Sub-Toxic Human Amylin Fragment Concentrations Promote the Survival and Proliferation of SH-SY5Y Cells via the Release of VEGF and HspB5 from Endothelial RBE4 Cells

 ,
,  and
and
Abstract
:
1. Introduction
2. Results
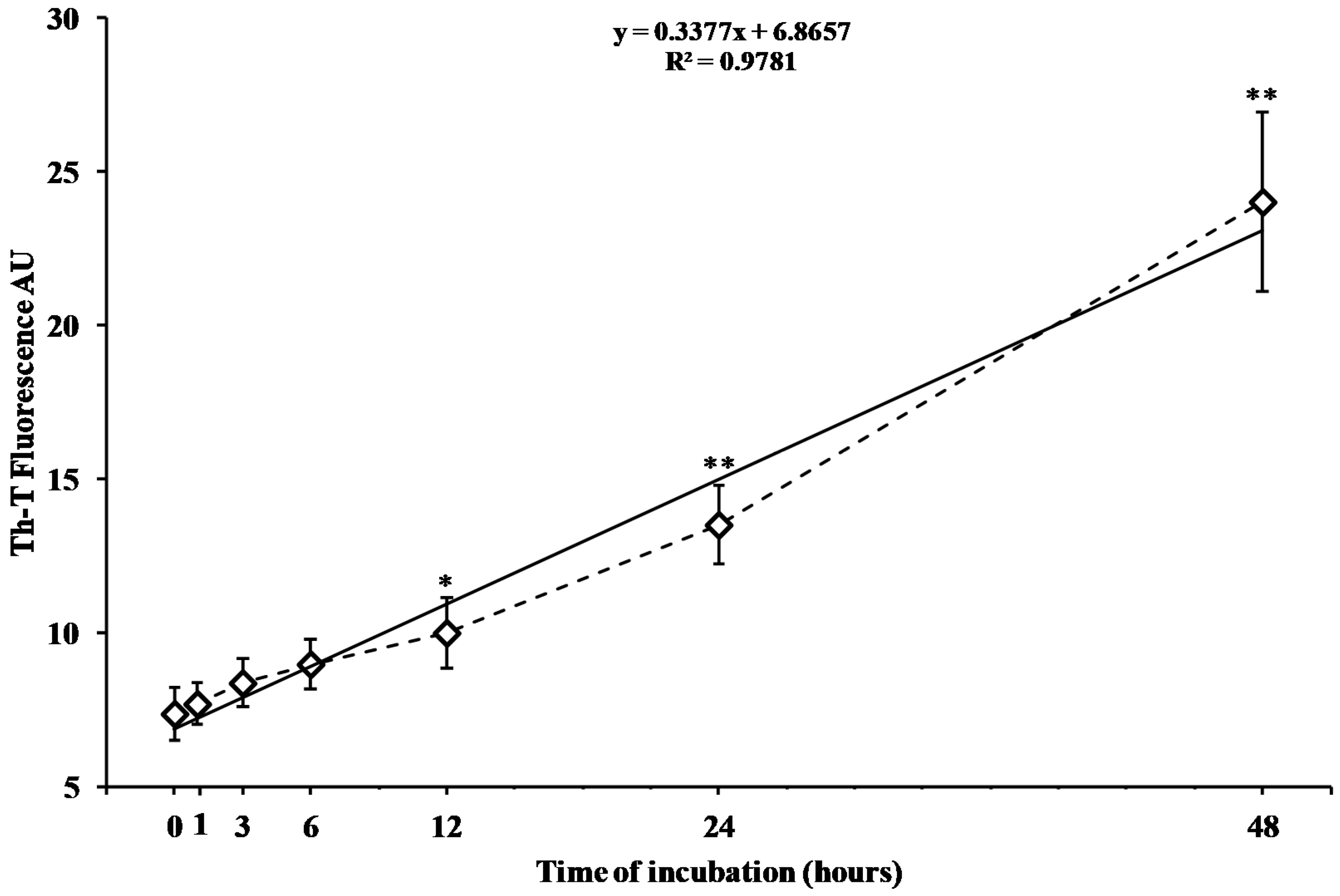
2.1. Time Course of In Vitro hA17–29 Aggregation
2.2. Effect of hA17–29 Fragment on RBE4 Cell Viability and Release in the Medium of Potentially Protective Proteins
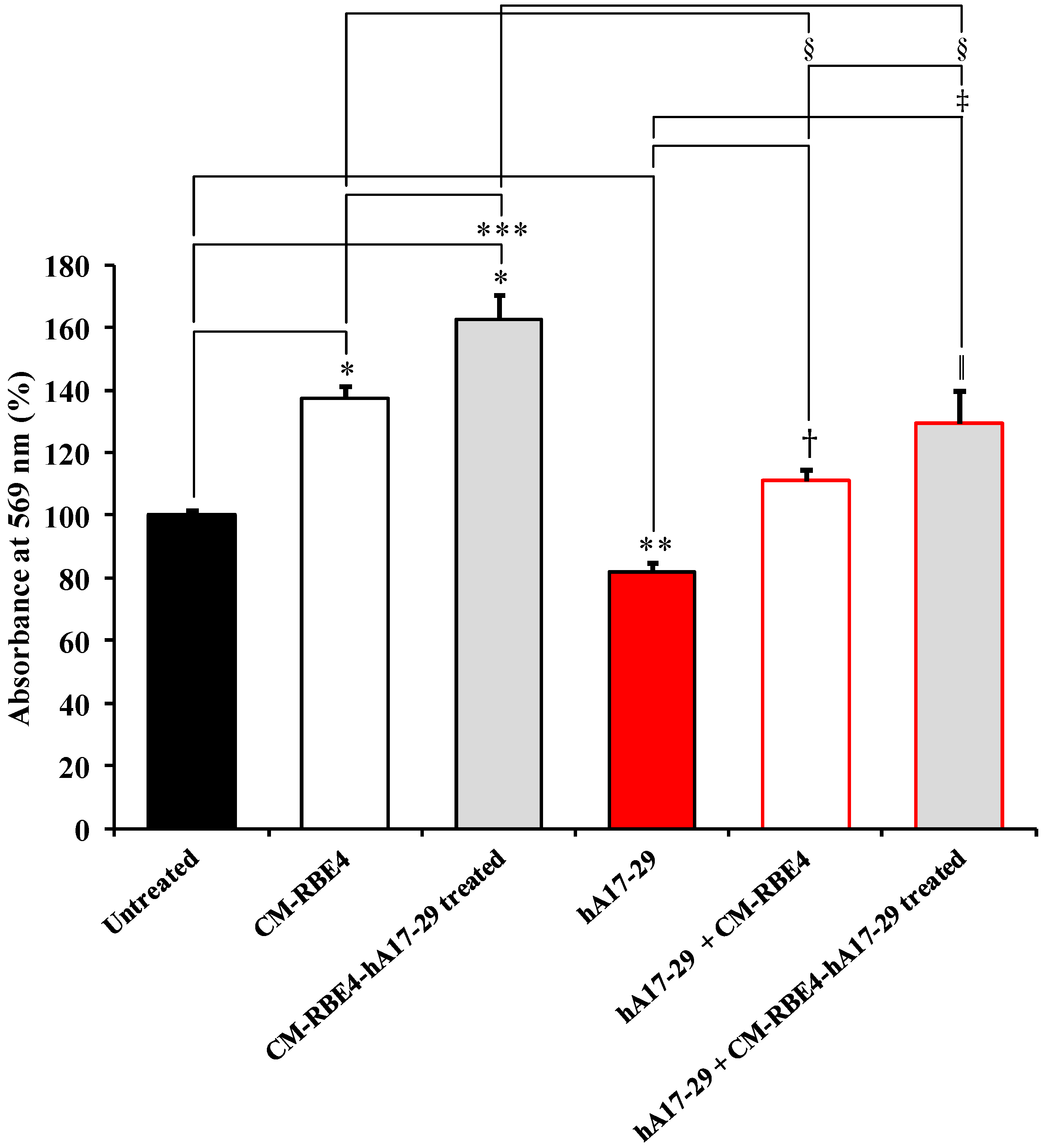
2.3. CM Derived from RBE4 Cells Counteracts SH-SY5Y Amyloid-Induced Toxicity
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Peptide Fragments Monomerization and Aggregation Studies
4.3. Rat Brain Endothelial (RBE4) and Human Neuroblastoma (SH-SY5Y) Cell Cultures
4.4. RBE4 Cells Stimulation and Quantification of HspB5 and VEGF in Conditioned Medium
4.5. RBE4 Conditioned Media Counteract SH-SY5Y Amyloid-Induced Toxicity: the Role of VEGF
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ahren, B.; Oosterwijk, C.; Lips, C.J.; Hoppener, J.W. Transgenic overexpression of human islet amyloid polypeptide inhibits insulin secretion and glucose elimination after gastric glucose gavage in mice. Diabetologia 1998, 41, 1374–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C. The physiology of amylin and insulin. Diabetes Educ. 2006, 32, 101S–104S. [Google Scholar] [CrossRef] [PubMed]
- Sanke, T.; Bell, G.I.; Sample, C.; Rubenstein, A.H.; Steiner, D.F. An islet amyloid peptide is derived from an 89-amino acid precursor by proteolytic processing. J. Biol. Chem. 1988, 263, 17243–17246. [Google Scholar] [PubMed]
- Marzban, L.; Trigo-Gonzalez, G.; Verchere, C.B. Processing of pro-islet amyloid polypeptide in the constitutive and regulated secretory pathways of beta cells. Mol. Endocrinol. 2005, 19, 2154–2163. [Google Scholar] [CrossRef] [PubMed]
- Kodali, R.; Wetzel, R. Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 2007, 17, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Jaikaran, E.T.; Clark, A. Islet amyloid and type 2 diabetes: From molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta 2001, 1537, 179–203. [Google Scholar] [CrossRef]
- Lorenzo, A.; Razzaboni, B.; Weir, G.C.; Yankner, B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijesekara, N.; Ahrens, R.; Sabale, M.; Wu, L.; Ha, K.; Verdile, G.; Fraser, P.E. Amyloid-beta and islet amyloid pathologies link Alzheimer’s disease and type 2 diabetes in a transgenic model. FASEB J. 2017, 31, 5409–5418. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Tascedda, F.; Merlo, S.; Benatti, C.; Spampinato, S.F.; Munafo, A.; Leggio, G.M.; Nicoletti, F.; Brunello, N.; Drago, F.; et al. Fluoxetine prevents abeta1-42-induced toxicity via a paracrine signaling mediated by transforming-growth-factor-beta1. Front. Pharmacol. 2016, 7, 389. [Google Scholar] [CrossRef] [PubMed]
- Polazzi, E.; Contestabile, A. Neuron-conditioned media differentially affect the survival of activated or unstimulated microglia: Evidence for neuronal control on apoptotic elimination of activated microglia. J. Neuropathol. Exp. Neurol. 2003, 62, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Lo, E.H. An oligovascular niche: Cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J. Neurosci. 2009, 29, 4351–4355. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.G.; Shenoy, A.K.Jr.; Clark, J.I. Interactions between important regulatory proteins and human alphab crystallin. Biochemistry 2007, 46, 6308–6317. [Google Scholar] [CrossRef] [PubMed]
- Kamradt, M.C.; Chen, F.; Cryns, V.L. The small heat shock protein alpha b-crystallin negatively regulates cytochrome c- and caspase-8-dependent activation of caspase-3 by inhibiting its autoproteolytic maturation. J. Biol. Chem. 2001, 276, 16059–16063. [Google Scholar] [CrossRef] [PubMed]
- Kamradt, M.C.; Lu, M.; Werner, M.E.; Kwan, T.; Chen, F.; Strohecker, A.; Oshita, S.; Wilkinson, J.C.; Yu, C.; Oliver, P.G.; et al. The small heat shock protein alpha b-crystallin is a novel inhibitor of trail-induced apoptosis that suppresses the activation of caspase-3. J. Biol. Chem. 2005, 280, 11059–11066. [Google Scholar] [CrossRef] [PubMed]
- Morozov, V.; Wawrousek, E.F. Caspase-dependent secondary lens fiber cell disintegration in alphaa-/alphab-crystallin double-knockout mice. Development 2006, 133, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of vegf and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Clauss, M.; Wiesnet, M.; Renz, D.; Schaper, W.; Karliczek, G.F. Hypoxia induces permeability in brain microvessel endothelial cells via vegf and no. Am. J. Physiol. 1999, 276, 812–820. [Google Scholar] [CrossRef]
- Fischer, S.; Wobben, M.; Marti, H.H.; Renz, D.; Schaper, W. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves vegf-mediated changes in the expression of zonula occludens-1. Microvasc. Res. 2002, 63, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, Z.; Ikezaki, K.; Samoto, K.; Inamura, T.; Fukui, M. Vegf and flt. Expression time kinetics in rat brain infarct. Stroke 1996, 27, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Herrán, E.; Perez-Gonzalez, R.; Igartua, M.; Pedraz, J.L.; Carro, E.; Hernandez, R.M. Vegf-releasing biodegradable nanospheres administered by craniotomy: A novel therapeutic approach in the app/ps1 mouse model of Alzheimer’s disease. J. Control. Release 2013, 170, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Kase, S.; He, S.; Sonoda, S.; Kitamura, M.; Spee, C.; Wawrousek, E.; Ryan, S.J.; Kannan, R.; Hinton, D.R. Alphab-crystallin regulation of angiogenesis by modulation of vegf. Blood 2010, 115, 3398–3406. [Google Scholar] [CrossRef] [PubMed]
- Van de Schootbrugge, C.; Bussink, J.; Span, P.N.; Sweep, F.C.; Grénman, R.; Stegeman, H.; Pruijn, G.J.; Kaanders, J.H.; Boelens, W.C. αB-crystallin stimulates VEGF secretion and tumor cell migration and correlates with enhanced distant metastasis in head and neck squamous cell carcinoma. BMC Cancer 2013, 13, 128. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.A.; Byzova, T.V. Alphab-crystallin: A novel vegf chaperone. Blood 2010, 115, 3181–3183. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, G.; Milardi, D.; Magri, A.; Attanasio, F.; Impellizzeri, G.; La Rosa, C.; Grasso, D.; Rizzarelli, E. Environmental factors differently affect human and rat iapp: Conformational preferences and membrane interactions of iapp17-29 peptide derivatives. Chemistry 2007, 13, 10204–10215. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Distefano, D.A.; Parlascino, P.; Fresta, C.G.; Lazzarino, G.; Lunte, S.M.; Nicoletti, V.G. Receptor-mediated toxicity of human amylin fragment aggregated by short- and long-term incubations with copper ions. Mol. Cell Biochem. 2017, 425, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Mazzaglia, A.; Micali, N.; Scolaro, L.M.; Attanasio, F.; Magri, A.; Pappalardo, G.; Villari, V. Aggregation properties of the peptide fragments derived from the 17–29 region of the human and rat iapp: A comparative study with two peg-conjugated variants of the human sequence. J. Phys. Chem. B 2010, 114, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Balbuena, P.; Li, W.; Magnin-Bissel, G.; Meldrum, J.B.; Ehrich, M. Comparison of two blood-brain barrier in vitro systems: Cytotoxicity and transfer assessments of malathion/oxon and lead acetate. Toxicol. Sci. 2010, 114, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Toimela, T.; Maenpaa, H.; Mannerstrom, M.; Tahti, H. Development of an in vitro blood-brain barrier model-cytotoxicity of mercury and aluminum. Toxicol. Appl. Pharmacol. 2004, 195, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Sinopoli, A.; Magri, A.; Milardi, D.; Pappalardo, M.; Pucci, P.; Flagiello, A.; Titman, J.J.; Nicoletti, V.G.; Caruso, G.; Pappalardo, G.; et al. The role of copper(ii) in the aggregation of human amylin. Metallomics 2014, 6, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Konarkowska, B.; Aitken, J.F.; Kistler, J.; Zhang, S.; Cooper, G.J. The aggregation potential of human amylin determines its cytotoxicity towards islet beta-cells. FEBS J. 2006, 273, 3614–3624. [Google Scholar] [CrossRef] [PubMed]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet amyloid polypeptide: Structure, function, and pathophysiology. J. Diabetes Res. 2016, 2016, 2798269. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.L.; Yarchoan, M.; Hwang, H.M.; Louneva, N.; Blair, J.A.; Palm, R.; Smith, M.A.; Lee, H.G.; Arnold, S.E.; Casadesus, G. Neuroprotective effects of the amylin analogue pramlintide on Alzheimer’s disease pathogenesis and cognition. Neurobiol. Aging 2014, 35, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Zhu, H. Amylin and its analogs: A friend or foe for the treatment of Alzheimer’s disease? Front. Aging Neurosci. 2014, 6, 186. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Liu, Y.; Qi, H.; Ma, Q.; Xu, L.; Chen, W.; Chen, G.; Xu, X. A novel brain neurovascular unit model with neurons, astrocytes and microvascular endothelial cells of rat. Int. J. Biol. Sci. 2013, 9, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Muchowski, P.J.; Bassuk, J.A.; Lubsen, N.H.; Clark, J.I. Human alphab-crystallin. Small heat shock protein and molecular chaperone. J. Biol. Chem. 1997, 272, 2578–2582. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, J.; Bova, M.P.; Ding, L.L.; Haley, D.A.; Stewart, P.L. Lens alpha-crystallin: Function and structure. Eye (Lond) 1999, 13, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.; McDermott, H.; Pike, I.; Spendlove, I.; Landon, M.; Mayer, R.J. Alpha b crystallin expression in non-lenticular tissues and selective presence in ubiquitinated inclusion bodies in human disease. J. Pathol. 1992, 166, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Rao, K.S.; Rao Ch, M. Ubiquitin-proteasome-mediated degradation and synthesis of myod is modulated by alphab-crystallin, a small heat shock protein, during muscle differentiation. Biochim. Biophys. Acta 2010, 1803, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.P.; Garland, D.L.; Green, D.E.; Tamm, E.R.; Giblin, F.J.; Wawrousek, E.F. AlphaB-crystallin in lens development and muscle integrity: A gene knockout approach. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2924–2934. [Google Scholar] [PubMed]
- Ito, H.; Kamei, K.; Iwamoto, I.; Inaguma, Y.; Kato, K. Regulation of the levels of small heat-shock proteins during differentiation of c2c12 cells. Exp. Cell Res. 2001, 266, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Renkawek, K.; Voorter, C.; Bosman, G.; van Workum, F.; De Jong, W. Expression of HspB5 in Alzheimer’s disease. Acta Neuropathol. 1994, 87, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Diokmetzidou, A.; Soumaka, E.; Kloukina, I.; Tsikitis, M.; Makridakis, M.; Varela, A.; Davos, C.H.; Georgopoulos, S.; Anesti, V.; Vlahou, A.; et al. Desmin and alphab-crystallin interplay in the maintenance of mitochondrial homeostasis and cardiomyocyte survival. J. Cell Sci. 2016, 129, 3705–3720. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.O.; Osmand, A.; Outeiro, T.F.; Muchowski, P.J.; Finkbeiner, S. Alphab-crystallin overexpression in astrocytes modulates the phenotype of the bachd mouse model of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Golenhofen, N.; Ness, W.; Wawrousek, E.F.; Drenckhahn, D. Expression and induction of the stress protein alpha-b-crystallin in vascular endothelial cells. Histochem. Cell Biol. 2002, 117, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Yin, B.; Song, E.; Chen, H.; Cheng, Y.; Zhang, X.; Bao, E.; Hartung, J. Aspirin upregulates alphab-crystallin to protect the myocardium against heat stress in broiler chickens. Sci. Rep. 2016, 6, 37273. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Kannan, R.; Spee, C.; Sreekumar, P.G.; Dou, G.; Hinton, D.R. Protection of retina by alphab crystallin in sodium iodate induced retinal degeneration. PLoS ONE 2014, 9, e98275. [Google Scholar]
- Mehlen, P.; Kretz-Remy, C.; Preville, X.; Arrigo, A.P. Human hsp27, drosophila hsp27 and human alphab-crystallin expression-mediated increase in glutathione is essential for the protective activity of these proteins against tnfalpha-induced cell death. EMBO J. 1996, 15, 2695–2706. [Google Scholar] [CrossRef] [PubMed]
- Raman, B.; Ban, T.; Sakai, M.; Pasta, S.Y.; Ramakrishna, T.; Naiki, H.; Goto, Y.; Rao, C.M. AlphaB-crystallin, a small heat-shock protein, prevents the amyloid fibril growth of an amyloid beta-peptide and beta2-microglobulin. Biochem. J. 2005, 392, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, S.; Li, D.; Liu, C. A Structural View of αB-crystallin Assembly and Amyloid Aggregation. Protein Pept. Lett. 2017, 24, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ward, N.; Boswell, M.; Katz, D.M. Secretion of brain-derived neurotrophic factor from brain microvascular endothelial cells. Eur. J. Neurosci. 2006, 23, 1665–1670. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Zhuang, M.; Wu, R. Secretion of nerve growth factor, brain-derived neurotrophic factor, and glial cell-line derived neurotrophic factor in co-culture of four cell types in cerebrospinal fluid-containing medium. Neural. Regen. Res. 2012, 7, 2907–2914. [Google Scholar] [PubMed]
- Meister, B.; Grunebach, F.; Bautz, F.; Brugger, W.; Fink, F.M.; Kanz, L.; Mohle, R. Expression of vascular endothelial growth factor (vegf) and its receptors in human neuroblastoma. Eur. J. Cancer 1999, 35, 445–449. [Google Scholar] [CrossRef]
- Roy Choudhury, S.; Karmakar, S.; Banik, N.L.; Ray, S.K. Targeting angiogenesis for controlling neuroblastoma. J. Oncol. 2012, 2012, 782020. [Google Scholar] [CrossRef] [PubMed]
- Restin, T.; Kajdi, M.E.; Schlapfer, M.; Roth, Z.; Z’graggen, B.R.; Booy, C.; Dumrese, C.; Beck-Schimmer, B. Sevoflurane protects rat brain endothelial barrier structure and function after hypoxia-reoxygenation injury. PLoS ONE 2017, 12, e0184973. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.C.; Moreira, P.I.; Oliveira, C.R.; Cardoso, S.M.; Pinton, P.; Pereira, C.F. Amyloid-beta disrupts calcium and redox homeostasis in brain endothelial cells. Mol. Neurobiol. 2015, 51, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, J.M.; Krum, J.M.; Ruhrberg, C. VEGF in the nervous system. Organogenesis 2010, 6, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.S.; Obeidat, A.; Giovanni, A.; Greene, L.A. Cell cycle regulators in neuronal death evoked by excitotoxic stress: Implications for neurodegeneration and its treatment. Neurobiol. Aging 2000, 21, 771–781. [Google Scholar] [CrossRef]
- Bonda, D.J.; Lee, H.P.; Kudo, W.; Zhu, X.; Smith, M.A.; Lee, H.G. Pathological implications of cell cycle re-entry in Alzheimer disease. Expert Rev. Mol. Med. 2010, 12, e19. [Google Scholar] [CrossRef] [PubMed]
- Moh, C.; Kubiak, J.Z.; Bajic, V.P.; Zhu, X.; Smith, M.A.; Lee, H.G. Cell cycle deregulation in the neurons of Alzheimer’s disease. Results Probl. Cell Differ. 2011, 53, 565–576. [Google Scholar] [PubMed]
- Dela Paz, N.G.; Walshe, T.E.; Leach, L.L.; Saint-Geniez, M.; D’Amore, P.A. Role of shear-stress-induced vegf expression in endothelial cell survival. J. Cell Sci. 2012, 125, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Lutgendorf, S.K.; Cole, S.; Costanzo, E.; Bradley, S.; Coffin, J.; Jabbari, S.; Rainwater, K.; Ritchie, J.M.; Yang, M.; Sood, A.K. Stress-related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines. Clin. Cancer Res. 2003, 9, 4514–4521. [Google Scholar] [PubMed]
- Boelens, W.C. Cell biological roles of alphab-crystallin. Prog. Biophys. Mol. Biol. 2014, 115, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.P.; Gangalum, R.K. Secretion of alphab-crystallin via exosomes: New clues to the function of human retinal pigment epithelium. Commun. Integr. Biol. 2011, 4, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, P.G.; Kannan, R.; Kitamura, M.; Spee, C.; Barron, E.; Ryan, S.J.; Hinton, D.R. αB crystallin is apically secreted within exosomes by polarized human retinal pigment epithelium and provides neuroprotection to adjacent cells. PLoS ONE 2010, 5, e12578. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Vukojevic, V.; Patel, A.; Soudy, R.; MacTavish, D.; Westaway, D.; Kaur, K.; Goncharuk, V.; Jhamandas, J. Role of microglial amylin receptors in mediating beta amyloid (abeta)-induced inflammation. J. Neuroinflammation 2017, 14, 199. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.M.; Senn, D.B.; Yuan, M.; Shine, J.P.; Yankner, B.A. Acceleration of amyloid beta-peptide aggregation by physiological concentrations of calcium. J. Biol. Chem. 2006, 281, 27916–27923. [Google Scholar] [CrossRef] [PubMed]
- Naldi, M.; Fiori, J.; Pistolozzi, M.; Drake, A.F.; Bertucci, C.; Wu, R.; Mlynarczyk, K.; Filipek, S.; De Simone, A.; Andrisano, V. Amyloid beta-peptide 25–35 self-assembly and its inhibition: A model undecapeptide system to gain atomistic and secondary structure details of the Alzheimer’s disease process and treatment. ACS Chem. Neurosci. 2012, 3, 952–962. [Google Scholar] [CrossRef] [PubMed]
- LeVine, H. Thioflavine t interaction with synthetic Alzheimer’s disease beta-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Fresta, C.G.; Chakraborty, A.; Wijesinghe, M.B.; Amorini, A.M.; Lazzarino, G.; Lazzarino, G.; Tavazzi, B.; Lunte, S.M.; Caraci, F.; Dhar, P.; et al. Non-toxic engineered carbon nanodiamond concentrations induce oxidative/nitrosative stress, imbalance of energy metabolism, and mitochondrial dysfunction in microglial and alveolar basal epithelial cells. Cell Death Dis. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, G.; Fresta, C.G.; Martinez-Becerra, F.; Antonio, L.; Johnson, R.T.; de Campos, R.P.S.; Siegel, J.M.; Wijesinghe, M.B.; Lazzarino, G.; Lunte, S.M. Carnosine modulates nitric oxide in stimulated murine raw 264.7 macrophages. Mol. Cell Biochem. 2017, 431, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Fresta, C.G.; Hogard, M.L.; Caruso, G.; Melo Costa, E.E.; Lazzarino, G.; Lunte, S.M. Monitoring carnosine uptake by raw 264.7 macrophage cells using microchip electrophoresis with fluorescence detection. Anal. Methods 2017, 9, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Lopalco, G.; Lucherini, O.M.; Vitale, A.; Talarico, R.; Lopalco, A.; Galeazzi, M.; Lapadula, G.; Cantarini, L.; Iannone, F. Putative role of serum amyloid-a and proinflammatory cytokines as biomarkers for behcet’s disease. Medicine 2015, 94, e1858. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Xue, X.; Wang, E.; Wallack, M.; Na, H.; Hooker, J.M.; Kowall, N.; Tao, Q.; Stein, T.D.; Wolozin, B.; et al. Amylin receptor ligands reduce the pathological cascade of Alzheimer’s disease. Neuropharmacology 2017, 119, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Patel, A.; Kimura, R.; Soudy, R.; Jhamandas, J.H. Amylin receptor: A potential therapeutic target for Alzheimer’s disease. Trends Mol. Med. 2017, 23, 709–720. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Treatment | Absorbance at 569 nm (%) in Absence of Anti-Flk1 | Absorbance at 569 nm (%) in Presence of Anti-Flk1 | Difference |
|---|---|---|---|
| CM-RBE4 | 134.27 (5.98) | 116.55 a (5.23) | −17.72 |
| CM-RBE4-hA17–29 | 158.88 (8.18) | 129.47 b (6.99) | −29.41 |
| hA17–29 + CM-RBE4 | 113.79 (4.75) | 98.90 d (4.18) | −14.88 |
| hA17–29 + CM-RBE4-hA17–29 | 126.38 (7.22) | 101.03 c,e (3.75) | −25.35 |
| Cell Treatment | VEGF Levels (pg/mL) | % of Increase Compared to CM-RBE4 |
|---|---|---|
| CM-RBE4 | 301.75 ± 47.72 | // |
| CM-RBE4-hA17–29 | 862.25 ± 72.2 a,b | +286 |
| CM-RBE4-Aβ25–35 | 709.50 ± 82.20 a | +235 |
| Cell Treatment | LDH Release (% of Controls) in Absence of Anti-Flk1 | LDH Release (% of Controls) in Presence of Anti-Flk1 |
|---|---|---|
| hA17–29 | 128.98 a (3.82) | NA |
| CM-RBE4 | 97.90 (2.31) | 102.75 (4.41) |
| CM-RBE4-hA17–29 | 99.97 (1.97) | 100.84 (2.73) |
| hA17–29 + CM-RBE4 | 111.92 b (2.54) | 116.09 (3.35) |
| hA17–29 + CM-RBE4-hA17–29 | 106.52 b (3.84) | 113.84 (5.18) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caruso, G.; Fresta, C.G.; Lazzarino, G.; Distefano, D.A.; Parlascino, P.; Lunte, S.M.; Lazzarino, G.; Caraci, F. Sub-Toxic Human Amylin Fragment Concentrations Promote the Survival and Proliferation of SH-SY5Y Cells via the Release of VEGF and HspB5 from Endothelial RBE4 Cells. Int. J. Mol. Sci. 2018, 19, 3659. https://doi.org/10.3390/ijms19113659
Caruso G, Fresta CG, Lazzarino G, Distefano DA, Parlascino P, Lunte SM, Lazzarino G, Caraci F. Sub-Toxic Human Amylin Fragment Concentrations Promote the Survival and Proliferation of SH-SY5Y Cells via the Release of VEGF and HspB5 from Endothelial RBE4 Cells. International Journal of Molecular Sciences. 2018; 19(11):3659. https://doi.org/10.3390/ijms19113659
Chicago/Turabian StyleCaruso, Giuseppe, Claudia G. Fresta, Giacomo Lazzarino, Donatella A. Distefano, Paolo Parlascino, Susan M. Lunte, Giuseppe Lazzarino, and Filippo Caraci. 2018. "Sub-Toxic Human Amylin Fragment Concentrations Promote the Survival and Proliferation of SH-SY5Y Cells via the Release of VEGF and HspB5 from Endothelial RBE4 Cells" International Journal of Molecular Sciences 19, no. 11: 3659. https://doi.org/10.3390/ijms19113659