Microduplication of 15q13.3 and Microdeletion of 18q21.32 in a Patient with Moyamoya Syndrome

 , , ,
, , ,  , ,
, , 
Abstract
:1. Introduction
2. Case Report
3. Results
4. Discussion
5. Materials and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kudo, T. Spontaneous occlusion of the circle of Willis. A disease apparently confined to Japanese. Neurology 1968, 18, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Takaku, A. Cerebrovascular ‘moyamoya’ disease. Disease showing abnormal net-like vessels in base of brain. Arch. Neurol. 1969, 20, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Phi, J.H.; Wang, K.C.; Lee, J.Y.; Kim, S.K. Moyamoya syndrome: A window of moyamoya disease. J. Korean Neurosurg. Soc. 2015, 57, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Guey, S.; Tournier-Lasserve, E.; Hervé, D.; Kossorotoff, M. Moyamoya disease and syndromes: From genetics to clinical management. Appl. Clin. Genet. 2015, 16, 49–68. [Google Scholar]
- Bersano, A.; Guey, S.; Bedini, G.; Nava, S.; Hervé, D.; Vajkoczy, P.; Tatlisumak, T.; Sareela, M.; van der Zwan, A.; Klijn, C.J.; et al. European Moyamoya Disease Initiative. Research Progresses in Understanding the Pathophysiology of Moyamoya Disease. Cerebrovas. Dis. 2016, 41, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Bedini, G.; Blecharz, K.G.; Nava, S.; Vajkoczy, P.; Alessandri, G.; Ranieri, M.; Acerbi, F.; Ferroli, P.; Riva, D.; Esposito, S.; et al. Vasculogenic and Angiogenic Pathways in Moyamoya Disease. Curr. Med. Chem. 2016, 23, 315–345. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Morito, D.; Takashima, S.; Mineharu, Y.; Kobayashi, H.; Hitomi, T.; Hashikata, H.; Matsuura, N.; Yamazaki, S.; Toyoda, A.; et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE 2011, 6, e22542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyatake, S.; Touho, H.; Miyake, N.; Ohba, C.; Doi, H.; Saitsu, H.; Taguri, M.; Morita, S.; Matsumoto, N. Sibling cases of moyamoya disease having homozygous and heterozygous c.14576G>A variant in RNF213 showed varying clinical course and severity. J. Hum. Genet. 2012, 57, 804–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guey, S.; Kraemer, M.; Hervé, D.; Ludwig, T.; Kossorotoff, M.; Bergametti, F.; Schwitalla, J.C.; Choi, S.; Broseus, L.; Callebaut, I.; et al. Rare RNF213 variants in the C-terminal region encompassing the RING-finger domain are associated with moyamoya angiopathy in Caucasians. Eur. J. Hum. Genet. 2017, 25, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Miskinyte, S.; Butler, M.G.; Hervé, D.; Sarret, C.; Nicolino, M.; Petralia, J.D.; Bergametti, F.; Arnould, M.; Pham, V.N.; Gore, A.V.; et al. Loss of BRCC3 deubiquitinating enzyme leads to abnormal angiogenesis and is associated with syndromic moyamoya. Am. J. Hum. Genet. 2011, 88, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Hervé, D.; Philippi, A.; Belbouab, R.; Zerah, M.; Chabrier, S.; Collardeau-Frachon, S.; Bergametti, F.; Essongue, A.; Berrou, E.; Krivosic, V.; et al. Loss of α1β1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am. J. Hum. Genet. 2014, 94, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Di Rocco, F.; Kossorotoff, M.; Zerah, M.; Boddaert, N.; Calmon, R.; Vidaud, D.; Cirillo, M.; Cinalli, G.; Mirone, G.; et al. Moyamoya syndrome in children with neurofibromatosis type 1: Italian-French experience. Am. J. Med. Genet. 2017, 173, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Ebisudani, Y.; Aoi, M.; Fukuhara, T. Adult-Onset Hemorrhagic Quasi-Moyamoya Disease with Unilateral Steno-occlusive Lesion in a Patient with Neurofibromatosis Type 1. J. Stroke Cerebrovas. Dis. 2018, 27, 1423–1424. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, V.; Kirkham, F.J. Noonan syndrome and moyamoya. Pediatr. Neurol. 1997, 16, 256–258. [Google Scholar] [CrossRef]
- Gupta, M.; Choudhri, O.A.; Feroze, A.H.; Do, H.M.; Grant, G.A.; Steinberg, G.K. Management of moyamoya syndrome in patients with Noonan syndrome. J. Clin. Neurosci. 2016, 28, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Shiihara, T.; Kato, M.; Mitsuhashi, Y.; Hayasaka, K. Costello syndrome showing moyamoya-like vasculopathy. Pediatr. Neurol. 2005, 32, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.; Soro, I.; Leitão, A.; Silva, M.L.; Leão, M. Moyamoya vascular pattern in Alagille syndrome. Pediatr. Neurol. 2012, 47, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Yokote, H.; Tsuura, M.; Nakai, K.; Ohshima, A.; Itakura, T. Marfan syndrome associated with moyamoya phenomenon and aortic dissection. Acta Neurochir. (Wien) 1999, 141, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Vinícius Ribeiro, E.; Silva, M.; Tavares Mendonça, F.; Garcia Dusi, R. Anesthetic management in a child with moya-moya disease and sickle cell anemia: Case report. Rev. Esp. Anestesiol. Reanim. 2016, 63, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Alamri, A.; Hever, P.; Cheserem, J.; Gradil, C.; Bassi, S.; Tolias, C.M. Encephaloduroateriosynangiosis (EDAS) in the management of Moyamoya syndrome in children with sickle cell disease. Br. J. Neurosurg. 2017, 15, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Xin, B.; Jones, S.; Puffenberger, E.G.; Hinze, C.; Bright, A.; Tan, H.; Zhou, A.; Wu, G.; Vargus-Adams, J.; Agamanolis, D.; et al. Homozygous mutation in SAMHD1 gene causes cerebral vasculopathy and early onset stroke. Proc. Natl. Acad. Sci. USA 2011, 108, 5372–5377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, V.; Bernardi, B.; Stafa, A.; Garone, C.; Franzoni, E.; Abinun, M.; Mitchell, P.; Mitra, D.; Friswell, M.; Nelson, J.; et al. Intracerebral large artery disease in Aicardi-Goutières syndrome implicates SAMHD1 in vascular homeostasis. Dev. Med. Child Neurol. 2010, 52, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Bober, M.B.; Khan, N.; Kaplan, J.; Lewis, K.; Feinstein, J.A.; Scott, C.I., Jr.; Steinberg, G.K. Majewski osteodysplastic primordial dwarfism type II (MOPD II): Expanding the vascular phenotype. Am. J. Med. Genet. A 2010, 152A, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Moftakhar, P.; Smith, E.R.; Choulakian, A.; Scott, R.M.; Pour, D. Moyamoya disease in children with congenital dwarfing conditions. Pediatr. Neurosurg. 2010, 46, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Codd, P.J.; Scott, R.M.; Smith, E.R. Seckel syndrome and moyamoya. J. Neurosurg. Pediatr. 2009, 3, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.E.; Egan, M.; Rodgers, S.; Harter, D.; Burnside, R.D.; Milla, S.; Pappas, J. Complex chromosome rearrangement of 6p25.3->p23 and 12q24.32->qter in a child with Moyamoya. Pediatrics 2013, 131, e1996–e2001. [Google Scholar] [CrossRef] [PubMed]
- Janczar, S.; Fogtman, A.; Koblowska, M.; Baranska, D.; Pastorczak, A.; Wegner, O.; Kostrzewska, M.; Laguna, P.; Borowiec, M.; Mlynarski, W. Novel severe hemophilia A and moyamoya (SHAM) syndrome caused by Xq28 deletions encompassing F8 and BRCC3 genes. Blood 2014, 123, 4002–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girirajan, S.; Mendoza-Londono, R.; Vlangos, C.N.; Dupuis, L.; Nowak, N.J.; Bunyan, D.J.; Hatchwell, E.; Elsea, S.H. Smith-Magenis syndrome and Moyamoya disease in a patient with del(17)(p11.2p13.1). Am. J. Med. Genet. A 2007, 143, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.; Deleyiannis, F.; Bernard, T.J.; Fenton, L.Z.; Somme, S.; Wilkinson, C.C. Moyamoya in a Patient with Smith-Magenis Syndrome. Pediatr. Neurosurg. 2017, 52, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.O.; Baek, H.J.; Woo, Y.J.; Choi, Y.Y.; Chung, T.W. Moyamoya syndrome in a child with trisomy 12p syndrome. Pediatr. Neurol. 2006, 35, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Prontera, P.; Rogaia, D.; Mencarelli, A.; Ottaviani, V.; Sallicandro, E.; Guercini, G.; Esposito, S.; Bersano, A.; Merla, G.; Stangoni, G. Juvenile Moyamoya and Craniosynostosis in a Child with Deletion 1p32p31: Expanding the Clinical Spectrum of 1p32p31 Deletion Syndrome and a Review of the Literature. Int. J. Mol. Sci. 2017, 18, 1998. [Google Scholar] [CrossRef] [PubMed]
- Kainth, D.S.; Chaudhry, S.A.; Kainth, H.S.; Suri, F.K.; Qureshi, A.I. Prevalence and characteristics of concurrent down syndrome in patients with moyamoya disease. Neurosurgery 2013, 72, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Spengos, K.; Kosmaidou-Aravidou, Z.; Tsivgoulis, G.; Vassilopoulou, S.; Grigori-Kostaraki, P.; Zis, V. Moyamoya syndrome in a Caucasian woman with Turner’s syndrome. Eur. J. Neurol. 2006, 13, e7–e8. [Google Scholar] [CrossRef] [PubMed]
- Szafranski, P.; Schaaf, C.P.; Person, R.E.; Gibson, I.B.; Xia, Z.; Mahadevan, S.; Wiszniewska, J.; Bacino, C.A.; Lalani, S.; Potocki, L.; et al. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: Benign or pathological? Hum. Mutat. 2010, 31, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Gillentine, M.A.; Schaaf, C.P. The human clinical phenotypes of altered CHRNA7 copy number. Biochem. Pharmacol. 2015, 97, 352–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti-Tronconi, B.M.; McLane, K.E.; Raftery, M.A.; Grando, S.A.; Protti, M.P. The nicotinic acetylcholine receptor: Structure and autoimmune pathology. Crit. Rev. Biochem. Mol. Biol. 1994, 29, 69–123. [Google Scholar] [CrossRef] [PubMed]
- Van Bon, B.W.; Mefford, H.C.; Menten, B.; Koolen, D.A.; Sharp, A.J.; Nillesen, W.M.; Innis, J.W.; de Ravel, T.J.; Mercer, C.L.; Fichera, M.; et al. Further delineation of the 15q13 microdeletion and duplication syndromes: A clinical spectrum varying from non-pathogenic to a severe outcome. J. Med. Genet. 2009, 46, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraemer, M.; Huynh, Q.B.; Wieczorek, D.; Balliu, B.; Mikat, B.; Boehringer, S. Distinctive facial features in idiopathic Moyamoya disease in Caucasians: A first systematic analysis. PeerJ 2018, 6, e4740. [Google Scholar] [CrossRef] [PubMed]
- Heeschen, C.; Weis, M.; Aicher, A.; Dimmeler, S.; Cooke, J.P. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J. Clin. Investig. 2002, 110, 527–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooq, I.S.; Yeo, G.S.; O’Rahilly, S. Binge eating as a phenotype of melanocortin 4 receptor gene mutations. N. Engl. J. Med. 2003, 349, 606–609. [Google Scholar]
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E.; Yu, J.; Funahashi, H.; Williams, T.; Ferreira, M.; Tang, V.; McGovern, R.A.; Kenny, C.D.; et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 2005, 123, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Huszar, D.; Lynch, C.A.; Fairchild-Huntress, V.; Dunmore, J.H.; Fang, Q.; Berkemeier, L.R.; Gu, W.; Kesterson, R.A.; Boston, B.A.; Cone, R.D.; et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 1997, 88, 131–141. [Google Scholar] [CrossRef]
- Ste Marie, L.; Miura, G.I.; Marsh, D.J.; Yagaloff, K.; Palmiter, R.D. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 12339–12344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wu, W.; Wu, Y.; Zheng, J.; Suo, T.; Tang, H.; Tang, J. RNF152, a novel lysosome localized E3 ligase with pro-apoptotic activities. Protein Cell 2010, 1, 656–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, R.; Howell, R.; Dagna Bricarelli, F.; Kristoffersson, U.; Cavani, S. Specific Constitutional Cytogenetic Guidelines of European Cytogeneticist Association E.C.A. Newsletter 2012, 30, 11–19. [Google Scholar]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T.; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standard and Guidelines for interpretation and reporting of postnatal constitutional of copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease Name or Mutated Gene | Genes/Chromosomes | Clinical Features |
|---|---|---|
| MA with autosomal dominant heritability | ||
| Type 1 Neurofibromatosis [12,13] | Neurofibromatosis 1 (NF1) |
|
| Noonan Syndrome [14,15] | Protein Tyrosine Phosphatase, non-receptor Type 1 (PTPN1) (12q24.13), Son Of Sevenless homolog 1 (Drosophila) (SOS1) (2p22.1), v-Raf-1 murine leukemia viral oncogene homolog 1 (RAF1), (3p25.2); more rarely: v-Ki-ras2 Kirsten Rat Sarcoma viral oncogene homolog (KRAS) (12p12.1), Neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS) (1p13.2), v-raf murine sarcoma viral oncogene homolog B1 (BRAF) (7q34), Mitogen-Activated Protein Kinase kinase 1 (MAP2K1) (15q22.31) |
|
| Costello Syndrome [16] | v-Ha-ras Harvey rat sarcoma viral oncogene homolog (HRAS) (11p15.5) | Prenatal features: prematurity, lymphatic dysplasia, macrosomia, fetal arrhythmias, etc. Neonatal period:
|
| Alagille Syndrome [17] | Jagged 1 (JAG 1) (20p12.2), NOTCH2 (1p12-p11) | Frequent symptoms:
|
| Marfan Syndrome [18] | Fibrillin 1 (FBN1) |
|
| MA with autosomal recessive heritability | ||
| Sickle Cell Disease [19,20] | Hemoglobin Beta (HBB) (11p15.5) |
|
| GUCY1A3 [11] | Guanylate Cyclase 1, soluble, Alpha 3 (GUCYIA3) (4q32.1) |
|
| SAMHDI [21,22] | SAM domain and HD domain 1 (SAMHD1) (20q11.3) | Aicardi-Goutières syndrome:
Congenital glaucoma, arthritis, chilblain lupus |
| Microcephalic Osteodysplastic Primordial Dwarfism, Type II (MOPD II)/ Majewski Syndrome [23,24] | Pericentrin (PCNT) (21q22.3) |
|
| Seckel Syndrome (microcephalic primordial dwarfism) [25] | Ataxia Telangiectasia and Rad3 related (ATR) (3q23), Retinoblastoma Binding Protein 8 (RBBP8) (18q11.2), Centromere Protein J (CENPJ) (13q12.12), Centrosomal Protein 152kDa (CEP152) (15q21.1), Centrosomal Protein 63Da (CEP63) (3q22.2), Ninein (GSK3B interacting protein) (NIN) (14q22.1) |
|
| Genomic disorders | ||
| 6p25.3-p23 del/dup and 12q24.32-qter dup [26] | On the 6p region: Interferon Regulatory Factor 4 (IRF4) and other 51 Online Mendelian Inheritance in Man (OMIM) genes including forkhead box C1 (FOXC1); on the 12q region: 22 OMIM genes not associated with genetic disorders. |
|
| Xq28 deletion [10,27] | Factor 8 (F8) (exons 1–6), FUN14 Domain Containing 2 (FUNDC2), mature T-cell proliferation 1 (MTCP1), nuclear gene encoding mitochondrial protein (MTCP1NB), BRCA1/BRCA2-containing complex, subunit 3 (BRCC3) | Frequent symptoms:
|
| Smith-Magenis Syndrome (del 17p11.2-p13) [28,29] | More than 25 genes including: Retinoic Acid Induced 1 (RAI1), Mediator complex subunit 9 (MED9), RAS, dexamethasone-induced 1 (RASD1), Folliculin (FLCN), Peripheral Myelin Protein 22 (PMP22), Cytochrome C Oxidase assembly homolog 10 (COX10), ElaC ribonuclease Z 2 (ELAC2), Zinc Finger protein 18 (ZNF18), Myosin, Heavy chain 1 (MYH1) |
|
| Trisomy 12p [30] | Genes included in the region of the rearrangement: 46, XX, rec(12)dup(12p)inv(12)(p11.2q24.3)mat |
|
| 1p32p31 Deletion [31] | OMIM genes included in the region of the rearrangement: Complement component 8, Alpha polypeptide (C8A), Complement component 8, alpha polypeptide (C8B), Tumor-Associated Calcium Signal Transducer 2 (TACSTD2), Angiopoietin-like 3 (ANGPTL3), Forkhead box D3 (FOXD3), ALG6, alpha-1,3-glucosyltransferase (ALG6), Phosphoglucomutase 1 (PGM1) |
|
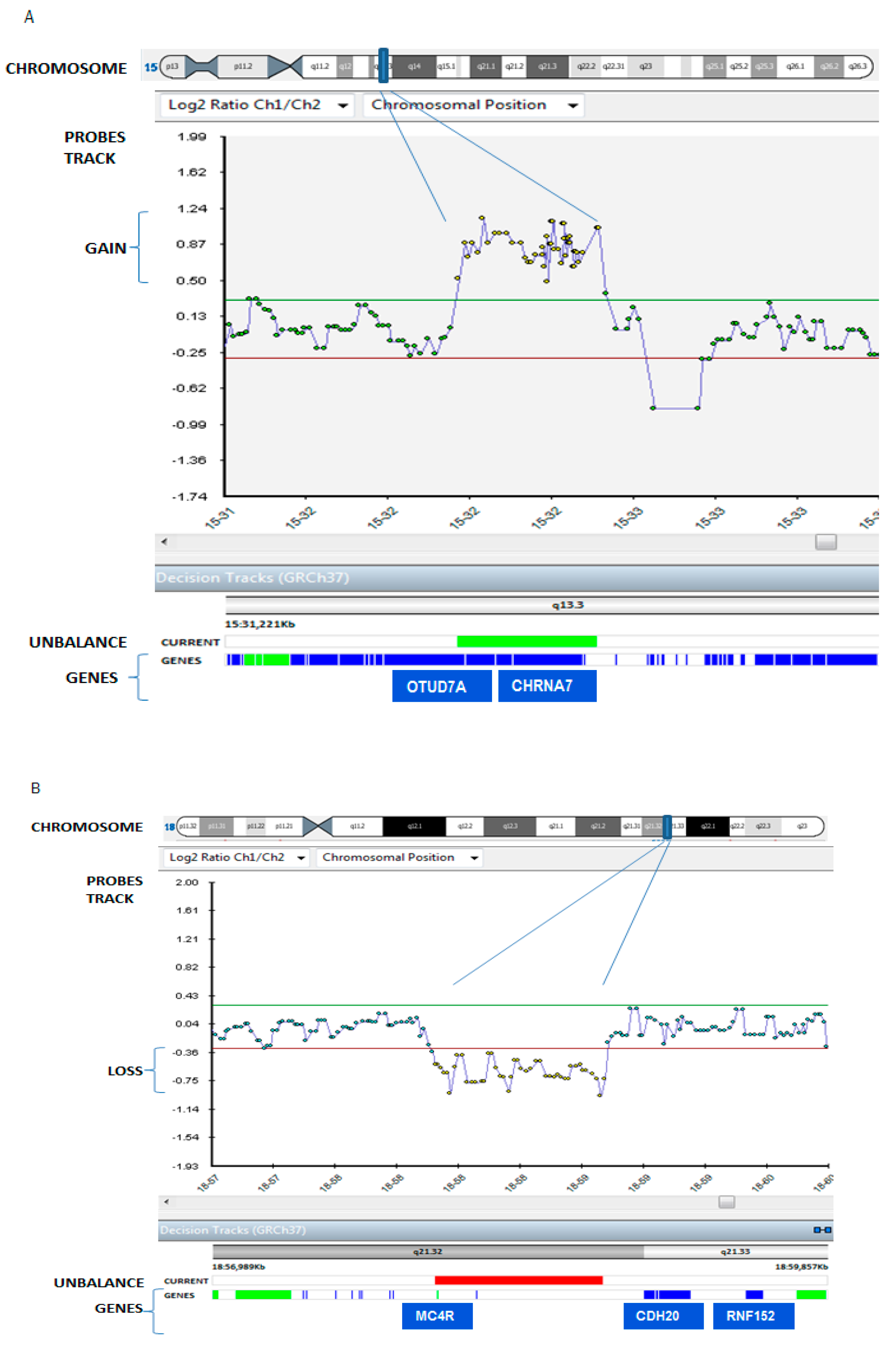
| 5q13.3 Duplication and 18 (Decipher ID: 263336) | Cholinergic Receptor, Nicotinic, alpha 7 (CHRNA7), Ovarian Tumor (OTU) domain containing 7A (OTUD7A) (our report) |
|
| Chromosome disorders | ||
| Down Syndrome [32] | 21 |
|
| Turner Syndrome [33] | X |
|
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sciacca, F.L.; Rizzo, A.; Bedini, G.; Capone, F.; Di Lazzaro, V.; Nava, S.; Acerbi, F.; Sebastiano, D.R.; Binelli, S.; Faragò, G.; et al. Microduplication of 15q13.3 and Microdeletion of 18q21.32 in a Patient with Moyamoya Syndrome. Int. J. Mol. Sci. 2018, 19, 3675. https://doi.org/10.3390/ijms19113675
Sciacca FL, Rizzo A, Bedini G, Capone F, Di Lazzaro V, Nava S, Acerbi F, Sebastiano DR, Binelli S, Faragò G, et al. Microduplication of 15q13.3 and Microdeletion of 18q21.32 in a Patient with Moyamoya Syndrome. International Journal of Molecular Sciences. 2018; 19(11):3675. https://doi.org/10.3390/ijms19113675
Chicago/Turabian StyleSciacca, Francesca Luisa, Ambra Rizzo, Gloria Bedini, Fioravante Capone, Vincenzo Di Lazzaro, Sara Nava, Francesco Acerbi, Davide Rossi Sebastiano, Simona Binelli, Giuseppe Faragò, and et al. 2018. "Microduplication of 15q13.3 and Microdeletion of 18q21.32 in a Patient with Moyamoya Syndrome" International Journal of Molecular Sciences 19, no. 11: 3675. https://doi.org/10.3390/ijms19113675
APA StyleSciacca, F. L., Rizzo, A., Bedini, G., Capone, F., Di Lazzaro, V., Nava, S., Acerbi, F., Sebastiano, D. R., Binelli, S., Faragò, G., Gioppo, A., Grisoli, M., Bruzzone, M. G., Ferroli, P., Pantaleoni, C., Caputi, L., Gomez, J. V., Parati, E. A., & Bersano, A. (2018). Microduplication of 15q13.3 and Microdeletion of 18q21.32 in a Patient with Moyamoya Syndrome. International Journal of Molecular Sciences, 19(11), 3675. https://doi.org/10.3390/ijms19113675