Transient Secondary Structures as General Target-Binding Motifs in Intrinsically Disordered Proteins


Abstract
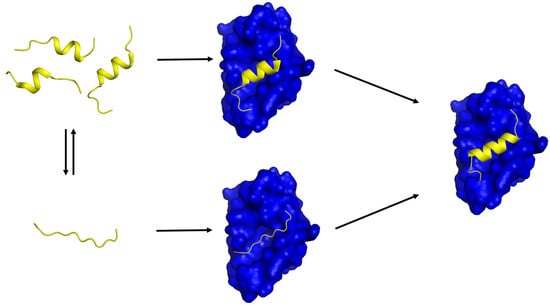
:
1. Introduction
2. Results
2.1. Favoring or Preferring Helical Conformation, Nascent Helix, and Helix-Forming
2.2. Local Structural Elements
2.3. Preformed, Pre-Ordered, Pre-Organized, and Pre-Structured
2.4. Transient Structure and Transient Order
2.5. Other Expressions
2.6. An overall Topology of IDPs
3. Discussion
4. Conclusion and Perspective
Funding
Conflicts of Interest
References
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondosm, S.E.; Dosztányi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered. Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins 2013, 1, e24157. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, D.; Tabaro, F.; Mičetić, I.; Necci, M.; Quaglia, F.; Oldfield, C.J.; Aspromonte, M.C.; Davey, N.E.; Davidović, R.; Dosztányi, Z.; et al. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acid Res. 2017, 45, D219–D227. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Davé, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological Unfoldomics of Uncontrolled Chaos: Intrinsically Disordered Proteins and Human Diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Kim, D.H.; Han, J.J.; Cha, E.J.; Lim, J.E.; Cho, Y.J.; Lee, C.; Han, K.H. Understanding pre-structured motifs (PreSMos) in intrinsically unfolded proteins. Curr. Protein Pept. Sci. 2012, 13, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Han, K.H. PreSMo target-binding signatures in intrinsically disordered proteins. Mol. Cells 2018, 41, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- Feany, M.B.; Bender, W.W. A Drosophila model of Parkinson’s disease. Nature 2009, 404, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, R.M. The steroid and thyroid hormone receptor superfamily. Science 1988, 240, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.E.; Palfreyman, J.W.; Preston, C.M. Identification of herpes implex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J. Mol. Biol. 1984, 180, 1–19. [Google Scholar] [CrossRef]
- Minezaki, Y.; Homma, K.; Kinjo, A.R.; Nishikawa, K. Human Transcription Factors Contain a High Fraction of Intrinsically Disordered Regions Essential for Transcriptional Regulation. J. Mol. Biol. 2006, 359, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 180, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.Y.; Cheong, J.Y.; Lim, W.; Jo, S.; Lee, Y.; Wang, H.J.; Han, K.H.; Cho, H. Prognostic significance of catalase expression and its regulatory effects on hepatitis B virus X protein (HBx) in HBV-related advanced hepatocellular carcinomas. Oncotarget 2014, 5, 12233–122346. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, I.; Pérez-Alvarado, G.C.; Parker, D.; Dyson, H.J.; Montminy, M.R.; Wright, P.E. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator: Coactivator interactions. Cell 1997, 91, 741–752. [Google Scholar] [CrossRef]
- Uesugi, M.; Nyanguile, O.; Lu, H.; Levine, A.J.; Verdine, G.L. Induced alpha helix in the VP16 activation domain upon binding to a human TAF. Science 1997, 277, 1310–1313. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.-X.; Jia, W.-H.; Bullock, B.P.; Habener, J.F.; Weiss, M.A. Transcriptional activator_coactivator recognition: Nascent folding of a kinase-inducible transactivation domain predicts its structure on coactivator binding. Biochemistry 1998, 37, 5858–5866. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, I.; Pérez-Alvarado, G.C.; Dyson, H.J.; Wright, P.E. Conformational preferences in the Ser133-phosphorylated and non-phosphorylated forms of the kinase inducible transactivation domain of CREB. FEBS Lett. 1998, 430, 317–322. [Google Scholar] [CrossRef]
- Noval, M.G.; Esperante, S.A.; Molina, I.G.; Chemes, L.B.; Prat-Gay, G.D. Intrinsic Disorder to Order Transitions in the Scaffold Phosphoprotein P from the Respiratory Syncytial Virus RNA Polymerase Complex. Biochemistry 2016, 55, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, C.M.; Wagner, G. The interaction of eIF4E with 4E-BP1 is an induced fit to a completely disordered protein. Protein Sci. 1998, 7, 1639–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Mok, K.H.; Muhandiram, R.; Park, K.H.; Suk, J.E.; Kim, D.H.; Chang, J.; Sung, Y.C.; Choi, K.Y.; Han, K.H. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J. Biol. Chem. 2000, 275, 29426–29432. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Georgieva, B.; Chabes, A.; Domkin, V.; Ippel, J.H.; Schleucher, J.; Wijmenga, S.; Thelander, L.; Rothstein, R. Mutational and structural analyses of the ribonucleotide reductase inhibitor Sml1 define its Rnr1 interaction domain whose inactivation allows suppression of mec1 and rad53 lethality. Mol. Cell. Biol. 2000, 20, 9076–9083. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational Properties of a-Synuclein in its Free and Lipid-associated States. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Chouard, T. Structural biology: Breaking the protein rules. Nature 2011, 471, 151–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daughdrill, G.W.; Hanely, L.J.; Dahlquist, F.W. The C-terminal half of the anti-sigma factor FlgM contains a dynamic equilibrium solution structure favoring helical conformations. Biochemistry 1998, 37, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.G.; Bayer, P.; Abo, A.; Kuhlmann, J.; Vetter, I.R.; Wittinghofer, A. The Cdc42/Rac interactive binding region motif of the Wiskott Aldrich Syndrome protein (WASP) is necessary but not sufficient for tight binding to Cdc42 and structure formation. J. Biol. Chem. 1998, 273, 18067–18076. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.; Munte, C.E.; Schorr, J.; Kellner, R.; Kalbitzer, H.R. Structure of the anchor-domain of myristoylated and nonmyristoylated HIV-1 Nef protein. J. Mol. Biol. 1999, 289, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Thapar, R.; Mueller, G.A.; Marzluff, W.F. The N-terminal domain of the Drosophila histone mRNA binding protein, SLBP, is intrinsically disordered with nascent helical structure. Biochemistry 2004, 43, 9390–9400. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.R.; Houben, K.; Lescop, E.; Blanchard, L.; Ruigrok, R.W.; Blackledge, M. Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: Application to the molecular recognition element of Sendai virus nucleoprotein. J. Am. Chem. Soc. 2008, 130, 8055–8061. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Qin, H.; Song, J. Intrinsically Unstructured Domain 3 of Hepatitis C Virus NS5A Forms a ‘‘Fuzzy Complex’’ with VAPB-MSP Domain Which Carries ALS-Causing Mutations. PLoS ONE 2012, 7, e39261. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Ma, D.; Yahr, T.L.; Chen, L. The transiently ordered regions in intrinsically disordered ExsE are correlated with structural elements involved in chaperone vinding. Biochem. Biophys. Res. Commun. 2012, 417, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.K.; Neuweiler, H.; Fersht, A.R. Long-range modulation of chain motions within the intrinsically disordered transactivation domain of tumor suppressor p53. J. Am. Chem. Soc. 2012, 134, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Schedlbauer, A.; Gandini, R.; Kontaxis, G.; Paulmichl, M.; Furst, J.; Konrat, R. The C-terminus of ICln is natively disordered but displays local structural preformation. Cell. Physiol. Biochem. 2011, 28, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Hazzard, J.; Sudhof, T.C.; Rizo, J. NMR analysis of the structure of synaptobrevin and of its interaction with syntaxin. J. Biomol. NMR 1999, 14, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Zitzewitz, J.A.; Ibarra-Molero, B.; Fishel, D.R.; Terry, K.L.; Matthews, C.R. Preformed secondary structure drives the association reaction of GCN4-p1, a model coiled-coil system. J. Mol. Biol. 2000, 296, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Ramelot, T.A.; Gentile, L.N.; Nicholson, L.K. Transient structure of the amyloid precursor protein cytoplasmic tail indicates preordering of structure for binding to cytosolic factors. Biochemistry 2000, 39, 2714–2725. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Gerstner, R.B.; Draper, D.E.; Torchia, D.A. Structural preordering in the N-terminal region of ribosomal protein S4 revealed by heteronuclear NMR spectroscopy. Biochemistry 2000, 39, 13602–13613. [Google Scholar] [CrossRef] [PubMed]
- Bienkiewicz, E.A.; Adkins, J.N.; Lumb, K.J. Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1). Biochemistry 2002, 41, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Kim, D.H.; Lee, S.H.; Chang, I.; Han, K.H. Pre-structured motifs in the natively unstructured preS1 surface antigen of hepatitis B virus. Protein Sci. 2007, 10, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Kim, D.H.; Lee, S.H.; Su, J.; Han, K.H. Structural investigation on the intrinsically disordered N-terminal region of HPV16 E7 protein. BMB Rep. 2016, 49, 431–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.H.; Lee, S.W.; Lee, E.J.; Choi, S.J.; Chung, S.S.; Lee, J.I.; Cho, J.M.; Seol, J.H.; Baek, S.H.; Kim, K.I.; et al. SUMO-specific protease SUSP4 positively regulates p53 by promoting Mdm2 self-ubiquitination. Nat. Cell Biol. 2006, 8, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Lee, C.; Lee, S.H.; Kim, K.T.; Han, J.J.; Cha, E.J.; Lim, J.E.; Cho, Y.J.; Hong, S.H.; Han, K.H. The Mechanism of p53 Rescue by SUSP4. Angew. Chem. Int. Ed. Engl. 2017, 56, 1278–1282. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Wright, A.; Han, K.H. An NMR study on the intrinsically disordered core transactivation domain of human glucocorticoid receptor. BMB Rep. 2017, 50, 522–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Lee, C.; Cho, Y.J.; Lee, S.H.; Cha, E.J.; Lim, J.E.; Sabo, T.M.; Griesinger, C.; Lee, D.; Han, K.H. A pre-structured helix in the intrinsically disordered 4EBP1. Mol. BioSyst. 2015, 11, 366–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moncoq, K.; Broutin, I.; Craescu, C.T.; Vachette, P.; Ducruix, A.; Durand, D. SAXS study of the PIR domain from the Grb14 molecular adaptor: A natively unfolded protein with a transient structure primer? Biophys. J. 2004, 87, 4056–4064. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.; Helander, S.; Lemak, A.; Farès, C.; Csizmok, V.; Carlsson, J.; Penn, L.Z.; Forman-Kay, J.D.; Arrowsmith, C.H.; Lundström, P.; et al. Transient structure and dynamics in the disordered c-Myc transactivation domain affect Bin1 binding. Nucleic Acids Res. 2012, 40, 6353–6366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuerstein, S.; Solyom, Z.; Aladag, A.; Favier, A.; Schwarten, M.; Hoffmann, S.; Willbold, D.; Brutscher, B. Transient structure and SH3 interaction sites in an intrinsically disordered fragment of the hepatitis C virus protein NS5A. J. Mol. Biol. 2012, 420, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Prives, C. Transcriptional regulation by p53: One protein, many possibilities. Cell Death Differ. 2006, 13, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Mittag, T.; Orlicky, S.; Choy, W.Y.; Tang, X.; Lin, H.; Sicheri, F.; Kay, L.E.; Tyers, M.; Forman-Kay, J.D. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 17772–17777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Lee, S.H.; Nam, K.H.; Chi, S.W.; Chang, I.; Han, K.H. Multiple hTAF(II)31-binding motifs in the intrinsically unfolded transcriptional activation domain of VP16. BMB Rep. 2009, 42, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Nováček, J.; Janda, L.; Dopitová, R.; Žídek, L.; Sklenář, V. Efficient protocol for backbone and side-chain assignments of large, intrinsically disordered proteins: Transient secondary structure analysis of 49.2 kDa microtubule associated protein 2c. J. Biomol. NMR 2013, 56, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Jonker, H.R.; Wechselberger, R.W.; Boelens, R.; Folkers, G.E.; Kaptein, R. Structural properties of the promiscuous VP16 activation domain. Biochemistry 2005, 25, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Csizmok, V.; Felli, I.C.; Tompa, P.; Banci, L.; Bertini, I. Structural and dynamic characterization of intrinsically disordered human securin by NMR spectroscopy. J. Am. Chem. Soc. 2008, 130, 16873–16879. [Google Scholar] [CrossRef] [PubMed]
- Lukhele, S.; Bah, A.; Lin, H.; Sonenberg, N.; Forman-Kay, J.D. Interaction of the eukaryotic initiation factor 4E with 4E-BP2 at a dynamic bipartite interface. Structure 2013, 21, 2186–2196. [Google Scholar] [CrossRef] [PubMed]
- Newcombe, E.A.; Ruff, K.M.; Sethi, A.; Ormsby, A.R.; Ramdzan, Y.M.; Fox, A.; Purcell, A.W.; Gooley, P.R.; Pappu, R.V.; Hatters, D.M. Tadpole-like Conformations of Huntingtin Exon 1 Are Characterized by Conformational Heterogeneity that Persists regardless of Polyglutamine Length. J. Mol. Biol. 2018, 430, 1442–1458. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Rajagopalan, S.; Settanni, G.; Marsh, R.J.; Armoogum, D.A.; Nicolaou, N.; Bain, A.J.; Lerner, E.; Haas, E.; Ying, L.; et al. Multiple conformations of full-length p53 detected with single-molecule fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 2009, 106, 20758–20763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Moncoq, K.; Broutin, I.; Larue, V.; Perdereau, D.; Cailliau, K.; Browaeys-Poly, E.; Burnol, A.F.; Ducruix, A. The PIR domain of Grb14 is an intrinsically unstructured protein: Implication in insulin signaling. FEBS Lett. 2003, 554, 240–246. [Google Scholar] [CrossRef]
- Mantsyzov, A.B.; Maltsev, A.S.; Ying, J.; Shen, Y.; Hummer, G.; Bax, A. A maximum entropy approach to the study of residue-specific backbone angle distributions in α-synuclein, an intrinsically disordered protein. Protein Sci. 2014, 23, 1275–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrali, M.G.; Schiavina, M.; Sainati, V.; Bermel, W.; Pierattelli, R.; Felli, I.C. 13C APSY-NMR for sequential assignment of intrinsically disordered proteins. J. Biomol. NMR 2018, 70, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Marcotrigiano, J.; Gingras, A.C.; Sonenberg, N.; Burley, S.K. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol. Cell 1999, 3, 707–716. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Borcherds, W.; Theillet, F.X.; Katzer, A.; Finzel, A.; Mishall, K.M.; Powell, A.T.; Wu, H.; Manieri, W.; Dieterich, C.; Selenko, P.; et al. Disorder and residual helicity alter p53-Mdm2 binding affinity and signaling in cells. Nat. Chem. Biol. 2014, 10, 1000–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iešmantavičius, V.; Dogan, J.; Jemth, P.; Teilum, K.; Kjaergaard, M. Helical propensity in an intrinsically disordered protein accelerates ligand binding. Angew. Chem. Int. Ed. Engl. 2014, 53, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Salamanova, E.; Costeira-Paulo, J.; Han, K.H.; Kim, D.H.; Nilsson, L.; Wright, A.P.H. A subset of functional adaptation mutations alter propensity for α-helical conformation in the intrinsically disordered glucocorticoid receptor tau1core activation domain. Biochim. Biophys. Acta 2018, 1862, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Dancheck, B.; Ragusa, M.J.; Allaire, M.; Forman-Kay, J.D.; Peti, W. Structural diversity in free and bound states of intrinsically disordered protein phosphatase 1 regulators. Structure 2010, 18, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Metallo, S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010, 14, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammoudeh, D.I.; Follis, A.V.; Prochownik, E.V.; Metallo, S.J. Multiple Independent Binding Sites for Small-Molecule Inhibitors on the Oncoprotein c-Myc. J. Am. Chem. Soc. 2009, 131, 7390–7401. [Google Scholar] [CrossRef] [PubMed]
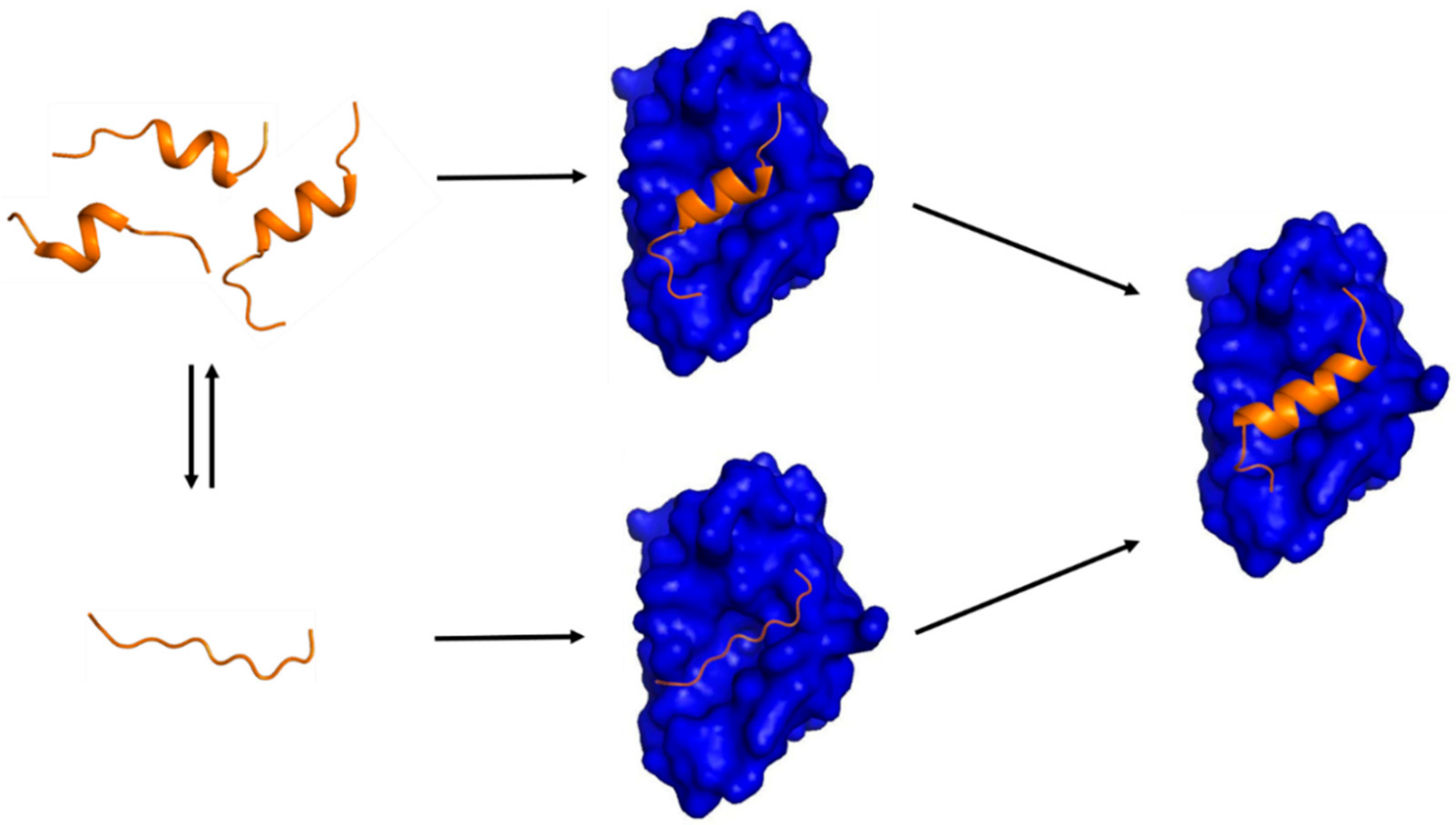

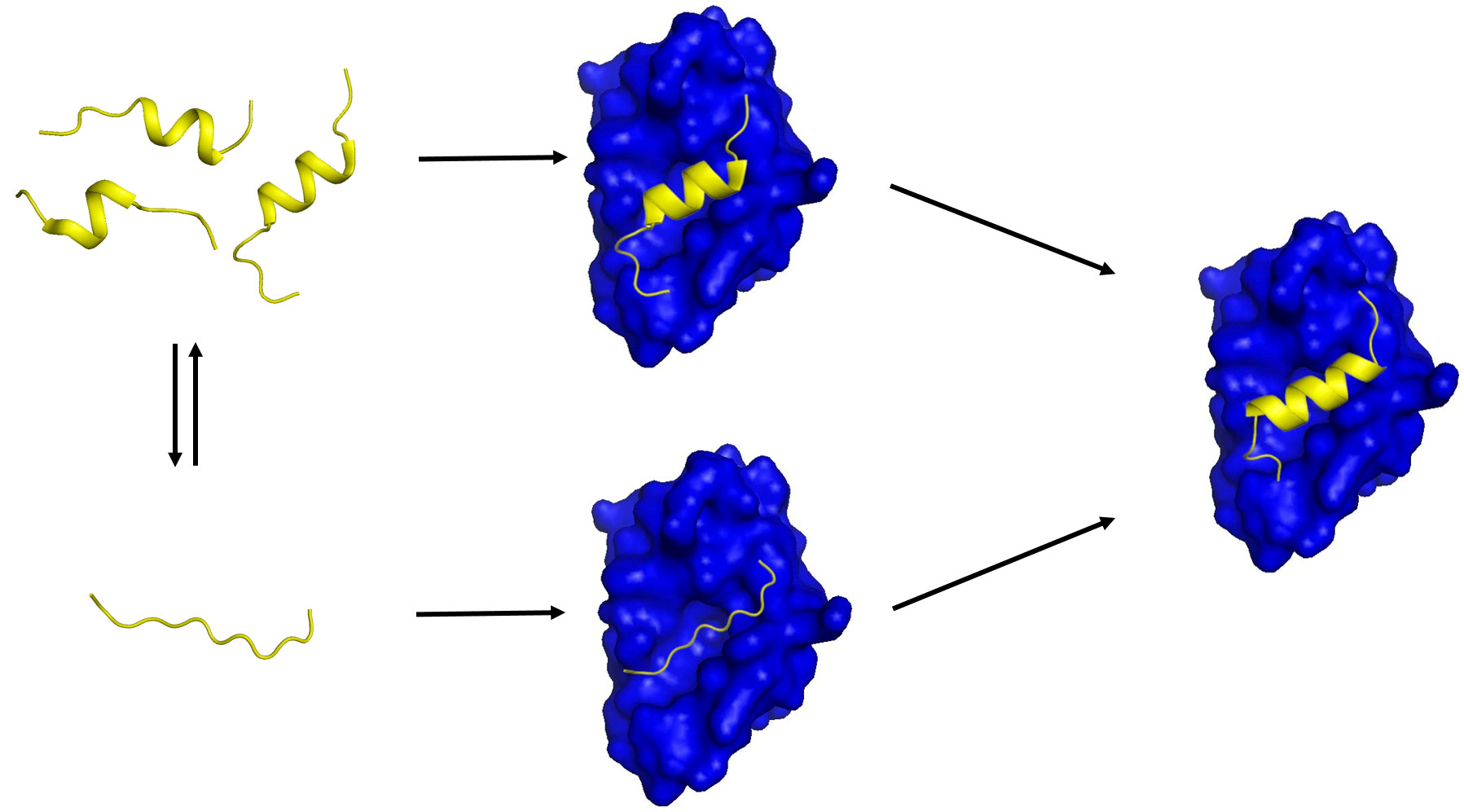{kind=link}
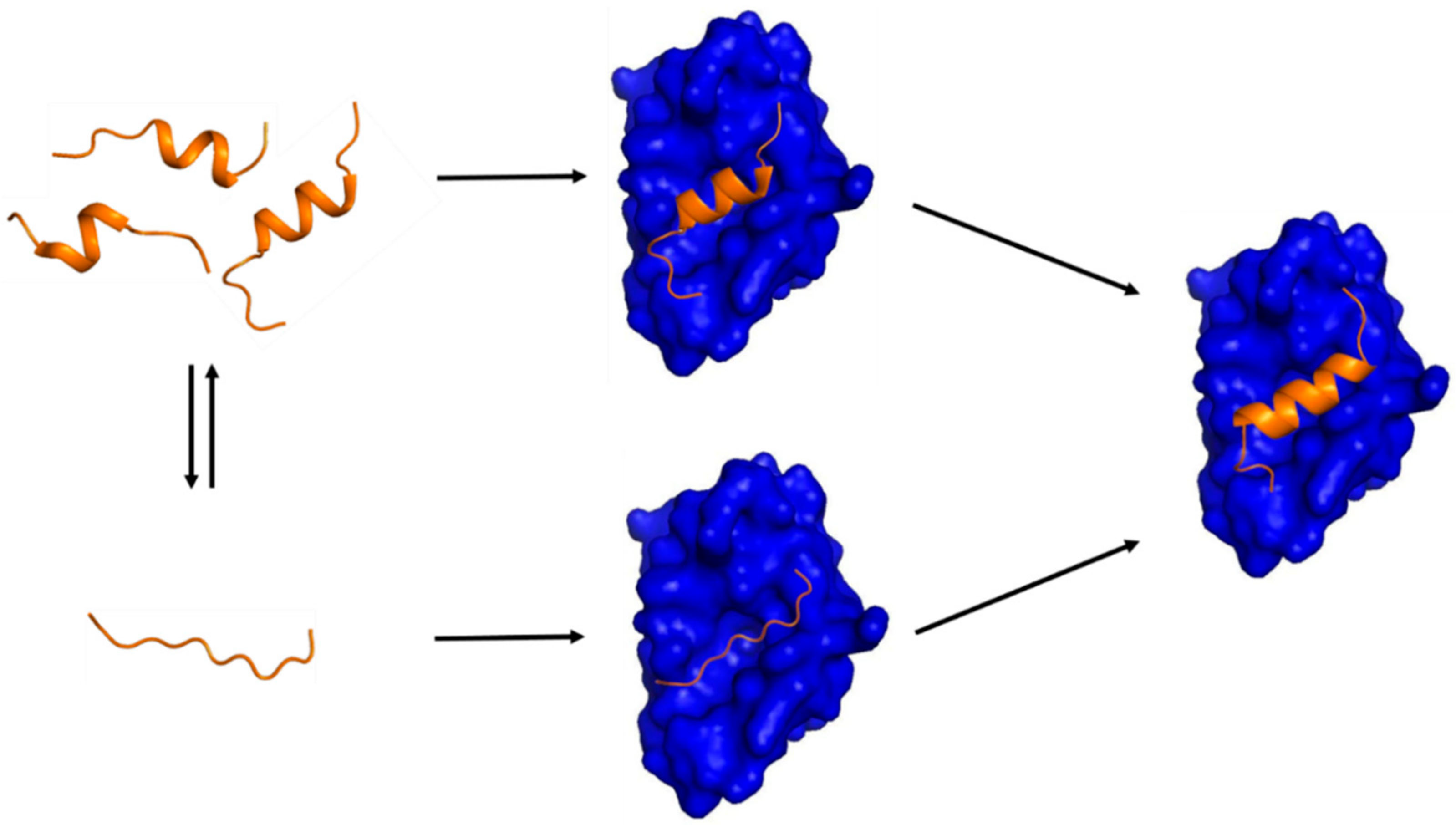{kind=link}
| CSI/SSP | Interproton NOE | 15N-1 HHet NOE | T1 | T2 T1ρ | 3JHNHα | Temp. Coeff. Backbone NH | HX Rate | Ref | |
|---|---|---|---|---|---|---|---|---|---|
| FlgM | O | O | O | O | 27 | ||||
| KID | O | O | 18 | ||||||
| GBD/CRIB WASP W7 | O | O | O | 28 | |||||
| HIV-1 Nef | O | O | O | O | 29 | ||||
| Synaptobrevin-2 | O | 36 | |||||||
| APPC | O | O | O | 38 | |||||
| p53 TAD | O | O | O | O | O | O | O | O | 23 |
| RPS4 | O | O | O | O | 39 | ||||
| α-Synuclein | O | 25 | |||||||
| Securin | O | O | O | O | 55 | ||||
| VP16 TAD | O | O | 54 | ||||||
| VP16 TAD | O | O | O | O | O | O | 52 | ||
| preS1 of HBV | O | O | O | O | O | O | O | 41 | |
| Sml1 | O | 24 | |||||||
| dSLBP | O | O | O | O | 30 | ||||
| NTAIL Sendai V. | O | 31 | |||||||
| nucleoprotein | |||||||||
| Sic1 | O | O | O | 51 | |||||
| c-Myc | O | O | O | O | O | 48 | |||
| ExsE | O | O | O | O | 33 | ||||
| MAP2c | O | 53 | |||||||
| NS5A HCV | O | O | 32 | ||||||
| NS5A HCV | O | O | O | O | 49 | ||||
| 4EBP2 | O | O | O | 56 | |||||
| 4EBP1 | O | O | O | O | O | O | 46 | ||
| ICIn | O | 35 | |||||||
| TAU | O | O | O | 7 | |||||
| E7 HPV | O | O | O | O | O | 42 | |||
| SUSP4 | O | O | O | O | O | O | 44 | ||
| hGR tau1c | O | O | O | O | O | O | 45 | ||
| Huntingtin Httex1 25Q | O | O | 57 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-H.; Han, K.-H. Transient Secondary Structures as General Target-Binding Motifs in Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2018, 19, 3614. https://doi.org/10.3390/ijms19113614
Kim D-H, Han K-H. Transient Secondary Structures as General Target-Binding Motifs in Intrinsically Disordered Proteins. International Journal of Molecular Sciences. 2018; 19(11):3614. https://doi.org/10.3390/ijms19113614
Chicago/Turabian StyleKim, Do-Hyoung, and Kyou-Hoon Han. 2018. "Transient Secondary Structures as General Target-Binding Motifs in Intrinsically Disordered Proteins" International Journal of Molecular Sciences 19, no. 11: 3614. https://doi.org/10.3390/ijms19113614
APA StyleKim, D.-H., & Han, K.-H. (2018). Transient Secondary Structures as General Target-Binding Motifs in Intrinsically Disordered Proteins. International Journal of Molecular Sciences, 19(11), 3614. https://doi.org/10.3390/ijms19113614