-uptake from cytosol back to SR and its exchange transport to the extracellular space. Table 4 surveys gene transcription assessments related to the voltage dependent L-type Ca2+ channels Cav1.2 (α1C subunit, encoded by Cacna1c), in which loss and gain of function CACNA1C-Q1916R [41] and CACNA1C-L762F mutations [42] result in pro-arrhythmic early and late repolarisation syndromes respectively, as well as Cav1.3 (α1D subunit, encoded by Cacna1d). We additionally included the voltage-dependent T-type, Cav3.1 (α1G subunit encoded by Cacna1g), and Cav3.2 (α1H subunit encoded by Cacna1h), that may function in normal pacemaker activity. Of accessory, nevertheless potentially regulatory subunits, we assayed the Ca2+ channel β2 subunit (encoded by Cacnb2), and α2/δ1 (Cacna2d1) and α2/δ2 subunit (Cacna2d2) of the voltage-dependent calcium channel complex. α2/δ2 is associated with increased peak amplitudes of N-, L- and T-type Ca2+ currents in the Xenopus oocyte expression system [43]; genetic knockout of α2δ1 is associated with reduced CaV2.2 levels and reduced Ca2+ current densities [44]. Ca2+ channels typically comprise complexes of α1, α2/δx, β, and γ subunits. Table 4 summarises mean gene-fold changes underlying channels related to Ca2+ homeostasis in Pgc-1α−/− compared to WT atria. At the functional group level, changes in the levels of gene expression in Pgc-1α−/− relative to WT were at p = 0.07. This was largely attributable to reduced expression in the regulatory Cacna2d2 (p = 0.005), Cacna2d1 (p = 0.065) subunits and Cacna1c and, therefore, Cav1.2 channel (p = 0.051) expression.
2.5. Genes Encoding Molecules Related to Intracellular Ca2+ Homeostasis
The depolarisation-triggered Ca2+ channel mediated entry of Ca2+ induces a release of sarcoplasmic reticular (SR) Ca2+ central to excitation contraction coupling by the cardiac isoform of the SR ryanodine receptor (RyR2) Ca2+ release channel, RyR2. Stress induced polymorphic ventricular tachycardia (PVT) and arrhythmogenic right ventricular dysplasia are conditions associated with RyR2 defects [45]. RyR3 also occurs in cardiac muscle; single nucleotide polymorphisms in this have pharmacogenetic associations with cardiac failure [46]. We assessed transcription in one of the three cardiac SERCA isoforms, Atp2a2, the principal cardiac sodium-calcium exchanger (solute carrier family 8 member A1 encoded by Slc8a1), and the cardiac isoform of the SR Ca2+ binding protein (encoded by Casq2) mutations in which cause stress-induced catecholaminergic polymorphic ventricular tachycardia type 2 (CPVT2), characterised by potentially fatal bidirectional ventricular tachycardia. Table 4 indicates that at the functional group level there were small reductions in the level of gene expression in Pgc-1α−/− relative to WT but only to a p = 0.24 significance level. Nevertheless, there were reductions in Casq2 expression (p = 0.05) suggesting reduced SR Ca2+ storage capacity, as the only change in molecular background bearing on Ca2+ homeostasis. RyR2 and RyR3 expression were relatively unchanged.
2.6. Genes Encoding Ion Channels Mediating Action Potential Recovery
The AP recovery phase that restores the transmembrane voltage to the resting potential depends on the action of outward K+ currents. These are mediated in murine hearts primarily by the voltage sensitive transient outward current Ito. This is carried by Kv1.4 (voltage-gated K+ channel subfamily A member 4, encoded by Kcna4) and Kv4.3 (voltage-gated K+ channel subfamily D member 3 encoded by Kcnd3), both of which are involved in mediating Ito1 (review: [16]). We also include assays for Kv11.1 (voltage-gated K+ channel subfamily H member 2, Kv11.1, encoded by Kcnh2) mediating the rapid K+ current, IKr, in view of its clinical importance. We also analyse the transcription of recently-characterised Ca2+-activated K+ channels (intermediate/small K+ conductance Ca2+ -activated K+ channel, subfamily N, member 1, KCa2.1, encoded by Kcnn1; intermediate/small K+ conductance Ca2+-activated channel, subfamily N, member 2, KCa2.2, encoded by Kcnn2). These are expressed differentially in the atria compared with the ventricles and thought to contribute to the AP repolarisation phase [47]. We include the two-predomain TWIK-related acid-sensitive potassium channel 1 (TASK-1) (K+ channel subfamily K member 3; K 2P 3.1, encoded by Kcnk3) [48]. Finally, we include the regulatory KCNE1 subunit, encoded by Kcne1l in view of its association with some human LQT syndromes. Table 5 indicates that at the functional group level, changes in the level of gene expression in Pgc-1α−/− compared to WT only showed a significance level of p = 0.20. Similarly at the individual gene level, there were no significant changes in the expression of individual genes between Pgc-1α−/− and WT even to a p < 0.1 significance level. These findings suggest an unchanged repolarisation function in the electrical activity of Pgc-1α−/− relative to WT.
2.7. Genes Encoding Receptors Mediating Cardiac Autonomic Responsiveness
The electrophysiological events above are modulated by autonomic, parasympathetic and sympathetic, inputs which accordingly potentially exert pro or anti-arrhythmic effects following activation of their relevant cardiac receptors. Parasympathetic stimulation has been associated with increased susceptibility to AF, but a canine AF model showed a possible compensatory muscarinic receptor, M2, M3 and M4 mAChR down-regulation [49]. Our current analysis includes Chrm2, which encodes the M2 muscarinic acetylcholine receptor. The remaining M1, M3, and M4 receptors are thought to occur in neural cells with M3 also occurring in smooth muscle, secretory cells and pancreas, rather than cardiac tissue.
Adrenergic receptors (AR) are similarly G-protein-coupled receptors critical to cardiac physiology. Although accounting for only a minor fraction of cardiac adrenoreceptors, cardiac α1-adrenoreceptors mediate important protective and adaptive functions in the heart, through Gq/11 signalling, in particular preventing pathological remodelling in heart failure [50]. We assessed transcription for all three, α1A, α1B and α1D (Adra1a, Adra1b, and Adra1d) adrenergic receptor subtypes. This is in addition to our inclusion of both β1 and β2- adrenergic receptors (Adrb1 and Adrb2), that modify both energy metabolism through cAMP, and stress related PI3K-Akt signalling. Table 6 summarises gene-fold changes underlying cholinergic and adrenergic receptors in Pgc-1α−/− compared to WT atria. At the level of the entire functional group level there was no significant (p = 0.174) change in the level of gene expression between Pgc-1α−/− and WT. At the individual gene level only the decrease in the expression of Adra1d encoding the α1D adrenergic receptor in Pgc-1α−/− compared to WT reached a significance level of p = 0.035 compatible with otherwise relatively little change in autonomic responsiveness; the latter including even β-adrenergic receptor function.
2.8. Genes Encoding Molecules Mediating Cyclic Nucleotide Signalling
The cellular capacity for response to autonomic activation depends upon G protein-coupled receptor/adenylyl cyclase/cyclic-3′,5′-adenosine monophosphate (cAMP) signalling. Of the nine isoforms of membrane-bound adenylyl cyclase, our qPCR included Adcy4 and Adcy5 encoding the enzymes adenylate cyclase types 4 and 5. Type 5 is one of the two major cardiac isoforms: its transgenic overexpression exacerbates cardiomyopathic changes with chronic catecholamine stimulation. Of eleven major phosphodiesterase subtypes, seven (PDE1, 2, 3, 4, 5, 8, and 9) are expressed in the heart, of which we have assayed transcription of Pde2a and Pde4d encoding cGMP-dependent 3′,5′-cyclic phosphodiesterase 2A and cAMP-specific 3′,5′-cyclic phosphodiesterase 4D, respectively. Of cAMP targets, we examined Prkaca encoding the phosphokinase holoenzyme forming the catalytic subunit α of protein kinase A, and the genes encoding regulatory subunits Prkar1a for cAMP-dependent protein kinase type I-α regulatory subunit, and Prkar2a and Prkar2b for the cAMP-dependent protein kinase type II-α and II-β regulatory subunits respectively [51]. Finally, we assay the major cardiac, calcium/calmodulin-dependent protein kinase, type II-δ, Camk2d, which mediates numerous cellular responses to Ca2+ signals [52]. Table 7 summarises gene -fold changes bearing on the cAMP pathway in Pgc-1α−/− compared to WT atria. The functional group level showed little difference in gene expression levels comparing Pgc-1α−/− with WT (p = 0.45). At the individual gene level, expression of Adcy4 encoding adenylyl cyclase type 4 but not cardiac type 5 was reduced (p = 0.017 and 0.71 respectively), and Prkar1a expression increased (p = 0.061) in Pgc-1α−/− compared to WT. These findings thus indicate relatively little alteration in gene expression bearing on cyclic nucleotide signalling.
2.9. Genes Encoding Fibrotic Change
Atrial fibrosis has been implicated in pro-arrhythmic atrial conduction disturbances in a range of models including those representing diabetes and age. Their markers examined here include the TGF-β1 isoform (out of TGF-β 1 to 4) for the cytokine transforming growth factor β (TGF-β) [53]. Electrical properties of homo- and heterotypic junctions involving mCx30.2 (encoded by Gjd3) may contribute to slow propagation velocity in nodal tissues and directional asymmetry of atrioventricular excitation spread [54]. Finally, we assay mRNA expression of Col1a1 encoding the major component of type I collagen, the fibrillar collagen found in most connective tissues and the collagen precursor Col3a1 encoding the collagen type III α1 chain [55]. Results for these genes are summarised in Table 8, and indicate likely insignificant decreases in expression of these (all p > 0.1), suggesting that fibrotic changes observed in these circumstances are unlikely to reflect alterations in background gene expression.
2.10. Genes Involving Developmental, Morphological or Other Background Properties
A final set of genes bearing on a number of nonexcitable physiological phenomena but nevertheless associated with pro- or anti-arrhythmic outcomes included Tbx3 of the transcriptional repressors Tbx2 and Tbx3, known to affect chamber-specific programs of gene expression that promote differentiation of distinct components of the cardiac conduction system [56]. Myh6 encodes MHC-α, particularly in human cardiac atria, the major thick filament protein, whose mutations are associated with late-onset hypertrophic cardiomyopathy, atrial septal defects, and sinus node disorder [16]. The nonspecific ion channel Trpc1 conducts both Ca2+ and Na+ with Ca2+ store depletion or activation of the phospholipase C system [57]. Finally, Nppa encodes atrial natriuretic peptide, a key autonomic nervous system and ion channel modulator, whose clinical mutations have been implicated in familial atrial fibrillation [58]. A final control assayed expression levels of Ppargc1a and Ppargc1b respectively encoding Pgc-1α and β, respectively. Table 8 suggests strongly significant alterations in Tbx3 transcription (p < 0.001), but insignificant differences in the remainder tested here (all p > 0.1).
2.11. Stratification of Gene Expression Differences between Pgc-1α and WT Changes by Magnitude and Significance
Table 9A stratifies particular changes stratified by statistical significance in categories corresponding to probability levels p < 0.001, 0.001 < p < 0.01, 0.01 < p < 0.05, and 0.05 < p < 0.1 respectively, in order to identify major features in the transcriptional background that may underlie particular physiological features resulting from the Pgc-1α−/− genotype. Notable is that all changes with a significance level p < 0.05 involved downregulatory although a number stratified between 0.1 < p < 0.05 involved upregulatory changes. At a functional group level, such a stratification implicated in descending order of significance: molecules related to pacemaker (p = 0.004), Na+-K+ ATPase activity (p = 0.01), Na+ channel (p = 0.073) and Ca2+ channel function (p = 0.07), implicating alterations in chronotropic response, energetically dependent aspects of cell ionic homeostasis, activation, and Ca2+ homeostasis, respectively.
Table 9.
Stratifications of mean gene fold changes in Pgc-1α−/− compared to WT atria.
Such findings could be compared with their effect sizes (Figure 1) for which the most marked downregulation, reflected in levels of Pgc-1α−/− normalised to WT expression, in descending order of effect size, occurred in Na+ channel (0.45 ± 0.06), pacemaker channel (0.45 ± 0.03), Na+-K+-ATPase (0.49 ± 0.05) and Ca2+ channel function (0.72 ± 0.13). All these concordantly yielded p < 0.05 in the tests for statistical significance, together giving results compatible with altered expression in molecules subserving metabolically mediated maintenance of ion gradients, SAN automaticity, AP activation and Ca2+ homeostasis. This contrasted with both high p-values and small effect sizes associated with molecules underlying cAMP signalling (1.10 ± 0.12), concerned with intracellular Ca2+ homeostasis (0.90 ± 0.07), K+ channels related to AP recovery (0.82 ± 0.12) and to resting potential maintenance (1.09 ± 0.19).
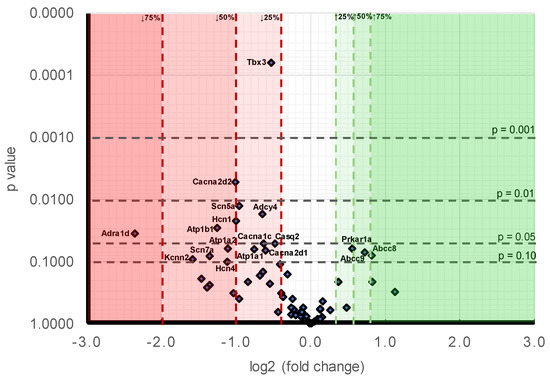
Figure 1.
Volcano plot comparing stratified statistical significances in expression levels and mean gene -fold changes in Pgc-1α−/− compared to WT atria for the collection of genes surveyed here. Horizontal stratifications correspond to probability levels p < 0.001, 0.001 < p < 0.01, 0.01 < p < 0.05 and 0.05 < p < 0.1 respectively. Ordinates stratify effect sizes indicating decreases/increases of 25%, 50%, and 75% respectively. Points falling within this stratification are labelled with the gene.
Figure 1 makes a comparison of the likelihood levels against the alterations of expressions. At the level of individual genes, in order of decreasing significance (Table 9B), the downregulated genes showing differences between Pgc-1α−/− and WT to a probability p < 0.01 were Tbx3 (p < 0.001) and Cacna2d2 (p = 0.005). The downregulated genes showing 0.01 < p < 0.05 were: Scn5a (p = 0.012), Adcy4 (p = 0.017), Hcn1 (p = 0.022), Atp1b1 (p = 0.028) and Adra1d (p = 0.035). The downregulated genes showing 0.05 < p < 0.1 were: Casq2 (p = 0.050), Cacna1c (p = 0.051), Atp1a2 (p = 0.062), Atp1a1 (p = 0.063), Cacna2d1 (p = 0.065), Scn7a (p = 0.081), Kcnn2 (p = 0.090), and Hcn4 (p = 0.100). Those genes appearing to be upregulated genes all fell within the limits <0.05 p < 0.1 were Prkar1a (p = 0.061), Abcc9 (p = 0.071) and Abcc8 (p = 0.078). A comparison of effect sizes (Table 9B) grouped the magnitudes of such alterations into: expression decreased by >75%: Adra1d (0.19 ± 0.08). Expression decreased by between 50–75%: Kcnn2 (0.33 ± 0.03), Scn7a (0.39 ± 0.09), Atp1b1 (0.42 ± 0.04), Hcn4 (0.46 ± 0.24) and Atp1a2 (0.46 ± 0.17). Expression decreased by between 25–50%: Cacna2d2 (0.50 ± 0.07), Hcn1 (0.50 ± 0.12), Scn5a (0.52 ± 0.04), Atp1a1 (0.59 ± 0.08), Adcy4 (0.64 ± 0.05), Cacna1c (0.65 ± 0.09), Cacna2d1 (0.66 ± 0.04), Tbx3 (0.69 ± 0.02), and Casq2 (0.72 ± 0.06). Expression increased by between 25–50%: Pkar1a (1.47 ± 0.14), Abcc9 (1.65 ± 0.24) and Abcc8 (1.77 ± 0.3).
2.12. Expression of Proteins Critical for Myocardial Conduction
Myocardial conduction velocity is related to the maximum rate of AP depolarisation (dV/dtmax), which is determined by the current through the Na+ channel. Additionally conduction velocity is related to the axial resistance to local circuit currents, largely determined by the conductance of gap junction proteins, typically connexins, as well as membrane capacitances [59]. The main murine connexin isoforms in the atria are Cx40 and Cx43 [60,61]. Figure 2 shows the average relative protein signal intensities of Nav1.5, Cx40, and Cx43 in Pgc-1α−/− and WT atria. Thus, compared to WT atria, the protein expression of Nav1.5 was significantly reduced in Pgc-1α−/− atria (p < 0.01). This is consistent with the qPCR results showing reduced gene expression of Scn5a, encoding Nav1.5 channel. When considering that peak Na+ current is a principal determinant of myocardial conduction velocity, this provides a potential molecular mechanism for the reduced conduction velocity observed in murine models of mitochondrial dysfunction [26,27]. However, the Pgc-1α−/− genotype was associated with no significant changes in the protein expression levels of Cx40 or Cx43.
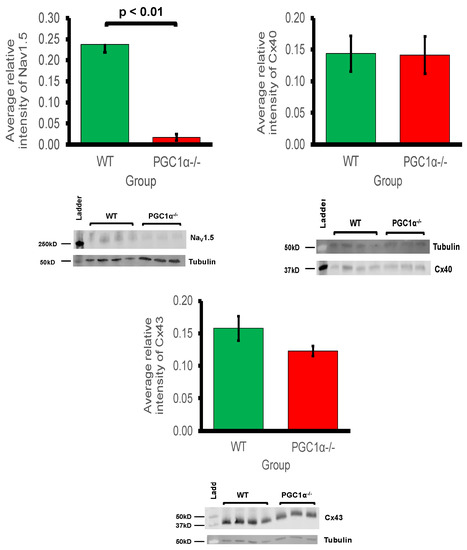
Figure 2.
Western blot analysis of atrial Nav1.5, Cx40 and Cx43 expression: 3 WT and 4 Pgc-1α−/− atria were used. Representative blots are shown and the housekeeping protein β-tubulin used as a loading control. The primary antibodies used included monoclonal rabbit anti-Nav1.5 (1:500); polyclonal goat anti-Cx40 (1:500); polyclonal rabbit anti-Cx43 (1:1000) and polyclonal goat anti-β-tubulin (1:1000). The secondary antibodies included donkey anti-goat IgG antibody in blots for Cx40 and β-tubulin (1:15,000), and donkey anti-rabbit IgG antibody in blots for Nav1.5 and Cx43 (1:10,000).
3. Discussion
Transcription factors play central roles in atrial remodelling which in turn is a major contributor to development of arrhythmic substrate leading to clinically important conditions such as atrial fibrillation. The present quantitative PCR study screened for changes in genes strategic to cardiac electrophysiological function potentially implicated in atrial arrhythmia that may potentially offer novel pharmacological targets. The studies used as model, the atria of murine hearts deficient in peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (Pgc-1α). The latter co-activator interacts with the transcription factor and nuclear receptor protein PPAR-α promoting transcription of fatty acid metabolism-related genes. Murine Pgc-1α−/− hearts thus potentially model a number of the energetic features of atrial fibrillation (AF), the most frequently clinically encountered cardiac arrhythmia. Although there remain significant gaps in our understanding of basic molecular targets in AF pathophysiology [62], recent metabolomic and proteomic studies implicated altered expression in molecules involved in metabolic pathways in human and experimental AF [62,63,64]. AF is accordingly associated with impaired mitochondrial function, decreased ATP production, and redox imbalance with increased reactive oxygen species (ROS) production [65]. The latter potentially injures genes and proteins central to cardiomyocyte function [62].
The present analysis examined expression of genes subclassified into functional groups corresponding to fundamental excitable processes in mouse [16], adapting similar approaches to rat hearts [30]. It surveyed Na+-K+-ATPase-mediated membrane transport, background K+ conductances underlying cardiomyocyte resting potentials, HCN and Na+ channels underlying cardiac automaticity and action potential activation and conduction, voltage-dependent Ca2+ current activation and intracellular Ca2+ homeostatic processes, and action potential recovery through voltage-dependent K+ currents. It also examined indicators for adrenergic and cholinergic modulation and their associated intracellular signalling processes, and longer term cellular and tissue changes including tissue fibrotic changes ultimately impacting upon AP generation and propagation.
Modelling studies predict that only large alterations in the related Na+/K+-ATPase activity would significantly alter cell background membrane potentials [32]. Atrial cardiomyocytes from patients with persistent AF did not show altered Na+-K+-ATPase current [66]. Pgc-1α−/− atria nevertheless did show decreased Atp1a1, Atp1a2, and Atp1b1 expression encoding regulatory and catalytic Na+/K+-ATPase subunits compared to levels in WT. However, Pgc-1α−/− atria showed little change in the expression of K+ channels underlying resting membrane potential maintenance, suggestive of normal background resting potential properties in Pgc-1α−/− relative to WT, findings reflected in related Pgc-1β−/− models [26]. By contrast, of genes encoding ion channels related to automaticity in and initiation and propagation of excitable activity, Pgc-1α−/− atria showed decreased transcription of pacemaker, Hcn1 and Hcn4, channels relative to WT. Such findings are relevant to the chronotropic background and its responses to autonomic activation. The findings recapitulate evidence of impaired canine sinoatrial function, and reduced sino-atrial node HCN2 and HCN4 mRNA expression and HCN related current densities in atrial tachypacing studies [67]. Previous ECG studies had demonstrated chronotropic incompetence at the level of the sino-atrial node in Pgc-1β−/− models of mitochondrial dysfunction following dobutamine challenge [23]. Pgc-1α−/− atria also showed decreases in cardiac Nav1.5 gene expression. This reduction was significant enough to be reflected at the protein level also. Previous studies in murine models of mitochondrial dysfunction have shown reduced Na+ current leading to decreased myocardial conduction velocity as a possible arrhythmogenic mechanism [26,27,28]. Thus, reduced Na+ current, arising from reduced genetic expression under conditions of mitochondrial dysfunction, is predictive of potentially proarrhythmic reductions in both excitability and AP propagation. This is in common with changes reported in murine models for Brugada Syndrome associated with loss of Na+ channel function [27]. Conduction velocity is also determined by gap channel conductance, primarily through connexin isoforms, but this study showed that protein expression levels of Cx40 and Cx43 were not significantly reduced in Pgc-1α−/− atria. This implies that the primary molecular mechanism for reduced conduction would be reduced Na+ channel expression, as opposed to altered gap junction proteins in Pgc-1α−/− atria.
Atrial cardiomyocytes show reduced L-type Ca2+ current, SR Ca2+ stores and cellular contractility with metabolic stress [68]. In AF, nuclear factor of activated T-cells (NFAT) is thought to down-regulate Cav1.2 channel α-subunit expression, through the Ca2+/calmodulin/calcineurin/NFAT pathway [69,70]. This attenuates L-type Ca2+ current and shortens atrial refractory periods [71]. In parallel with this, the qPCR analysis suggested widespread changes in molecules mediating cellular Ca2+ homeostasis. Altered transcription in genes encoding ion channel molecules related to membrane Ca2+ current manifested as reduced expression of Cacna1c, Cacna2d2, and Cacna2d1. These encode Cav1.2 channels and channel regulatory subunits, respectively. L-type Ca2+ channel (Cav1.2) α1c subunit was implicated as an important regulator of reentrant spiral dynamics and as a major component of AF-related electrical remodelling [72]. In addition loss-of-function CACNA1C-Q1916R is thought to contribute to early repolarisation syndrome-related sudden cardiac death [41], and gain of function CACNA1C-L762F with development of LQTS through slower channel inactivation and increased sustained and window current [42]. α2/δ2 is associated with increased peak amplitudes of N-, L- and T-type Ca2+ currents expressed in Xenopus oocytes [43]; genetic knockout of α2δ-1 is associated with reduced CaV2.2 levels and reduced Ca2+ current densities [44]. Mutations in the regulatory Cacna2d1 are also associated with cardiac deficiencies, including Brugada and short QT syndromes [73].
Finally, the present observations of reduced Casq2-encoded calsequestrin 2 expression, important in SR Ca2+ storage capacity, but relatively unchanged Ryr2 and Ryr3 expression mediating RyR-mediated SR Ca2+ release is compatible with potentially pro-arrhythmic diastolic releases of SR Ca2+ associated with either RyR2 [74] or Casq2-mediated [75] atrial pro-arrhythmic changes. The latter are associated with some catecholaminergic polymorphic ventricular tachycardic syndromes. In contrast to the relatively targeted gene functional changes of molecules involved in cellular electrophysiological activation and excitation contraction coupling, we did not observe changes in expression in channels known to be involved in action potential recovery through generation of voltage-dependent K+ currents.
Surveys of indicators for adrenergic and cholinergic modulation and their associated intracellular signalling processes demonstrated no significant changes in the cardiac β-adrenergic receptor or atrial parasympathetic receptor, compatible with relatively little change in the transcriptional background to autonomic responsiveness in Pgc-1α−/− compared to WT. Furthermore, changes observed in intracellular autonomic signalling were confined to neuronal adenylyl cyclise type 4, but not the cardiac type 5. Nevertheless, there was decreased expression of Adra1d. The latter encodes α1D adrenergic receptors thought to mediate Gq/11 signalling important in protective and adaptive functions against pathologic remodelling in heart failure [50]. Finally, an examination of transcriptional features of molecules involved in longer term cellular and tissue, including fibrotic changes ultimately impacting AP generation and propagation (cf. [55,76]) did not reveal major change.
The findings provide a genomic background for future therapeutic explorations directed at atrial arrhythmias directed at its associated alterations in cellular energetic processes. The therapeutic directions suggested by such findings could suggest explorations of management strategies aimed directly at the ion channels indicated as affected here. The latter could include pacemaker based HCN modulation [67,77,78]. They could also involve pharmacotherapeutic manoeuvres directed at expected alterations in ion channel functions, particularly bearing on the alterations in Ca2+ homeostasis suggested here.
Alternatively, possibilities for more upstream therapies are suggested by both clinical studies and experimental results. Studies in rabbit models associated AF with reduced protein expression of sirtuin1, PGC-1α, and PPAR-α. AF patients showed reduced atrial tissue mast cell protease 1 (mCPT-1) and glucose transporter type 4 (GLUT4) protein expression, indicating reduced FA oxidation and glucose transport, compared to control patients in sinus-rhythm. Rabbits in AF showed a similar decrease in these molecules. The PPAR-α agonist fenofibrate then restored expression of mCPT-1 and GLUT4 and activation of the PPAR-α/sirtuin1/PGC-1α pathway, and suppressed AF inducibility [13].
At the level of oxidative function, the bioactive polyphenol resveratrol is associated with diverse benefits including antioxidant cardioprotection, possibly through inducing mitochondrial biogenesis via PGC-1α activation [29]. Resveratrol activates 5’ AMP-activated protein kinase (AMPK)/sirtuin1 signalling [79], modulating cardiac metabolism. In addition, the multifunctional small-molecule resveratrol-derivative, C1 alters function in multiple ion channels mediating ultrarapid and acetylcholine-activated K+ currents, as well as Na+ currents. It showed antioxidant properties in human and rat atrial cardiomyocytes. It also reduced AF sustainability in atrial tachypaced dogs [80]. Resveratrol also modified remodelling changes associated with AF in a rabbit heart failure (HF) model by activating PI3K/AKT/eNOS signaling and reducing AF susceptibility and triggered activity. This occurred by preventing atrial electrical, contractile, and fibrotic remodelling [81]. It similarly prevented cardiomyopathy and restored cardiac function in mdx mice used to model Duchenne muscular dystrophy [82], possibly through attenuating the expression of the p300 coactivator which is a key contributor to cardiac hypertrophy and fibrosis [82].
The present analysis permits several physiological interpretations relevant to arrhythmogenesis; however this is limited to findings directly related to whole atrial tissue. There is increasing evidence to suggest that the cardiac fibroblast plays an important role in the excitation and propagation of electrical activity. It has been shown that contrary to the classical view, arrhythmogenic activity is not only specific to the cardiac myocyte but is related to heterocellular electrotonic interactions between cardiomyocytes and other cardiac related cells such as the cardiac myofibroblast. Such heterogeneity in electrical properties exist due the varying resting membrane potential of the cardiac myofibroblast relative to that of the cardiac myocyte [83,84]. Additionally, our present work used mice that were of advanced age where some level of fibrotic development may have occurred. It has been shown that in fibrotic hearts, proarrhythmic myofibroblast-cardiomyocyte crosstalk in vitro is mediated by TGF-β1 [83]. It is likely that selectively targeting the cardiac myofibroblast may be a useful anti-arrhythmic strategy as well. Given the important role cardiac myofibroblasts play in cardiac electrical properties, future studies should attempt at isolating and differentiating the gene expression and protein profile of native cardiac myofibroblasts and comparing these to corresponding features of native cardiac myocytes. Future studies should be directed at in-vivo electrophysiological interrogation of the Pgc-1α−/− murine atria including both native cardiac myocytes and cardiac myofibroblasts with particular focus on the identifying functional alterations that may correlate with the molecular findings in the present work. It would be additionally useful for future work studying ventricular gene and protein expression profile in the Pgc-1α−/− murine to allow comparison between atria and ventricular findings. Such information, whilst potentially not directly comparable due to the variation in tissue electrical properties between atria and ventricles, would allow for elucidation of other mitochondrial dysfunction mediated arrhythmogenic mechanisms.
4. Methods
4.1. Animals
All experimental protocols were approved under the UK Home Office regulations (Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012) following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB) and conducted under a designated project license number PPL70/8726, dated 4 February 2016. All procedures complied with the UK Home Office regulations (Animals (Scientific Procedures) Act 1986). We also followed the Guide for the Care and Use of Laboratory Animals, U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996). An animal house maintained at 21 °C was used for the mice, with 12-h light/dark cycles. The mice had sterile chow (RM3 Maintenance Diet, SDS, Witham, Essex, UK) and free access to water, bedding and environmental stimuli. Mice were sacrificed by cervical dislocation and no anaesthetic or surgical procedures were required. Wild-type (WT) C57/B6 and Pgc-1α−/− (The Jackson Laboratory, Bar Harbor, ME, USA) adult mice were bred for the experimental protocols. Mice were bred on a C57/B6 background to avoid possible strain-related confounds. The mice were divided into WT and Pgc-1α−/−, and all were between the ages of 12–24 months old.
4.2. Quantitative PCR
In this gene expression study, there were n = 3 mice in both the WT and Pgc-1α−/− groups. RNA was extracted from fresh frozen murine atria, stored at −80 °C, with the Qiagen RNeasy mini Plus kit (Qiagen, Manchester, UK)). The atrial tissue was weighed and quickly minced on ice. A third of the tissue, about 30 mg, was used in the next step of the RNA isolation protocol. Tissue pieces were taken from the ice and homogenised in RLT buffer supplemented with beta-mercaptoethanol with a Stuart handheld homogeniser until completely smooth. Genomic DNA was eliminated by centrifugation through a column supplied with the kit prior to extraction of the RNA according to the manufacturer’s protocol. RNA integrity was assessed by using an Agilent bioanalyser (Agilent Technologies, Santa Clara, CA, USA) to obtain RNA integrity numbers (RIN) according to the manufacturer’s protocol. RNA samples with RINs above 8 were used for the study. The RNA was used to prepare cDNA with High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA, USA) according to the manufacturer’s instructions. The efficiency of the cDNA protocol was tested by preparing the cDNA from a serial dilution of the RNA and then these cDNA samples were run on a qPCR confirming equal efficiency over a range of RNA concentrations. cDNA was also confirmed negative for genomic DNA contamination. Each custom-made card contained 64 pre-validated assays in triplicate with a reaction volume of 1 μL. The cards were run exactly according to instructions specific for the cards. Briefly, the cDNA (100 ng/well) was mixed with 2× Mastermix from Thermo Fisher (Waltham, MA, USA), 100 μL was loaded in each well slot on the cards, the cards were then spun down and sealed and run on a Quant 7 cycler (Applied Biosystems, Waltham, MA, USA). The amplification conditions were: 50 °C for 2 min and 95 °C for 10 min for the initial DNA melting and inactivation of RT reaction, followed by 40 cycles of 95 °C for 15 s and 60 °C for 60 s. Analysis of the Taqman array card data was performed by using the Quant studio software (Applied Biosystems, Waltham, MA, USA) and Microsoft Excel (Microsoft Corporation, Redmond, WA, USA) by calculating fold changes with the ΔCt method as previously described [31]. The threshold was set at 0.2 fluorescence units and the baseline range was set to automatic assignment. A combination of HPRT, Gapdh and ActinB were used as reference genes and amplifications were calculated with the regression threshold and baseline subtraction curve fit auto settings with the BioRad CFX manager software. The statistical analysis involved use of paired Student’s t-test to compare for differences in expression of functional gene groups and use of unpaired Student’s t-test to compare for differences in expression of individual genes between WT and Pgc-1α−/− atria, for a stratification of values of p.
4.3. Western Blots
Murine atria were extracted and homogenised in 450µl of lysis buffer (150 mM NaCl, 25 mM tris(hydroxymethyl)aminomethane (tris), pH 7–8, 1% Triton-X100 detergent, 5 mM ethylenediaminetetraacetic acid (EDTA) and Roche® cOmplete™ mini protease inhibitor (Merck KGaA, Darmstadt, Germany)). After a 20-min centrifugation at 12,000 RPM, the clear lysate was obtained and a bicinchoninic acid (BCA) assay performed to assess for protein content (Thermo Scientific Microplate BCA Protein Assay Kit #23252: manufacturer recommended protocol). The samples underwent sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) using loading buffer (12.8 mL tris, pH 6.8, 3.2 g sodium dodecyl sulphate (SDS), 1.85 g dithiothreitol (DTT), 16 mL 100% glycerol, bromophenol blue, 11.2 mL H2O) in the ratio of 3:1 volume of clear lysate to loading buffer. The mixtures were heated at 70 °C for 5 min and loaded into Mini-Protean TGX™ (Bio-Rad, Watford, UK), 4–15% acrylamide gradient, precast gel wells (20 µg for Nav1.5 blots and 30 µg for Cx40 and Cx43 blots). Samples were run at 120 V for 30 min, then 250 V for 20 min and compared to β-tubulin as a loading control. Semi-dry transfer of the proteins was carried out onto polyvinylidene fluoride (PVDF) membranes (Immobilon™ PVDF membrane, Merck KGaA, Darmstadt, Germany) using Trans-Blot® Turbo™ kit (BioRad, Watford, UK). The transfer settings were 1.3 A current and 25 V potential for 10 min. The PVDF membranes were then blocked with Odyssey® blocking buffer (Li-Cor Biosciences, Cambridge, UK) for one hour at room temperature, rinsed with PBS-T (0.1% Tween) and incubated with primary antibody diluted in Odyssey® blocking buffer diluted 33% in PBS-T overnight at 4 °C. The primary antibodies used were Nav1.5 (Cell Signalling Technology, London, UK), Cx43 (Sigma-Aldrich Company Ltd, Gillingham, UK), Cx40 (Santa Cruz Biotechnology, Dallas, TX, USA) and β-tubulin (Abcam, Cambridge, UK). The membranes were washed three times and then incubated with secondary antibodies diluted in Odyssey® blocking buffer diluted 33% in PBS-T at room temperature for 45 min. Imaging of the blots utilised the Odyssey® Fc imaging system (Li-Cor Biosciences, Cambridge, UK), which measured emission from the secondary antibodies at 600 and 800 nm. Image Studio™ software (Image Studio 4.0, Li-Cor Biosciences, Cambridge, UK) was used to quantify the protein band intensity and subtract the background signal, and then express this relative to the control, β-tubulin, signal. Statistical analysis was performed using the unpaired Student’s t-test.
Author Contributions
Conceptualization: C.L.-H.H. and K.J.; data curation: K.R.C., C.E.E., H.V., and S.A.; formal analysis: K.R.C. and C.E.E.; funding acquisition: C.L.-H.H. and K.J.; investigation: K.R.C., C.E.E., H.V., and S.A.; methodology: K.R.C., C.E.E., H.V., and S.A.; project administration: C.L.-H.H. and K.J.; resources: C.L.-H.H., and K.J.; software: K.R.C. and C.E.E.; supervision: C.L.-H.H. and K.J.; validation: K.R.C. and C.E.E.; visualization: C.L.-H.H. and K.J.; writing—original draft: K.R.C., C.L.-H.H., and K.J.; writing—review and editing: K.R.C., C.E.E., H.V., S.A., C.L.-H.H., and K.J.
Funding
Medical Research Council (MR/M001288/1), Wellcome Trust (105727/Z/14/Z), British Heart Foundation (PG/14/79/31102 and PG/15/12/31280), Sudden Adult Death Syndrome (SADS) UK Society, and the Fundamental Research Grant Scheme (FRGS/2014/SKK01/PERDANA/02/1) (Ministry of Education, Malaysia).
Acknowledgments
We would like to acknowledge the Department of Preclinical Veterinary Scince, University of Surrey for additional support in kind.
Conflicts of Interest
The authors declare no conflict of interest. The funding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Miyasaka, Y.; Barnes, M.E.; Gersh, B.J.; Cha, S.S.; Bailey, K.R.; Abhayaratna, W.P.; Seward, J.B.; Tsang, T.S. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation 2006, 114, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Vianna, C.R.; Huntgeburth, M.; Coppari, R.; Choi, C.S.; Lin, J.; Krauss, S.; Barbatelli, G.; Tzameli, I.; Kim, Y.B.; Cinti, S.; et al. Hypomorphic mutation of PGC-1β causes mitochondrial dysfunction and liver insulin resistance. Cell Metab. 2006, 4, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Greaves, L.C.; Reeve, A.K.; Turnbull, D. The ageing mitochondrial genome. Nucleic Acids Res. 2007, 35, 7399–7405. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; West, J.A.; Murray, A.J.; Griffin, J.L. Comprehensive Metabolic Profiling of Age-Related Mitochondrial Dysfunction in the High-Fat-Fed ob/ob Mouse Heart. J. Proteome Res. 2015, 14, 2849–2862. [Google Scholar] [CrossRef] [PubMed]
- Akar, F.G.; O’Rourke, B. Mitochondria are sources of metabolic sink and arrhythmias. Pharmacol. Ther. 2011, 131, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.E.; Rahban, Y.; Sandhu, H.; Hiremath, P.G.; Ayalon, N.; Qin, F.; Perez, A.J.; Downing, J.; Gopal, D.M.; Cheng, S.; et al. Preclinical Alterations in Myocardial Microstructure in People with Metabolic Syndrome. Obesity 2017, 25, 1516–1522. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, J.; Mehl, I.R.; Chong, L.W.; Nofsinger, R.R.; Evans, R.M. PGC-1β controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc. Natl. Acad. Sci. USA 2007, 104, 5223–5228. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann-Freestone, M.; Madsen, P.L.; Manners, D.; Blamire, A.M.; Buckingham, R.E.; Styles, P.; Radda, G.K.; Neubauer, S.; Clarke, K. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation 2003, 107, 3040–3046. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P.H.; et al. Transcriptional coactivator PGC-1α controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Y.; Tzeng, T.F.; Liu, I.M. Zerumbone, a Bioactive Sesquiterpene, Ameliorates Diabetes-Induced Retinal Microvascular Damage through Inhibition of Phospho-p38 Mitogen-Activated Protein Kinase and Nuclear Factor-kappaB Pathways. Molecules 2016, 21, 1708. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Z.; Hou, T.T.; Yuan, Y.; Hang, P.Z.; Zhao, J.J.; Sun, L.; Zhao, G.Q.; Zhao, J.; Dong, J.M.; Wang, X.B.; et al. Fenofibrate inhibits atrial metabolic remodelling in atrial fibrillation through PPAR-alpha/sirtuin 1/PGC-1α pathway. Br. J. Pharmacol. 2016, 173, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Kim, S.H.; Ariel, D.; Abbasi, F.; Lamendola, C.; Cardell, J.; Xu, S.; Patel, S.; Tomasso, V.; Mojaddidi, H.; et al. Does enhanced insulin sensitivity improve sleep measures in patients with obstructive sleep apnea: A randomized, placebo-controlled pilot study. Sleep Med. 2016, 22, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.F.; Young, K.C.; Bai, C.H.; Yu, B.C.; Ma, C.T.; Chien, Y.C.; Chiang, C.L.; Liao, C.S.; Lai, H.W.; Tsao, C.W. Rosiglitazone regulates anti-inflammation and growth inhibition via PTEN. Biomed. Res. Int. 2014, 2014, 787924. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.L. Murine Electrophysiological Models of Cardiac Arrhythmogenesis. Physiol. Rev. 2017, 97, 283–409. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Xu, H.; London, B.; Nerbonne, J.M. Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. J. Physiol. 1999, 521 Pt 3, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Nakaya, Y. T wave polarity related to the repolarization process of epicardial and endocardial ventricular surfaces. Am. Heart J. 1984, 108, 290–295. [Google Scholar] [CrossRef]
- Liu, G.; Iden, J.B.; Kovithavongs, K.; Gulamhusein, R.; Duff, H.J.; Kavanagh, K.M. In vivo temporal and spatial distribution of depolarization and repolarization and the illusive murine T wave. J. Physiol. 2004, 555, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Voigt, N.; Nattel, S.; Dobrev, D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ. Res. 2014, 114, 1483–1499. [Google Scholar] [CrossRef] [PubMed]
- Grace, A.A.; Roden, D.M. Systems biology and cardiac arrhythmias. Lancet 2012, 380, 1498–1508. [Google Scholar] [CrossRef]
- Ahmad, S.; Valli, H.; Chadda, K.R.; Cranley, J.; Jeevaratnam, K.; Huang, C.L. Ventricular pro-arrhythmic phenotype, arrhythmic substrate, ageing and mitochondrial dysfunction in peroxisome proliferator activated receptor-gamma coactivator-1β deficient (Pgc-1β−/−) murine hearts. Mech. Ageing Dev. 2018, 173, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Valli, H.; Salvage, S.C.; Grace, A.A.; Jeevaratnam, K.; Huang, C.L. Age-dependent electrocardiographic changes in Pgc-1β deficient murine hearts. Clin. Exp. Pharmacol. Physiol. 2018, 45, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Gurung, I.S.; Medina-Gomez, G.; Kis, A.; Baker, M.; Velagapudi, V.; Neogi, S.G.; Campbell, M.; Rodriguez-Cuenca, S.; Lelliott, C.; McFarlane, I.; et al. Deletion of the metabolic transcriptional coactivator Pgc-1β induces cardiac arrhythmia. Cardiovasc. Res. 2011, 92, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Mutikainen, M.; Tuomainen, T.; Naumenko, N.; Huusko, J.; Smirin, B.; Laidinen, S.; Kokki, K.; Hynynen, H.; Yla-Herttuala, S.; Heinaniemi, M.; et al. Peroxisome proliferator-activated receptor-gamma coactivator 1 α1 induces a cardiac excitation-contraction coupling phenotype without metabolic remodelling. J. Physiol. 2016, 594, 7049–7071. [Google Scholar] [CrossRef] [PubMed]
- Valli, H.; Ahmad, S.; Chadda, K.R.; Al-Hadithi, A.; Grace, A.A.; Jeevaratnam, K.; Huang, C.L. Age-dependent atrial arrhythmic phenotype secondary to mitochondrial dysfunction in Pgc-1β deficient murine hearts. Mech. Ageing Dev. 2017, 167, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Valli, H.; Ahmad, S.; Fraser, J.A.; Jeevaratnam, K.; Huang, C.L. Pro-arrhythmic atrial phenotypes in incrementally paced murine Pgc-1β−/− hearts: Effects of age. Exp. Physiol. 2017, 102, 1619–1634. [Google Scholar] [CrossRef] [PubMed]
- Valli, H.; Ahmad, S.; Jiang, A.Y.; Smyth, R.; Jeevaratnam, K.; Matthews, H.R.; Huang, C.L. Cardiomyocyte ionic currents in intact young and aged murine Pgc-1β−/− atrial preparations. Mech. Ageing Dev. 2018, 169, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Komen, J.C.; Thorburn, D.R. Turn up the power—Pharmacological activation of mitochondrial biogenesis in mouse models. Br. J. Pharmacol. 2014, 171, 1818–1836. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, A.J.; Logantha, S.J.; Hao, G.; Yanni, J.; Fedorenko, O.; Sinha, A.; Gilbert, S.H.; Benson, A.P.; Buckley, D.L.; Anderson, R.H.; et al. Functional, anatomical, and molecular investigation of the cardiac conduction system and arrhythmogenic atrioventricular ring tissue in the rat heart. J. Am. Heart Assoc. 2013, 2, e000246. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.A.; Huang, C.L. Quantitative techniques for steady-state calculation and dynamic integrated modelling of membrane potential and intracellular ion concentrations. Prog. Biophys. Mol. Biol. 2007, 94, 336–372. [Google Scholar] [CrossRef] [PubMed]
- Dhamoon, A.S.; Jalife, J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm 2005, 2, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.G. KATP channels as molecular sensors of cellular metabolism. Nature 2006, 440, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Jeevaratnam, K.; Chadda, K.R.; Huang, C.L.; Camm, A.J. Cardiac Potassium Channels: Physiological Insights for Targeted Therapy. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fu, X.; Zhang, Z.; Yu, B. Knockdown of cardiac Kir3.1 gene with siRNA can improve bradycardia in an experimental sinus bradycardia rat model. Mol. Cell. Biochem. 2017, 429, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Stolting, G.; Fischer, M.; Fahlke, C. CLC channel function and dysfunction in health and disease. Front. Physiol. 2014, 5, 378. [Google Scholar] [CrossRef] [PubMed]
- Thollon, C.; Bedut, S.; Villeneuve, N.; Coge, F.; Piffard, L.; Guillaumin, J.P.; Brunel-Jacquemin, C.; Chomarat, P.; Boutin, J.A.; Peglion, J.L.; et al. Use-dependent inhibition of hHCN4 by ivabradine and relationship with reduction in pacemaker activity. Br. J. Pharmacol. 2007, 150, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Jeevaratnam, K.; Guzadhur, L.; Goh, Y.M.; Grace, A.A.; Huang, C.L. Sodium channel haploinsufficiency and structural change in ventricular arrhythmogenesis. Acta Physiol. 2016, 216, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Goddard, C.; Liu, J.; Leoni, A.L.; Royer, A.; Fung, S.S.; Xiao, G.; Ma, A.; Zhang, H.; Charpentier, F.; et al. Sinus node dysfunction following targeted disruption of the murine cardiac sodium channel gene Scn5a. J. Physiol. 2005, 567, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shen, Y.; Xie, J.; Bao, H.; Cao, Q.; Wan, R.; Xu, X.; Zhou, H.; Huang, L.; Xu, Z.; et al. A mutation in the CACNA1C gene leads to early repolarization syndrome with incomplete penetrance: A Chinese family study. PLoS ONE 2017, 12, e0177532. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Boczek, N.J.; Ye, D.; Miyake, C.Y.; De la Uz, C.M.; Allen, H.D.; Ackerman, M.J.; Kim, J.J. Novel long QT syndrome-associated missense mutation, L762F, in CACNA1C-encoded L-type calcium channel imparts a slower inactivation tau and increased sustained and window current. Int. J. Cardiol. 2016, 220, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Sekido, Y.; Maximov, A.; Saad, M.; Forgacs, E.; Latif, F.; Wei, M.H.; Lerman, M.; Lee, J.H.; Perez-Reyes, E.; et al. Functional properties of a new voltage-dependent calcium channel α2δ auxiliary subunit gene (CACNA2D2). J. Biol. Chem. 2000, 275, 12237–12242. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Bauer, C.S.; Nieto-Rostro, M.; Margas, W.; Ferron, L.; Chaggar, K.; Crews, K.; Ramirez, J.D.; Bennett, D.L.; Schwartz, A.; et al. α2δ-1 gene deletion affects somatosensory neuron function and delays mechanical hypersensitivity in response to peripheral nerve damage. J. Neurosci. 2013, 33, 16412–16426. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Corrado, D.; Basso, C. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Orphanet J. Rare Dis. 2007, 2, 45. [Google Scholar] [CrossRef] [PubMed]
- Lynch, A.I.; Irvin, M.R.; Boerwinkle, E.; Davis, B.R.; Vaughan, L.K.; Ford, C.E.; Aissani, B.; Eckfeldt, J.H.; Arnett, D.K.; Shrestha, S. RYR3 gene polymorphisms and cardiovascular disease outcomes in the context of antihypertensive treatment. Pharmacogenomics J. 2013, 13, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tuteja, D.; Zhang, Z.; Xu, D.; Zhang, Y.; Rodriguez, J.; Nie, L.; Tuxson, H.R.; Young, J.N.; Glatter, K.A.; et al. Molecular identification and functional roles of a Ca2+-activated K+ channel in human and mouse hearts. J. Biol. Chem. 2003, 278, 49085–49094. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, A.; Veale, E.L.; Nagy, B.M.; Nagaraj, C.; Kwapiszewska, G.; Antigny, F.; Lambert, M.; Humbert, M.; Czirjak, G.; Enyedi, P.; et al. TASK-1 (KCNK3) channels in the lung: From cell biology to clinical implications. Eur. Respir. J. 2017, 50, 1700754. [Google Scholar] [CrossRef] [PubMed]
- James, A.F.; Hancox, J.C. More types than one: Multiple muscarinic receptor coupled K+ currents undergo remodelling in an experimental model of atrial fibrillation. Br. J. Pharmacol. 2007, 152, 981–983. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, T.D.; Jensen, B.C.; Baker, A.J.; Simpson, P.C. Cardiac alpha1-adrenergic receptors: Novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol. Rev. 2014, 66, 308–333. [Google Scholar] [CrossRef] [PubMed]
- Turnham, R.E.; Scott, J.D. Protein kinase A catalytic subunit isoform PRKACA; History, function and physiology. Gene 2016, 577, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Miyamoto, S.; Brown, J.H. Cardiomyocyte calcium and calcium/calmodulin-dependent protein kinase II: Friends or foes? Recent Prog. Horm. Res. 2004, 59, 141–168. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Zhang, Y.; Zhang, X.; Nirmalan, M.; Davies, L.; Konstantinou, D.; Yin, F.; Dobrzynski, H.; Wang, X.; Grace, A.; et al. TGF-beta1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circ. Arrhythm. Electrophysiol. 2011, 4, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Kreuzberg, M.M.; Sohl, G.; Kim, J.S.; Verselis, V.K.; Willecke, K.; Bukauskas, F.F. Functional properties of mouse connexin30.2 expressed in the conduction system of the heart. Circ. Res. 2005, 96, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; Jin, J.; Shen, W.; Tsui, H.; Shi, Y.; Wang, Y.; Zhang, Y.; Hao, G.; Wu, J.; Chen, S.; et al. Mkk4 is a negative regulator of the transforming growth factor beta 1 signaling associated with atrial remodeling and arrhythmogenesis with age. J. Am. Heart Assoc. 2014, 3, e000340. [Google Scholar] [CrossRef] [PubMed]
- Sylva, M.; van den Hoff, M.J.; Moorman, A.F. Development of the human heart. Am. J. Med. Genet. A 2014, 164, 1347–1371. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Z.; Beech, D.J. TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circ. Res. 2001, 88, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.J.; Gollob, M.H. The role of atrial natriuretic peptide in modulating cardiac electrophysiology. Heart Rhythm 2012, 9, 610–615. [Google Scholar] [CrossRef] [PubMed]
- King, J.H.; Huang, C.L.; Fraser, J.A. Determinants of myocardial conduction velocity: Implications for arrhythmogenesis. Front. Physiol. 2013, 4, 154. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, P.; Yamada, K.A.; Baertschi, A.J.; Green, K.; Kanter, E.M.; Saffitz, J.E.; Kleber, A.G. Relative contributions of connexins 40 and 43 to atrial impulse propagation in synthetic strands of neonatal and fetal murine cardiomyocytes. Circ. Res. 2006, 99, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Kanagaratnam, P.; Rothery, S.; Patel, P.; Severs, N.J.; Peters, N.S. Relative expression of immunolocalized connexins 40 and 43 correlates with human atrial conduction properties. J. Am. Coll. Cardiol. 2002, 39, 116–123. [Google Scholar] [CrossRef]
- Harada, M.; Melka, J.; Sobue, Y.; Nattel, S. Metabolic Considerations in Atrial Fibrillation- Mechanistic Insights and Therapeutic Opportunities. Circ. J. 2017, 81, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Mayr, M.; Yusuf, S.; Weir, G.; Chung, Y.L.; Mayr, U.; Yin, X.; Ladroue, C.; Madhu, B.; Roberts, N.; De Souza, A.; et al. Combined metabolomic and proteomic analysis of human atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 585–594. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.I.; Cardin, S.; Wait, R.; Chung, Y.L.; Vijayakumar, M.; Maguy, A.; Camm, A.J.; Nattel, S. Proteomic and metabolomic analysis of atrial profibrillatory remodelling in congestive heart failure. J. Mol. Cell. Cardiol. 2010, 49, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Workman, A.J.; Kane, K.A.; Rankin, A.C. Characterisation of the Na, K pump current in atrial cells from patients with and without chronic atrial fibrillation. Cardiovasc. Res. 2003, 59, 593–602. [Google Scholar] [CrossRef]
- Roubille, F.; Tardif, J.C. New therapeutic targets in cardiology: Heart failure and arrhythmia: HCN channels. Circulation 2013, 127, 1986–1996. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Tadevosyan, A.; Qi, X.; Xiao, J.; Liu, T.; Voigt, N.; Karck, M.; Kamler, M.; Kodama, I.; Murohara, T.; et al. Atrial Fibrillation Activates AMP-Dependent Protein Kinase and its Regulation of Cellular Calcium Handling: Potential Role in Metabolic Adaptation and Prevention of Progression. J. Am. Coll. Cardiol. 2015, 66, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Melnyk, P.; Gaspo, R.; Wang, Z.; Nattel, S. Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circ. Res. 1999, 84, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.Y.; Yeh, Y.H.; Xiao, L.; Burstein, B.; Maguy, A.; Chartier, D.; Villeneuve, L.R.; Brundel, B.J.; Dobrev, D.; Nattel, S. Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ. Res. 2008, 103, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Wakili, R.; Voigt, N.; Kaab, S.; Dobrev, D.; Nattel, S. Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Investig. 2011, 121, 2955–2968. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Xu, X.; Yuan, S.; Tan, L.; Gao, L.; Ma, S.; Zhang, S.; Ma, Z.; Jiang, W.; Liu, F.; et al. Oxidized Low-density Lipoprotein (ox-LDL) Cholesterol Induces the Expression of miRNA-223 and L-type Calcium Channel Protein in Atrial Fibrillation. Sci. Rep. 2016, 6, 30368. [Google Scholar] [CrossRef] [PubMed]
- Risgaard, B.; Jabbari, R.; Refsgaard, L.; Holst, A.G.; Haunso, S.; Sadjadieh, A.; Winkel, B.G.; Olesen, M.S.; Tfelt-Hansen, J. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin. Genet. 2013, 84, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fraser, J.A.; Jeevaratnam, K.; Hao, X.; Hothi, S.S.; Grace, A.A.; Lei, M.; Huang, C.L. Acute atrial arrhythmogenicity and altered Ca2+ homeostasis in murine RyR2-P2328S hearts. Cardiovasc. Res. 2011, 89, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, A.V.; Kalyanasundaram, A.; Lou, Q.; Hage, L.T.; Hansen, B.J.; Belevych, A.E.; Mohler, P.J.; Knollmann, B.C.; Periasamy, M.; Gyorke, S.; et al. Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex1. Eur. Heart J. 2015, 36, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Van Veen, T.A.; Stein, M.; Royer, A.; Le Quang, K.; Charpentier, F.; Colledge, W.H.; Huang, C.L.; Wilders, R.; Grace, A.A.; Escande, D.; et al. Impaired impulse propagation in Scn5a-knockout mice: Combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation 2005, 112, 1927–1935. [Google Scholar] [CrossRef] [PubMed]
- Azene, E.M.; Xue, T.; Marban, E.; Tomaselli, G.F.; Li, R.A. Non-equilibrium behavior of HCN channels: Insights into the role of HCN channels in native and engineered pacemakers. Cardiovasc. Res. 2005, 67, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Lieu, D.K.; Chan, Y.C.; Lau, C.P.; Tse, H.F.; Siu, C.W.; Li, R.A. Overexpression of HCN-encoded pacemaker current silences bioartificial pacemakers. Heart Rhythm 2008, 5, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Takemura, G.; Goto, K.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; Watanabe, T.; Morishita, K.; Kawasaki, M.; et al. Resveratrol reverses remodeling in hearts with large, old myocardial infarctions through enhanced autophagy-activating AMP kinase pathway. Am. J. Pathol. 2013, 182, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Baczko, I.; Liknes, D.; Yang, W.; Hamming, K.C.; Searle, G.; Jaeger, K.; Husti, Z.; Juhasz, V.; Klausz, G.; Pap, R.; et al. Characterization of a novel multifunctional resveratrol derivative for the treatment of atrial fibrillation. Br. J. Pharmacol. 2014, 171, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.; Chang, S.L.; Hsiao, Y.W.; Singhal, R.; Liu, S.H.; Leha, T.; Lin, W.Y.; Hsu, C.P.; Chen, Y.C.; Chen, Y.J.; et al. Resveratrol, a red wine antioxidant, reduces atrial fibrillation susceptibility in the failing heart by PI3K/AKT/eNOS signaling pathway activation. Heart Rhythm 2015, 12, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Kuno, A.; Hori, Y.S.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol improves cardiomyopathy in dystrophin-deficient mice through SIRT1 protein-mediated modulation of p300 protein. J. Biol. Chem. 2013, 288, 5963–5972. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Maguy, A.; De Simone, S.A.; Miragoli, M.; Jousset, F.; Rohr, S. TGF-beta1 (Transforming Growth Factor-beta1) Plays a Pivotal Role in Cardiac Myofibroblast Arrhythmogenicity. Circ. Arrhythm. Electrophysiol. 2017, 10, e004567. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Salvarani, N.; Rohr, S. Myofibroblasts induce ectopic activity in cardiac tissue. Circ. Res. 2007, 101, 755–758. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).