Molecular Regulation of Catalpol and Acteoside Accumulation in Radial Striation and non-Radial Striation of Rehmannia glutinosa Tuberous Root

Abstract
1. Introduction
2. Results
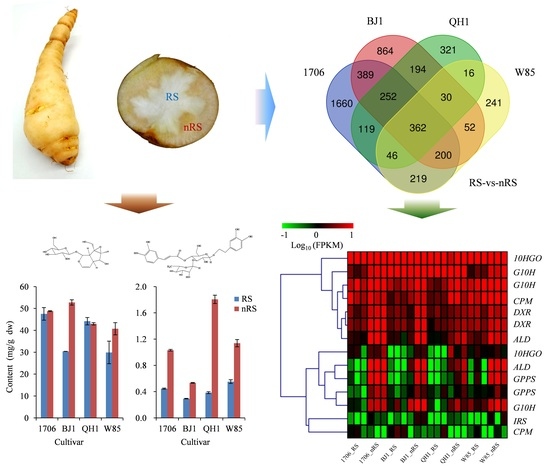
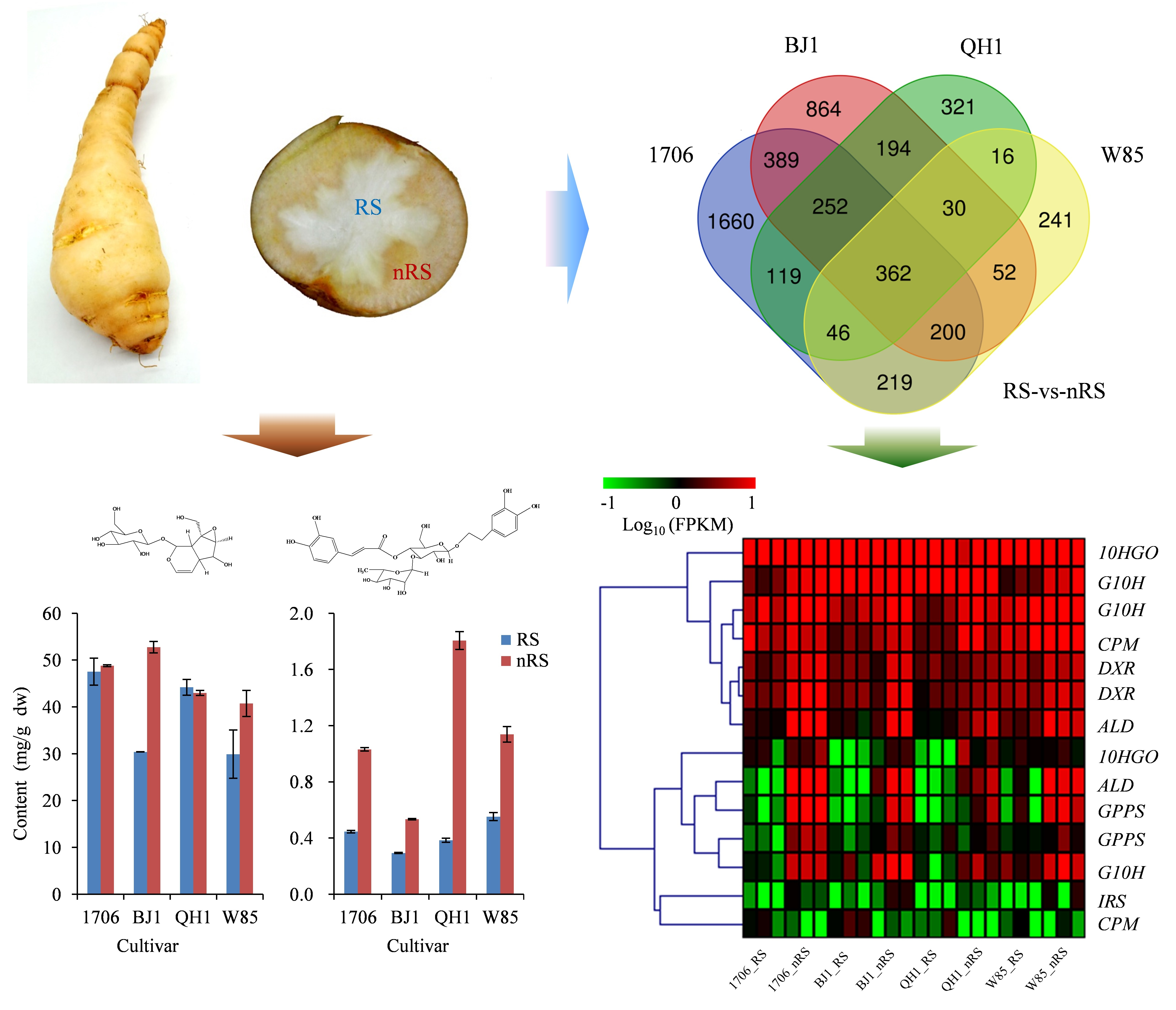
2.1. Quality Characteristics of Radial Striation and Non-Radial Striation of R. glutinosa
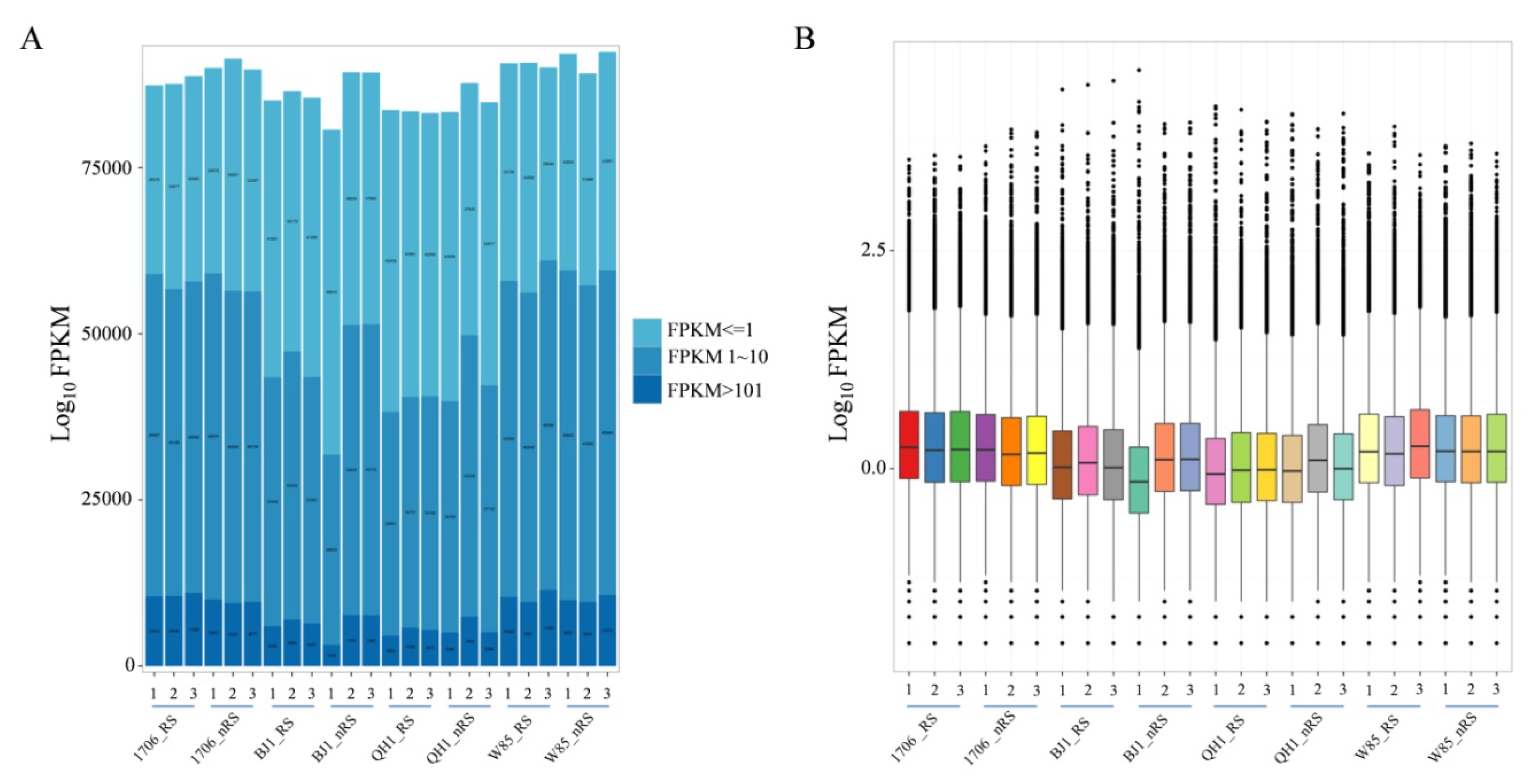
2.2. R. glutinosa Transcriptome Sequencing and Unigene Assembly
2.3. Functional Annotation of R. glutinosa Unigenes
2.4. Expression Analysis for Radial Striation and Non-Radial Striation of Four R. glutinosa Cultivars
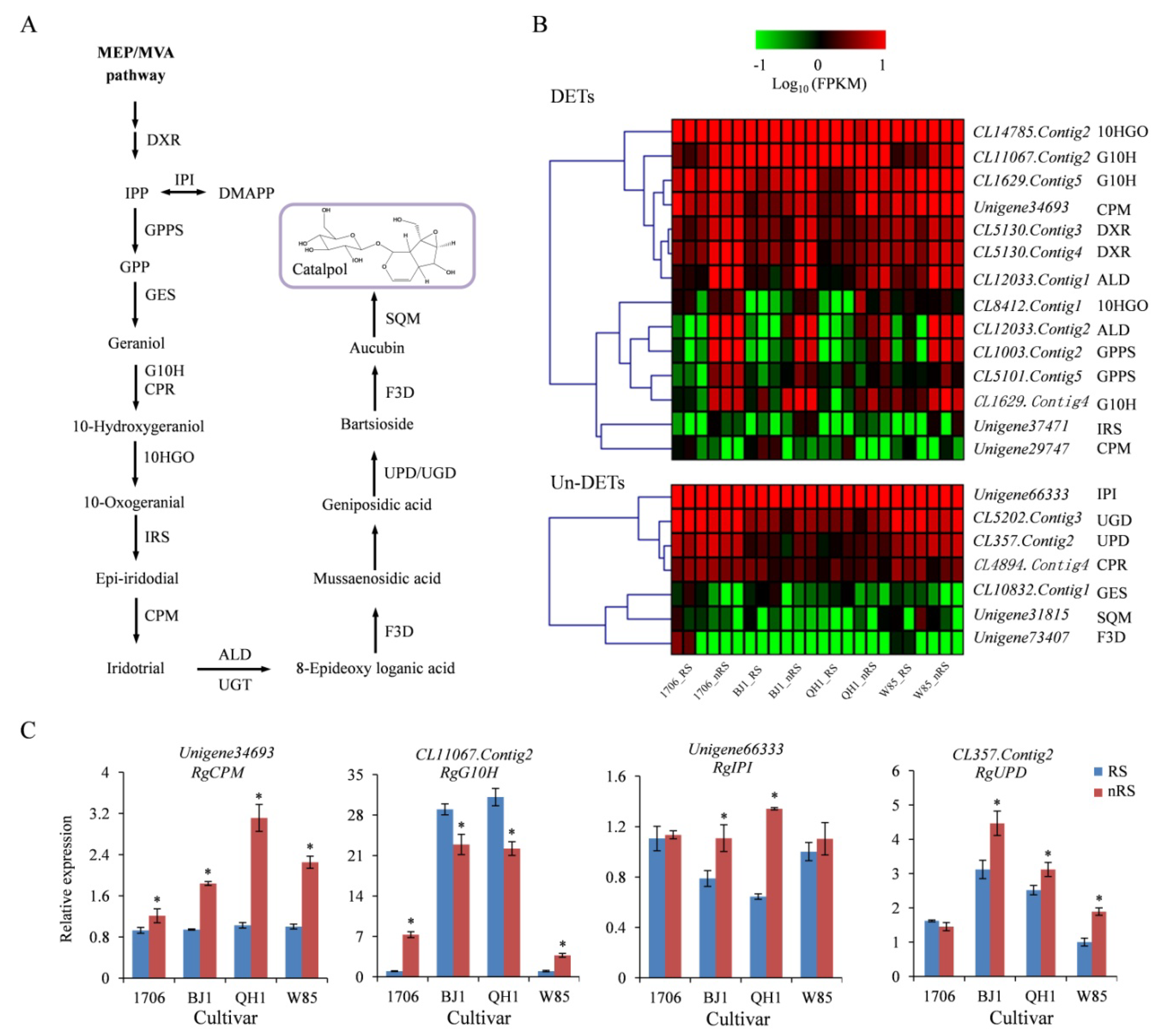
2.5. Genes Related to Catalpol Biosynthesis in R. glutinosa
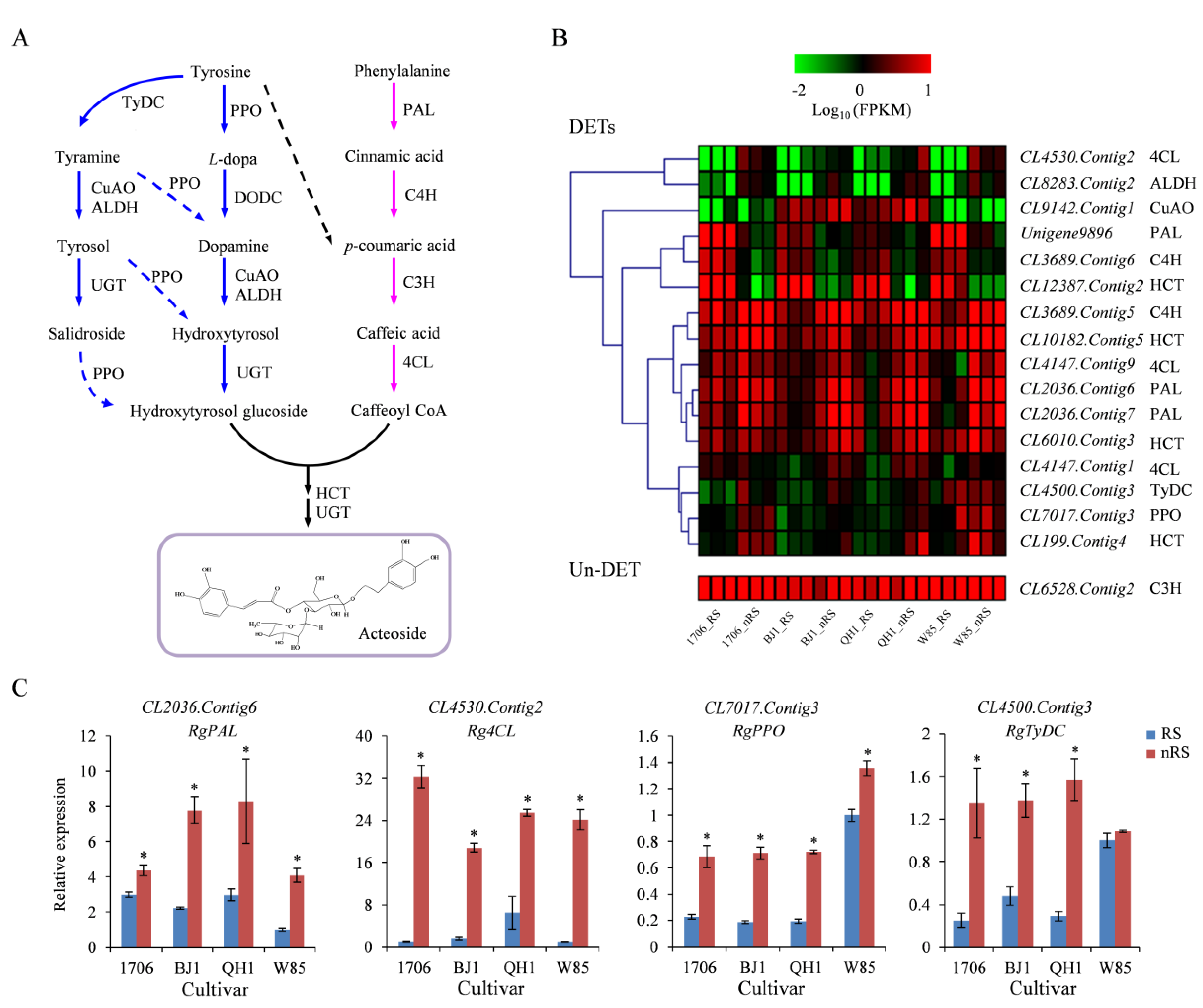
2.6. Genes Related to Acteoside Biosynthesis in R. glutinosa
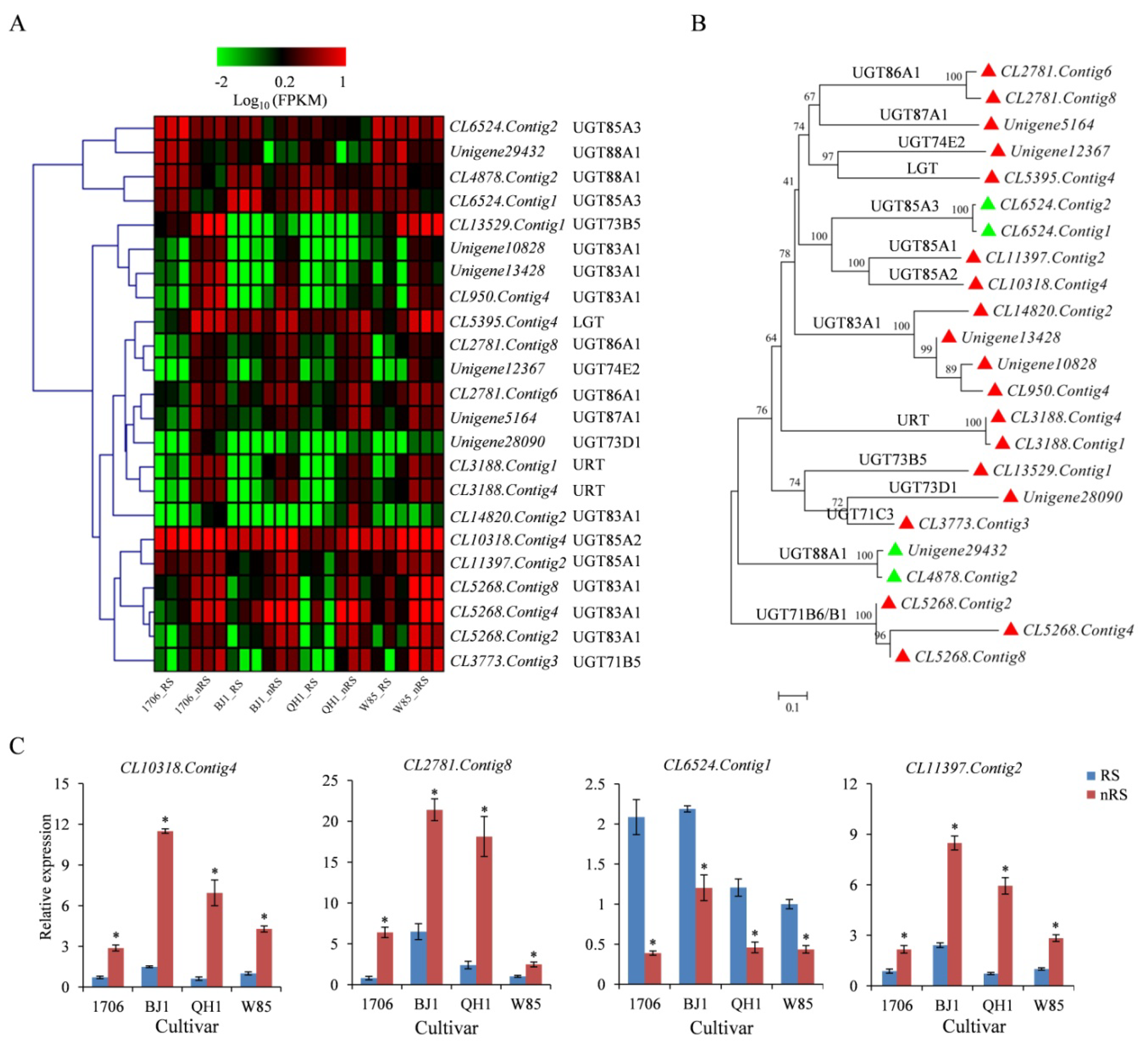
2.7. UDP-Dependent Glycosyltransferases (UGT) Gene
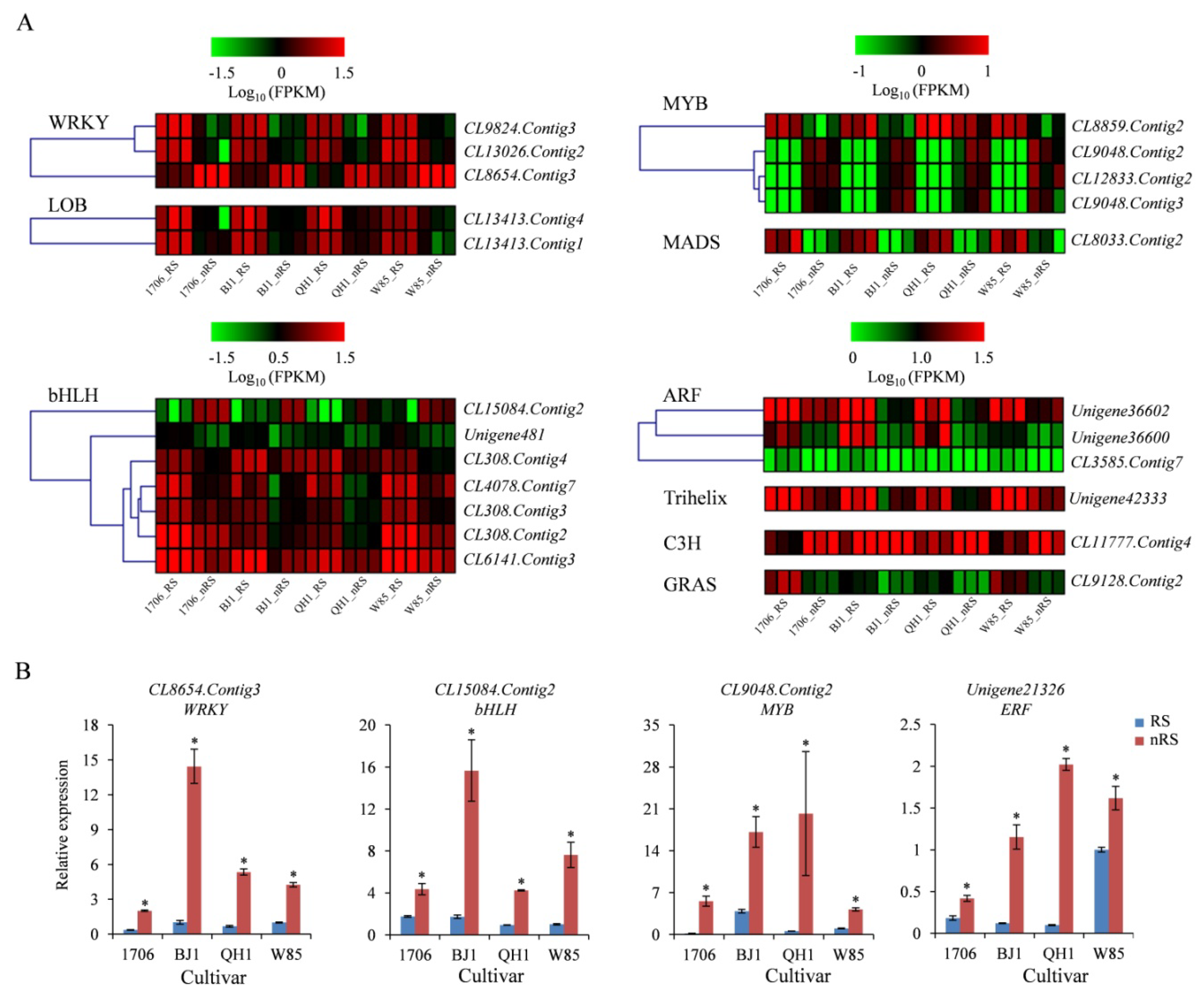
2.8. Transcription Factor Genes
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Analysis of Quality Characteristics of Radial Striation and Non-Radial Striation of R. glutinosa
4.3. RNA Isolation and Illumina Sequencing
4.4. Sequence Assembly and Annotation
4.5. Differential Gene Expression Analysis
4.6. Quantitative RT-PCR Analysis
4.7. Data Submission
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TCM | Traditional Chinese medicine |
| RS | Radial striation |
| nRS | Non-radial striation |
| TF | Transcription factor |
| UGTs | UDP-dependent glycosyltransferases |
| PhGs | Phenylethanoid glycosides |
| IGs | Iridoid glycosides |
| PE | Paired-end |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| NT | NCBI nucleotide sequences |
| GO | Gene Ontology |
| KOG | EuKaryotic Ortholog Groups |
| ORF | Open reading frame |
| CDS | Coding DNA sequences |
| FPKM | Reads Per Kilobase of exon model per Million mapped reads |
| DETs | Differentially expressed transcripts |
| FDR | False discovery rate |
References
- Zhang, R.; Li, M.; Jia, Z. Rehmannia glutinosa: Review of botany, chemistry and pharmacology. J. Ethnopharmacol. 2008, 117, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, L.; Luo, H.; Hao, Z.; Wang, Y.; Zhang, C.; Chen, R.; Yu, D. Chemical constituents from root tubers of Rehmannia glutinosa. Chin. Tradit. Herb. Drugs 2014, 45, 16–22. [Google Scholar] [CrossRef]
- Li, M.; Wang, X.; Zhang, Z.; Zhang, J.; Zhao, X.; Zheng, X.; Feng, W. Three new alkaloids and a new iridoid glycoside from the roots of Rehmannia glutinosa. Phytochem. Lett. 2017, 21, 157–162. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, A.; Jiang, B.; Bao, Y.; Wang, J.; An, L. Further pharmacological evidence of the neuroprotective effect of catalpol from Rehmannia glutinosa. Phytomedicine 2008, 15, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pei, L.; Zhang, Y.; Cheng, Y.; Niu, C.; Cui, Y.; Wang, G. Ethanol extract of Rehmannia glutinosa exerts antidepressant-like effects on a rat chronic unpredictable mild stress model by involving monoamines and BDNF. Metab. Brain Dis. 2018, 33, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Song, L.; Li, M.; Liu, G.; Chu, Y.; Ma, L.; Huang, L. Genetic variation and metabolic pathway intricacy govern the active compound content and quality of the Chinese medicinal plant Lonicera japonica thunb. BMC Genom. 2012, 13, 195. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Peters, R.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Chen, S. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Luo, H.; Ji, A.; Zhang, X.; Song, J.; Chen, S. Global identification of the full-Length transcripts and alternative splicing related to phenolic acid biosynthetic genes in Salvia miltiorrhiza. Front. Plant Sci. 2016, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Alagna, F.; Mariotti, R.; Panara, F.; Caporali, S.; Urbani, S.; Veneziani, G.; Baldoni, L. Olive phenolic compounds: Metabolic and transcriptional profiling during fruit development. BMC Plant Biol. 2012, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, C.; Wang, H.; Zhang, L. Assaying of content of catalpol in Rehmanniag glutinosa from different origins. Chin. Tradit. Herb. Drugs 2002, 33, 126–128. [Google Scholar]
- Ji, X.; Sun, P.; Qi, J.; Liao, D.; Li, X. Study on distribution and dynamic accumulation of catalpol and total iridoid in fresh Rehmannia glutinosa. China J. Chin. Mater. Med. 2014, 39, 466–470. [Google Scholar] [CrossRef]
- Wang, F.; Wang, L.; Zhi, J.; Zhang, M.; Yang, C.; Huang, Y.; Xie, C.; Zhang, Z. Changes in dynamic accumulation of acteoside from different Rehmannia glutinosa cultivars. Chin. J. Exp. Tradit. Med. Formulae 2017, 23, 78–83. [Google Scholar] [CrossRef]
- Xie, C.; Zhang, M.; Li, Y.; Geng, X.; Wang, F.; Zhang, Z. Comparison of chemical quality characteristics between radial striations and non-radial striations in tuberous root of Rehmannia glutinosa. China J. Chin. Mater. Med. 2017, 42, 4172–4179. [Google Scholar] [CrossRef]
- Jiang, B.; Shen, R.; Bi, J.; Tian, X.; Hinchliffe, T.; Xia, Y. Catalpol: A potential therapeutic for neurodegenerative diseases. Curr. Med. Chem. 2015, 22, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hu, X.; Zeng, Y.; Li, Y.; Wu, H.; Qiu, R.; He, Z. Advanced research on acteoside for chemistry and bioactivities. J. Asian Nat. Prod. Res. 2011, 13, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Song, S.; Zhou, L.; Zhang, B.; Qi, J.; Li, X. Transcriptome analysis reveals putative genes involved in iridoid biosynthesis in Rehmannia glutinosa. Int. J. Mol. Sci. 2012, 13, 13748–13763. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, X.; Wang, W.; Duan, H. De novo transcriptome sequencing-based discovery and expression analyses of verbascoside biosynthesis-associated genes in Rehmannia glutinosa tuberous roots. Mol. Breed. 2016, 36, 139. [Google Scholar] [CrossRef]
- Wang, F.; Zhi, J.; Zhang, Z.; Wang, L.; Suo, Y.; Xie, C.; Li, M.; Zhang, B.; Du, J.; Gu, L.; et al. Transcriptome analysis of salicylic acid treatment in Rehmannia glutinosa hairy roots using RNA-seq technique for identification of genes involved in acteoside biosynthesis. Front. Plant Sci. 2017, 8, 787. [Google Scholar] [CrossRef] [PubMed]
- Damtoft, S. Biosynthesis of catalpol. Phytochemistry 1994, 35, 1187–1189. [Google Scholar] [CrossRef]
- Jensen, S.R.; Franzyk, H.; Wallander, E. Chemotaxonomy of the Oleaceae: Iridoids as taxonomic markers. Phytochemistry 2002, 60, 213–231. [Google Scholar] [CrossRef]
- Shitiz, K.; Sharma, N.; Pal, T.; Sood, H.; Chauhan, R. NGS transcriptomes and enzyme inhibitors unravel complexity of picrosides biosynthesis in Picrorhiza kurroa Royle ex. Benth. PLoS ONE 2015, 10, e0144546. [Google Scholar] [CrossRef] [PubMed]
- Saimaru, H.; Orihara, Y. Biosynthesis of acteoside in cultured cells of Olea europaea. J. Nat. Med. 2010, 64, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Dare, A.; Yauk, Y.; Tomes, S.; McGhie, T.; Rebstock, R.; Cooney, J.; Atkinson, R. Silencing a phloretin-specific glycosyltransferase perturbs both general phenylpropanoid biosynthesis and plant development. Plant J. 2017, 91, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Baldauf, S.; Lim, E.; Bowles, D. Phylogenetic analysis of the UDP-glycosyltransferase multigene family of Arabidopsis thaliana. J. Biol. Chem. 2001, 276, 4338–4343. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, F.; Dossa, K.; Wang, Z.; Ke, T. Genome-wide analysis of UDP-glycosyltransferase super family in Brassica rapa and Brassica oleracea reveals its evolutionary history and functional characterization. BMC Genom. 2017, 18, 474. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, J.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.; Samaha, R.; et al. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Qi, L.; Yang, J.; Wu, C.; Liu, Y.; Huang, L. A Scutellaria baicalensis, R2R3-MYB gene, SbMYB8, regulates flavonoid biosynthesis and improves drought stress tolerance in transgenic tobacco. Plant Cell Tissue Organ Cult. 2015, 120, 961–972. [Google Scholar] [CrossRef]
- Yan, T.; Li, L.; Xie, L.; Chen, M.; Shen, Q.; Pan, Q.; Fu, X.; Shi, P.; Tang, Y.; Huang, H.; et al. A novel HD-ZIP IV/MIXTA complex promotes glandular trichome initiation and cuticle development in Artemisia annua. New Phytol. 2018, 218, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Suttipanta, N.; Pattanaik, S.; Kulshrestha, M.; Patra, B.; Singh, S.; Yuan, L. The transcription factor CrWRKY1 positively regulates the terpenoid indole alkaloid biosynthesis in Catharanthus roseus. Plant Physiol. 2011, 157, 2081–2093. [Google Scholar] [CrossRef] [PubMed]
- Zhai, R.; Wang, Z.; Zhang, S.; Meng, G.; Song, L.; Wang, Z.; Li, P.; Ma, F.; Xu, L. Two MYB transcription factors regulate flavonoid biosynthesis in pear fruit (Pyrus bretschneideri Rehd.). J. Exp. Bot. 2016, 67, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Singh, S.; Patra, B.; Sui, X.; Pattanaik, S.; Yuan, L. A differentially regulated AP2/ERF transcription factor gene cluster acts downstream of a MAP kinase cascade to modulate terpenoid indole alkaloid biosynthesis in Catharanthus roseus. New Phytol. 2016, 213, 1017–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Li, G.; Hu, P.; Zhao, X.; Li, L.; Wei, W.; Feng, J.; Zhou, H. Identification of basic/helix-loop-helix transcription factors reveals candidate genes involved in anthocyanin biosynthesis from the strawberry white-flesh mutant. Sci. Rep. 2018, 8, 2721. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, Y.; Li, X.; Gu, L.; Wang, F.; Feng, F.; Tian, Y.; Wang, F.; Wang, X.; Lin, W.; et al. Analysis of integrated multiple ‘omics’ datasets reveals the mechanisms of initiation and determination in the formation of tuberous roots in Rehmannia glutinosa. J. Exp. Bot. 2015, 66, 5837–5851. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.; Losada, J.; Niklas, K. Phloem networks in leaves. Curr. Opin. Plant Biol. 2018, 43, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Li, Y.; Zhang, M.; Geng, X.; Wang, F.; Zhang, Z. Feature analysis of active principle between radial striations (Xylem) and non-radial striations (phloem and periderm) in tuberous root of different germplasms Rehmanniae radix. Chin. J. Exp. Tradit. Med. Formulae 2018, 20, 75–83. [Google Scholar] [CrossRef]
- Yang, F.; Yu, H.; Chai, X.; Peng, S.; Yang, J.; Wu, D.; Du, J.; Wang, Y. Illumination on “reserving phloem and discarding xylem” and quality evaluation of Radix polygalae by determining oligosaccharide esters, saponins, and xanthones. Molecules 2018, 23, 836. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Kamochi, H.; Suzuki, H.; Nakamura, M.; Takahashi, H.; Hatada, T.; Saito, K.; Yamazaki, M. De novo transcriptome assembly and characterization of nine tissues of Lonicera japonica to identify potential candidate genes involved in chlorogenic acid, luteolosides, and secoiridoid biosynthesis pathways. J. Nat. Med. 2017, 71, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhao, W.; Guo, Q.; Zhang, W.; Ye, Z. Study on chemical components distribution in Paeoniae Radix Alba and its processing methods. China J. Chin. Mater. Med. 2015, 40, 1953–1959. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, X.; Chen, J.; Xu, H.; Li, J.; Zhang, Z. Differential miRNA expression in Rehmannia glutinosa plants subjected to continuous cropping. BMC Plant Biol. 2011, 11, 53. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, Y.; Chen, X.; Wang, F.; Lin, W.; Yi, Y.; Zeng, L.; Yang, S.; Zhang, Z. Transcriptome/degradome-wide identification of R. glutinosa miRNAs and their targets: The role of miRNA activity in the replanting disease. PLoS ONE 2013, 8, e68531. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Xiao, X.; Duan, L.; Guo, Y.; Qi, J.; Liao, D.; Zhao, C.; Liu, Y.; Zhou, L.; Li, X. Dynamic transcriptional profiling provides insights into tuberous root development in Rehmannia glutinosa. Front. Plant Sci. 2015, 6, 396. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.; Heger, A.; Ponting, C. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, Y.; Feng, F.; Zhang, B.; Chen, S.; Yang, C.; Gu, L.; Wang, L.; Zhang, J.; Chen, A.; et al. Differential proteomic analysis of replanted Rehmannia glutinosa roots by iTRAQ reveals molecular mechanisms for formation of replant disease. BMC Plant Biol. 2017, 17, 116. [Google Scholar] [CrossRef] [PubMed]
- Caputi, L.; Malnoy, M.; Goremykin, V.; Nikiforova, S.; Martens, S. A genome-wide phylogenetic reconstruction of family 1 UDP-glycosyltransferases revealed the expansion of the family during the adaptation of plants to life on land. Plant J. 2012, 69, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Hosokawa, M.; Hoshino, A.; Kitamura, Y.; Morita, Y.; Park, K.; Yazawa, S.; Nakashima, A.; Deguchi, A.; Tatsuzawa, F.; et al. A bHLH transcription factor, DvIVS, is involved in regulation of anthocyanin synthesis in dahlia (Dahlia variabilis). J. Exp. Bot. 2011, 62, 5105–5116. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Duan, J.; Huo, D.; Shi, Q.; Niu, L.; Zhang, Y. Transcriptomic analysis reveals transcription factors related to leaf anthocyanin biosynthesis in Paeonia qiui. Molecules 2017, 22, 2186. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Pollier, J.; Vanden, B.; Lopez, V.; Franco, Z.; Goossens, A. The bHLH transcription factors TSAR1 and TSAR2 regulate triterpene saponin biosynthesis in Medicago truncatula. Plant Physiol. 2015, 170, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Feng, C.; Tang, X.; Xu, L.; Tao, X.; Zhang, L.; Li, G. Content determination of phenylethanoid glycosides and acteoside in Plantago Herba from different producing areas. Chin. J. Inf. Tradit. Chin. Med. 2017, 24, 84–86. [Google Scholar] [CrossRef]
- Grabherr, M.; Haas, B.; Yassour, M.; Levin, J.; Thompson, D.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.; Salzberg, S.; Wold, B.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Suo, Y.; Wei, H.; Li, M.; Xie, C.; Wang, L.; Chen, X.; Zhang, Z. Identification and characterization of 40 isolated Rehmannia glutinosa MYB family genes and their expression profiles in response to shading and continuous cropping. Int. J. Mol. Sci. 2015, 16, 15009–15030. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Raw Reads | Total Clean Reads | Total Clean Bases (Gb) | Clean Reads Q20 (%) | Clean Reads Q30 (%) | Clean Reads Ratio (%) |
|---|---|---|---|---|---|---|
| 1706_RS1 | 35,930,376 | 29,939,804 | 4.49 | 97.89 | 93.55 | 83.33 |
| 1706_RS2 | 35,930,108 | 30,066,360 | 4.51 | 97.91 | 93.56 | 83.68 |
| 1706_RS3 | 35,930,118 | 30,416,510 | 4.56 | 97.91 | 93.6 | 84.65 |
| 1706_nRS1 | 35,930,582 | 30,106,740 | 4.52 | 97.98 | 93.78 | 83.79 |
| 1706_nRS2 | 35,931,096 | 29,693,676 | 4.45 | 98.1 | 93.9 | 82.64 |
| 1706_nRS3 | 34,205,142 | 28,499,086 | 4.27 | 97.91 | 93.59 | 83.32 |
| BJ1_RS1 | 35,931,216 | 29,457,812 | 4.42 | 97.91 | 93.35 | 81.98 |
| BJ1_RS2 | 35,931,386 | 29,683,162 | 4.45 | 97.81 | 93.18 | 82.61 |
| BJ1_RS3 | 35,931,264 | 29,768,646 | 4.47 | 97.86 | 93.27 | 82.85 |
| BJ1_nRS1 | 35,931,542 | 29,720,528 | 4.46 | 98.16 | 94.02 | 82.71 |
| BJ1_nRS2 | 35,930,546 | 29,537,004 | 4.43 | 97.83 | 93.39 | 82.21 |
| BJ1_nRS3 | 35,930,574 | 29,910,368 | 4.49 | 97.82 | 93.36 | 83.24 |
| QH1_RS1 | 35,930,624 | 29,870,448 | 4.48 | 97.86 | 93.42 | 83.13 |
| QH1_RS2 | 35,929,752 | 29,930,562 | 4.49 | 97.59 | 92.8 | 83.3 |
| QH1_RS3 | 35,930,062 | 29,617,920 | 4.44 | 97.74 | 93.13 | 82.43 |
| QH1_nRS1 | 35,930,688 | 29,542,912 | 4.43 | 97.8 | 93.29 | 82.22 |
| QH1_nRS2 | 37,564,468 | 30,470,168 | 4.57 | 98.07 | 93.81 | 81.11 |
| QH1_nRS3 | 37,564,500 | 30,466,862 | 4.57 | 98.05 | 93.69 | 81.11 |
| W85_RS1 | 35,930,236 | 30,246,190 | 4.54 | 97.9 | 93.56 | 84.18 |
| W85_RS2 | 35,930,102 | 30,523,230 | 4.58 | 97.95 | 93.69 | 84.95 |
| W85_RS3 | 35,930,692 | 30,136,144 | 4.52 | 97.9 | 93.59 | 83.87 |
| W85_nRS1 | 33,750,468 | 28,498,496 | 4.27 | 97.91 | 93.62 | 84.44 |
| W85_nRS2 | 35,716,050 | 29,601,754 | 4.44 | 97.88 | 93.52 | 82.88 |
| W85_nRS3 | 34,297,508 | 29,453,820 | 4.42 | 97.98 | 93.78 | 85.88 |
| Transcription Factor | Total of Unigene | 1706 | BJ1 | QH1 | W85 | All | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Up | Down | Up | Down | Up | Down | Up | Down | Up | Down | ||
| WRKY | 184 | 17 | 11 | 11 | 10 | 6 | 8 | 4 | 11 | 1 | 2 |
| TCP | 39 | 2 | 2 | 1 | 2 | 1 | 0 | 1 | 1 | 0 | 0 |
| MADS | 119 | 3 | 2 | 0 | 2 | 1 | 3 | 0 | 1 | 0 | 1 |
| C2C2-GATA | 62 | 2 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| AP2-EREBP | 286 | 35 | 9 | 17 | 8 | 6 | 2 | 2 | 1 | 0 | 0 |
| C2H2 | 93 | 4 | 5 | 1 | 2 | 0 | 2 | 1 | 1 | 0 | 0 |
| bHLH | 235 | 17 | 28 | 9 | 25 | 3 | 16 | 5 | 8 | 1 | 6 |
| MYB | 513 | 19 | 21 | 15 | 24 | 9 | 6 | 8 | 3 | 3 | 1 |
| Trihelix | 104 | 0 | 2 | 0 | 2 | 0 | 1 | 0 | 1 | 0 | 1 |
| HSF | 53 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 |
| C2C2-CO-like | 26 | 1 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| NAC | 141 | 2 | 16 | 1 | 3 | 1 | 0 | 0 | 8 | 0 | 0 |
| C3H | 299 | 10 | 2 | 2 | 1 | 3 | 0 | 2 | 0 | 1 | 0 |
| C2C2-Dof | 94 | 2 | 2 | 0 | 7 | 1 | 0 | 0 | 1 | 0 | 0 |
| ARF | 155 | 0 | 15 | 0 | 9 | 0 | 8 | 0 | 5 | 0 | 3 |
| FAR1 | 139 | 1 | 0 | 0 | 0 | 3 | 0 | 0 | 2 | 0 | 0 |
| SBP | 91 | 9 | 0 | 1 | 1 | 1 | 0 | 3 | 0 | 0 | 0 |
| ABI3VP1 | 120 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 |
| LOB | 49 | 0 | 6 | 0 | 7 | 0 | 6 | 0 | 3 | 0 | 2 |
| FHA | 61 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| BES1 | 47 | 1 | 2 | 1 | 5 | 1 | 3 | 0 | 0 | 0 | 0 |
| zf-HD | 45 | 0 | 2 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| EIL | 22 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| OFP | 30 | 8 | 1 | 4 | 0 | 0 | 0 | 3 | 0 | 0 | 0 |
| GRAS | 154 | 8 | 2 | 0 | 3 | 0 | 1 | 0 | 2 | 0 | 1 |
| Bzip | 40 | 0 | 2 | 0 | 4 | 0 | 0 | 0 | 1 | 0 | 0 |
| TCP | 39 | 2 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 |
| Total of DET | 145 | 139 | 66 | 119 | 37 | 58 | 33 | 50 | 6 | 17 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhi, J.; Li, Y.; Zhang, Z.; Yang, C.; Geng, X.; Zhang, M.; Li, X.; Zuo, X.; Li, M.; Huang, Y.; et al. Molecular Regulation of Catalpol and Acteoside Accumulation in Radial Striation and non-Radial Striation of Rehmannia glutinosa Tuberous Root. Int. J. Mol. Sci. 2018, 19, 3751. https://doi.org/10.3390/ijms19123751
Zhi J, Li Y, Zhang Z, Yang C, Geng X, Zhang M, Li X, Zuo X, Li M, Huang Y, et al. Molecular Regulation of Catalpol and Acteoside Accumulation in Radial Striation and non-Radial Striation of Rehmannia glutinosa Tuberous Root. International Journal of Molecular Sciences. 2018; 19(12):3751. https://doi.org/10.3390/ijms19123751
Chicago/Turabian StyleZhi, Jingyu, Yajing Li, Zhongyi Zhang, Chaofei Yang, Xiaotong Geng, Miao Zhang, Xinrong Li, Xin Zuo, Mingjie Li, Yong Huang, and et al. 2018. "Molecular Regulation of Catalpol and Acteoside Accumulation in Radial Striation and non-Radial Striation of Rehmannia glutinosa Tuberous Root" International Journal of Molecular Sciences 19, no. 12: 3751. https://doi.org/10.3390/ijms19123751
APA StyleZhi, J., Li, Y., Zhang, Z., Yang, C., Geng, X., Zhang, M., Li, X., Zuo, X., Li, M., Huang, Y., Wang, F., & Xie, C. (2018). Molecular Regulation of Catalpol and Acteoside Accumulation in Radial Striation and non-Radial Striation of Rehmannia glutinosa Tuberous Root. International Journal of Molecular Sciences, 19(12), 3751. https://doi.org/10.3390/ijms19123751