Prediction of Signal Peptides in Proteins from Malaria Parasites

 , , and
, , and
Abstract
1. Introduction
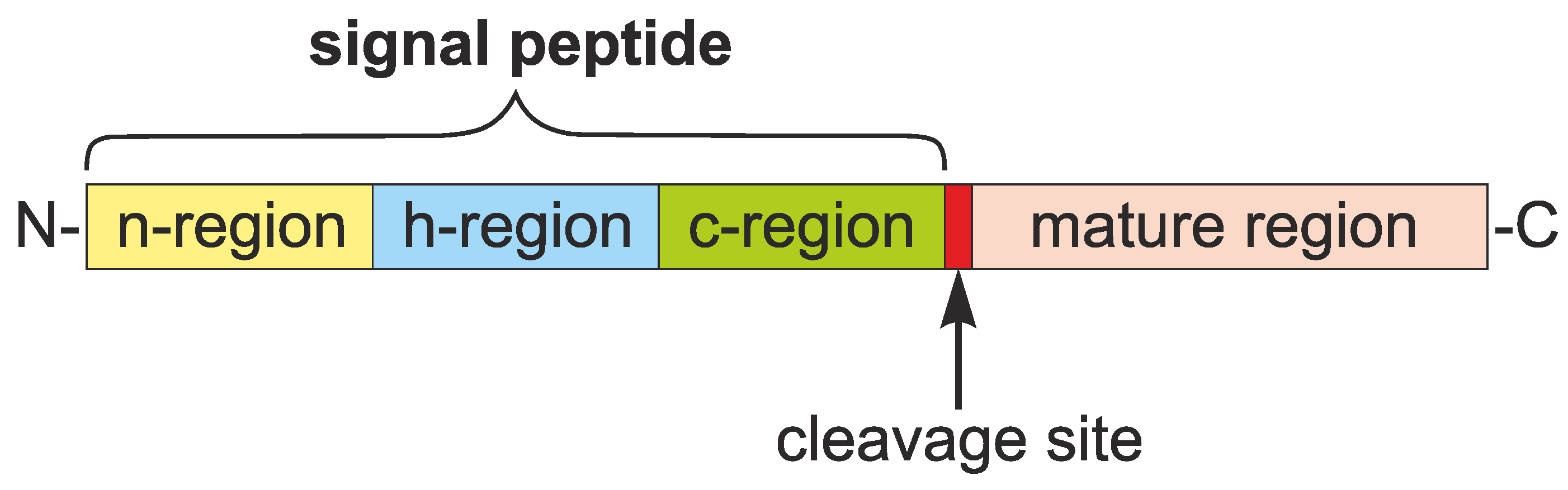
1.1. Roles and Features of Signal Peptides
1.2. Software Predicting Signal Peptides
2. Results and Discussion
2.1. Performance of SignalHsmm Algorithm
2.2. Comparison of Amino Acid Encodings
2.3. Benchmark Tests
2.4. Specific Composition of Plasmodiidae Signal Peptides
3. Materials and Methods
3.1. Overview
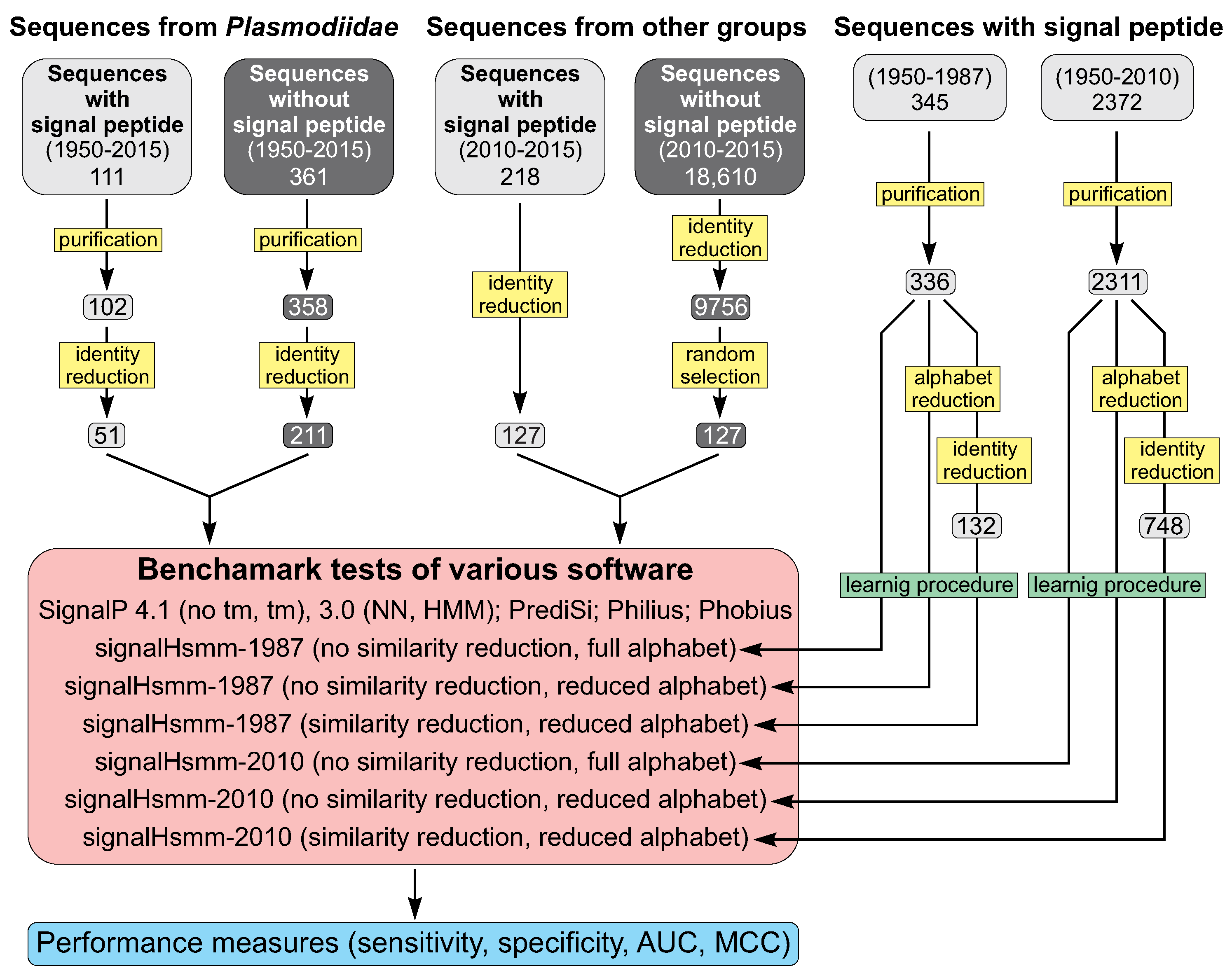
3.2. Data Selection
3.3. Sequence Identity Reduction in Studied Sequence Sets
3.4. Clustering of Amino Acids into Groups
3.5. Hidden Semi-Markov Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SP | Signal Peptide |
| HSMM | Hidden Semi-Markov Model |
| HMM | Hidden Markov Model |
| AUC | Area Under the Curve |
| MCC | Matthews Correlation Coefficient |
References
- Rapoport, T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007, 450, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.J.; Schultz, L.D. Mutations of the alpha-galactosidase signal peptide which greatly enhance secretion of heterologous proteins by yeast. Gene 1991, 101, 105–111. [Google Scholar] [CrossRef]
- Chan, D.; Ho, M.S.P.; Cheah, K.S.E. Aberrant signal peptide cleavage of collagen X in Schmid metaphyseal chondrodysplasia. Implications for the molecular basis of the disease. J. Biol. Chem. 2001, 276, 7992–7997. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Leng, Q.; Mixson, A.J. Alteration in the IL-2 signal peptide affects secretion of proteins in vitro and in vivo. J. Gene Med. 2005, 7, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wilkinson, G.F.; Willars, G.B. Role of the signal peptide in the synthesis and processing of the glucagon-like peptide-1 receptor. Br. J. Pharmacol. 2010, 159, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Ladunga, I. PHYSEAN: PHYsical SEquence ANalysis for the identification of protein domains on the basis of physical and chemical properties of amino acids. Bioinformatics 1999, 15, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Izard, J.W.; Kendall, D.A. Signal peptides: Exquisitely designed transport promoters. Mol. Microbiol. 1994, 13, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.; Schröder, B.; Fluhrer, R. Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP-like proteases. Biochim. Biophys. Acta 2013, 1828, 2828–2839. [Google Scholar] [CrossRef] [PubMed]
- von Heijne, G.; Gavel, Y. Topogenic signals in integral membrane proteins. Eur. J. Biochem. 1988, 174, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Paetzel, M.; Karla, A.; Strynadka, N.C.; Dalbey, R.E. Signal peptidases. Chem. Rev. 2002, 102, 4549–4580. [Google Scholar] [CrossRef] [PubMed]
- Palzkill, T.; Le, Q.Q.; Wong, A.; Botstein, D. Selection of functional signal peptide cleavage sites from a library of random sequences. J. Bacteriol. 1994, 176, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Szczesna-Skorupa, E.; Browne, N.; Mead, D.; Kemper, B. Positive charges at the NH2 terminus convert the membrane-anchor signal peptide of cytochrome P-450 to a secretory signal peptide. Proc. Natl. Acad. Sci. USA 1988, 85, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tan, S.; Berry, J.O.; Li, P.; Ren, N.; Li, S.; Yang, G.; Wang, W.B.; Qi, X.T.; Yin, L.P. An Uncleaved signal peptide directs the Malus xiaojinensis iron transporter protein Mx IRT1 into the ER for the PM Secretory Pathway. Int. J. Mol. Sci. 2014, 15, 20413–20433. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.S.; Bernstein, H.D. The surprising complexity of signal sequences. Trends Biochem. Sci. 2006, 31, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Moeller, L.; Taylor-Vokes, R.; Fox, S.; Gan, Q.; Johnson, L.; Wang, K. Wet-milling transgenic maize seed for fraction enrichment of recombinant subunit vaccine. Biotechnol. Prog. 2010, 26, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Kerzerho, J.; Schneider, A.; Favry, E.; Castelli, F.A.; Maillère, B. The signal peptide of the tumor-shared antigen midkine hosts CD4+ T cell epitopes. J. Biol. Chem. 2013, 288, 13370–13377. [Google Scholar] [CrossRef] [PubMed]
- Neto Ade, M.; Alvarenga, D.A.; Rezende, A.M.; Resende, S.S.; Ribeiro Rde, S.; Fontes, C.J.; Carvalho, L.H.; de Brito, C.F. Improving N-terminal protein annotation of Plasmodium species based on signal peptide prediction of orthologous proteins. Malar. J. 2012, 11, 375. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, C.J.; Kalanon, M.; McFadden, G.I. Protein targeting to the malaria parasite plastid. Traffic 2008, 9, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Foth, B.J.; McFadden, G.I. The apicoplast: A plastid in Plasmodium falciparum and other Apicomplexan parasites. Int. Rev. Cytol. 2003, 224, 57–110. [Google Scholar] [PubMed]
- Lim, L.; McFadden, G.I. The evolution, metabolism and functions of the apicoplast. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 749–763. [Google Scholar] [CrossRef] [PubMed]
- McFadden, G.I. The apicoplast. Protoplasma 2011, 248, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Heiny, S.R.; Pautz, S.; Recker, M.; Przyborski, J.M. Protein traffic to the Plasmodium falciparum apicoplast: Evidence for a sorting branch point at the Golgi. Traffic 2014, 15, 1290–1304. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, J.; Wilson, E.H.; Masek, K.; Hunter, C.A.; Striepen, B. Apicoplast fatty acid synthesis is essential for organelle biogenesis and parasite survival in Toxoplasma gondii. Proc. Natl. Acad. Sci. USA 2006, 103, 13192–13197. [Google Scholar] [CrossRef] [PubMed]
- Fichera, M.E.; Roos, D.S. A plastid organelle as a drug target in apicomplexan parasites. Nature 1997, 390, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.A.; D’Ombrain, M.C.; McFadden, G.I. The apicoplast as an antimalarial drug target. Drug. Resist. Updat. 2001, 4, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Gornicki, P. Apicoplast fatty acid biosynthesis as a target for medical intervention in apicomplexan parasites. Int. J. Parasitol. 2003, 33, 885–896. [Google Scholar] [CrossRef]
- Garcia-Estrada, C.; Prada, C.F.; Fernandez-Rubio, C.; Rojo-Vazquez, F.; Balana-Fouce, R. DNA topoisomerases in apicomplexan parasites: Promising targets for drug discovery. Proc. Biol. Sci. 2010, 277, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Vandomme, A.; Fréville, A.; Cailliau, K.; Kalamou, H.; Bodart, J.F.; Khalife, J.; Pierrot, C. PhosphoTyrosyl phosphatase activator of Plasmodium falciparum: Identification of its residues involved in binding to and activation of PP2A. Int. J. Mol. Sci. 2014, 15, 2431–2453. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.L.; Fidock, D.A.; Bogyo, M. Protein degradation systems as antimalarial therapeutic targets. Trends Parasitol. 2017, 33, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Díaz, M.B.; Ebert, D.; Salinas, Y.; Pradhan, A.; Lehane, A.M.; Myrand-Lapierre, M.E.; O’Loughlin, K.G.; Shackleford, D.M.; de Almeida, M.J.; Carrillo, A.K.; et al. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc. Natl. Acad. Sci. USA 2014, 111, E5455–E5462. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.B.; Morrisey, J.M.; Zhang, Z.; Das, S.; Daly, T.M.; Otto, T.D.; Spillman, N.J.; Wyvratt, M.; Siegl, P.; Marfurt, J.; et al. Pyrazoleamide compounds are potent antimalarials that target Na+ homeostasis in intraerythrocytic Plasmodium falciparum. Nat. Commun. 2014, 5, 5521. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.A.; Lotharius, J.; Marsh, K.; White, J.; Dayan, A.; White, K.L.; Njoroge, J.W.; El Mazouni, F.; Lao, Y.; Kokkonda, S.; et al. A Long-Duration Dihydroorotate Dehydrogenase Inhibitor (DSM265) for Prevention and Treatment of Malaria. Sci. Transl. Med. 2015, 7, 296ra111. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.W.; Zhang, T.H.; Zhang, J.N.; Huang, Y. Prediction of signal peptide cleavage sites with subsite-coupled and template matching fusion algorithm. Mol. Inform. 2014, 33, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Chen, Y.; Chen, L.; Guo, G.; Fan, Y.; Kong, X. Signal-BNF: A Bayesian network fusing approach to predict signal peptides. J. Biomed. Biotechnol. 2012, 2012, 492174. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.B.; Chou, K.C. Signal-3L: A 3-layer approach for predicting signal peptides. Biochem. Biophys. Res. Commun. 2007, 363, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Hiller, K.; Grote, A.; Scheer, M.; Münch, R.; Jahn, D. PrediSi: Prediction of signal peptides and their cleavage positions. Nucleic Acids Res. 2004, 32, W375–W379. [Google Scholar] [CrossRef] [PubMed]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, S.M.; Kall, L.; Riffle, M.E.; Bilmes, J.A.; Noble, W.S. Transmembrane topology and signal peptide prediction using dynamic bayesian networks. PLoS Comput. Biol. 2008, 4, e1000213. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, J.D.; Nielsen, H.; von Heijne, G.; Brunak, S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004, 340, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.; Krogh, A. Prediction of signal peptides and signal anchors by a hidden Markov model. In Proceedings of the International Conference on Intelligent Systems for Molecular Biology, Toronto, ON, Canada, 19–23 July 2008; Volume 6, pp. 122–130. [Google Scholar]
- Maetschke, S.; Towsey, M.; Bodén, M. BLOMAP: An Encoding of Amino Acids which Improves Signal Peptide Cleavage Site Prediction. In Proceedings of the 3rd Asia-Pacific Bioinformatics Conference, Singapore, 17–21 January 2005; Imperial College Press: London, UK, 2005; pp. 141–150. [Google Scholar]
- Murphy, L.R.; Wallqvist, A.; Levy, R.M. Simplified amino acid alphabets for protein fold recognition and implications for folding. Protein Eng. 2000, 13, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.L.; Kondev, J.; Theriot, J.A.; Phillips, R. Reduced amino acid alphabets exhibit an improved sensitivity and selectivity in fold assignment. Bioinformatics 2009, 25, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- von Heijne, G. A new method for predicting signal sequence cleavage sites. Nucleic Acids Res. 1986, 14, 4683–4690. [Google Scholar] [CrossRef] [PubMed]
- Cilingir, G.; Broschat, S.L.; Lau, A.O. ApicoAP: The first computational model for identifying apicoplast-targeted proteins in multiple species of Apicomplexa. PLoS ONE 2012, 7, e36598. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Williams, A.H.; Hane, J.K.; Singh, K.B.; Taylor, J.M. Evaluation of Secretion Prediction Highlights Differing Approaches Needed for Oomycete and Fungal Effectors. Front. Plant Sci. 2015, 6, 1168. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Li, X.X.; Yang, H.; Li, Y.H.; Xue, W.W.; Chen, Y.Z.; Tao, L.; Zhu, F. Assessing the performances of protein function prediction algorithms from the perspectives of identification accuracy and false discovery rate. Int. J. Mol. Sci. 2018, 19, 183. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.; Engelbrecht, J.; Brunak, S.; von Heijne, G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Burdukiewicz, M.; Sobczyk, P.; Rödiger, S.; Duda-Madej, A.; Mackiewicz, P.; Kotulska, M. Amyloidogenic Motifs Revealed by N-Gram Analysis. Sci. Rep. 2017, 7, 12961. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.M. Size; Academic Press: New York, NY, USA, 1972; pp. 1–38. [Google Scholar]
- Fasman, G.D. Proteins, 3rd ed.; CRC Press: Cleveland, OH, USA, 1976; Volume 1. [Google Scholar]
- Goldsack, D.E.; Chalifoux, R.C. Contribution of the free energy of mixing of hydrophobic side chains to the stability of the tertiary structure of proteins. J. Theor. Biol. 1973, 39, 645–651. [Google Scholar] [CrossRef]
- Zimmerman, J.M.; Eliezer, N.; Simha, R. The characterization of amino acid sequences in proteins by statistical methods. J. Theor. Biol. 1968, 21, 170–201. [Google Scholar] [CrossRef]
- Cid, H.; Bunster, M.; Canales, M.; Gazitua, F. Hydrophobicity and structural classes in proteins. Protein Eng. 1992, 5, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D. Three-dimensional structure of membrane and surface proteins. Annu. Rev. Biochem. 1984, 53, 595–623. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Ponnuswamy, P.K.; Prabhakaran, M.; Manavalan, P. Hydrophobic packing and spatial arrangement of amino acid residues in globular proteins. Biochim. Biophys. Acta 1980, 623, 301–316. [Google Scholar] [CrossRef]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Radzicka, A.; Pedersen, L.; Wolfenden, R. Influences of solvent water on protein folding: Free energies of solvation of cis and trans peptides are nearly identical. Biochemistry 1988, 27, 4538–4541. [Google Scholar] [CrossRef] [PubMed]
- Argos, P.; Rao, J.K.; Hargrave, P.A. Structural prediction of membrane-bound proteins. Eur. J. Biochem. 1982, 128, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.Y.; Fasman, G.D. Prediction of the secondary structure of proteins from their amino acid sequence. Adv. Enzymol. Relat. Areas Mol. Biol. 1978, 47, 45–148. [Google Scholar] [PubMed]
- Prabhakaran, M. The distribution of physical, chemical and conformational properties in signal and nascent peptides. Biochem. J. 1990, 269, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2008, 36, D202–D205. [Google Scholar] [CrossRef] [PubMed]
- Rabiner, L.R. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE 1989, 77, 257–286. [Google Scholar] [CrossRef]
- Yu, S.Z. Hidden semi-Markov models. Artif. Intell. 2010, 174, 215–243. [Google Scholar] [CrossRef]
- Koski, T. Hidden Markov Models for Bioinformatics; Computational Biology; Springer: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Durbin, R.; Eddy, S.R.; Krogh, A.; Mitchison, G. Biological Sequence Analysis. Probabilistic Models of Proteins and Nucleic Acids; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measure | Mean | SD |
|---|---|---|
| AUC | 0.9682 | 0.0023 |
| Sensitivity | 0.9407 | 0.0008 |
| Specificity | 0.9272 | 0.0050 |
| MCC | 0.8681 | 0.0049 |
| Group | Amino Acids |
|---|---|
| 1 | D, E, H, K, N, Q, R |
| 2 | G, P, S, T, Y |
| 3 | F, I, L, M, V, W |
| 4 | A, C |
| Group | Amino Acids |
|---|---|
| 1 | A, E, K, Q, R |
| 2 | D, G, N, P, S, T |
| 3 | C, H, I, L, M, V |
| 4 | F, W, Y |
| Sensitivity | Specificity | MCC | AUC | |
|---|---|---|---|---|
| SignalP 4.1 (no tm) [33] | 0.8235 | 0.9100 | 0.6872 | 0.8667 |
| SignalP 4.1 (tm) [33] | 0.6471 | 0.9431 | 0.6196 | 0.7951 |
| SignalP 3.0 (NN) [40] | 0.8824 | 0.9052 | 0.7220 | 0.8938 |
| SignalP 3.0 (HMM) [40] | 0.6275 | 0.9194 | 0.5553 | 0.7734 |
| PrediSi [37] | 0.3333 | 0.9573 | 0.3849 | 0.6453 |
| Philius [39] | 0.6078 | 0.9336 | 0.5684 | 0.7707 |
| Phobius [38] | 0.6471 | 0.9289 | 0.5895 | 0.7880 |
| signalHsmm-2010 | 0.9804 | 0.8720 | 0.7409 | 0.9262 |
| signalHsmm-2010 (ident. 50%) | 1.0000 | 0.8768 | 0.7621 | 0.9384 |
| signalHsmm-2010 (raw aa) | 0.8431 | 0.9005 | 0.6853 | 0.8718 |
| signalHsmm-1987 | 0.9216 | 0.8910 | 0.7271 | 0.9063 |
| signalHsmm-1987 (ident. 50%) | 0.9412 | 0.8768 | 0.7194 | 0.9090 |
| signalHsmm-1987 (raw aa) | 0.7647 | 0.9052 | 0.6350 | 0.8350 |
| Property Name | Amino Acid Scale |
|---|---|
| Size | Size [52] |
| Size | Molecular weight [53] |
| Size | Residue volume [54] |
| Size | Bulkiness [55] |
| Hydrophobicity | Normalized hydrophobicity scales for -proteins [56] |
| Hydrophobicity | Consensus normalized hydrophobicity scale [57] |
| Hydrophobicity | Hydropathy index [58] |
| Hydrophobicity | Surrounding hydrophobicity in -helix [59] |
| Polarity | Polarity [60] |
| Polarity | Mean polarity [61] |
| Occurrence in -helices | Signal sequence helical potential [62] |
| Occurrence in -helices | Normalized frequency of N-terminal helix [63] |
| Occurrence in -helices | Relative frequency in -helix [64] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burdukiewicz, M.; Sobczyk, P.; Chilimoniuk, J.; Gagat, P.; Mackiewicz, P. Prediction of Signal Peptides in Proteins from Malaria Parasites. Int. J. Mol. Sci. 2018, 19, 3709. https://doi.org/10.3390/ijms19123709
Burdukiewicz M, Sobczyk P, Chilimoniuk J, Gagat P, Mackiewicz P. Prediction of Signal Peptides in Proteins from Malaria Parasites. International Journal of Molecular Sciences. 2018; 19(12):3709. https://doi.org/10.3390/ijms19123709
Chicago/Turabian StyleBurdukiewicz, Michał, Piotr Sobczyk, Jarosław Chilimoniuk, Przemysław Gagat, and Paweł Mackiewicz. 2018. "Prediction of Signal Peptides in Proteins from Malaria Parasites" International Journal of Molecular Sciences 19, no. 12: 3709. https://doi.org/10.3390/ijms19123709
APA StyleBurdukiewicz, M., Sobczyk, P., Chilimoniuk, J., Gagat, P., & Mackiewicz, P. (2018). Prediction of Signal Peptides in Proteins from Malaria Parasites. International Journal of Molecular Sciences, 19(12), 3709. https://doi.org/10.3390/ijms19123709