Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
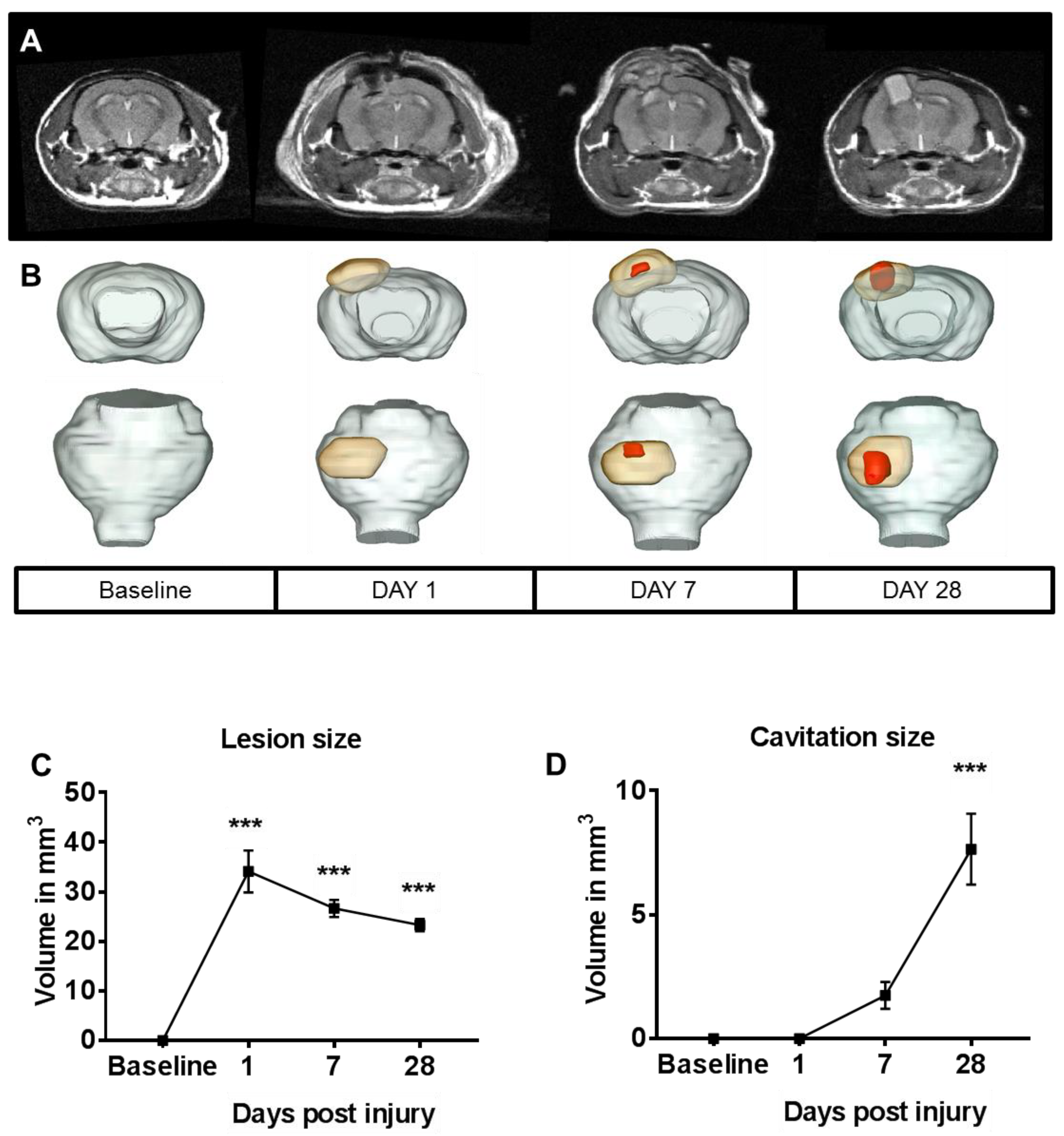
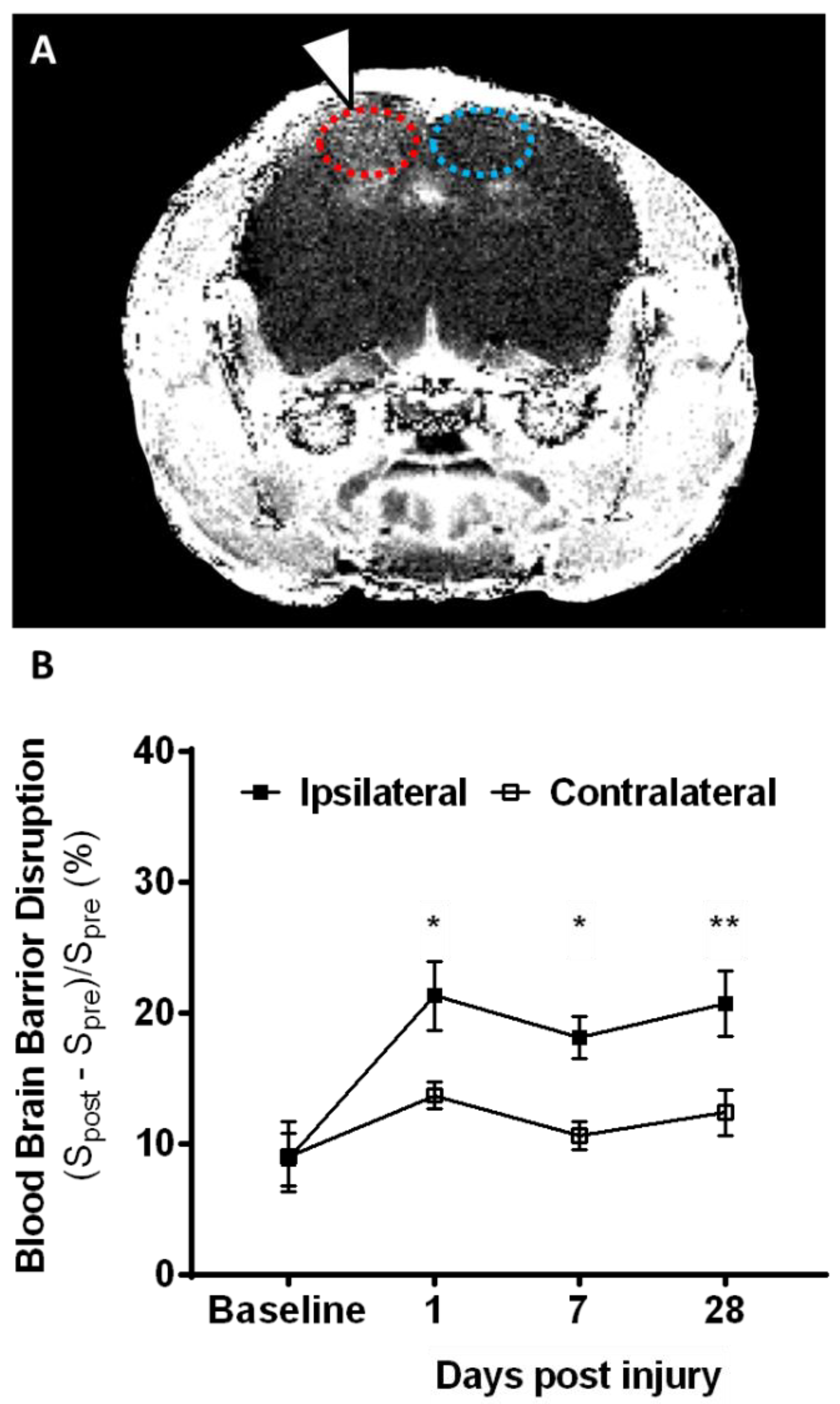
2.1. Lesion Progression after Contusion Injury in Aged Animals
2.2. Microglia Numbers Increase Chronically after Contusion Injury
2.3. Contusion Injury Exacerbates Microglia Phagocytic Activity in the Aged Brain
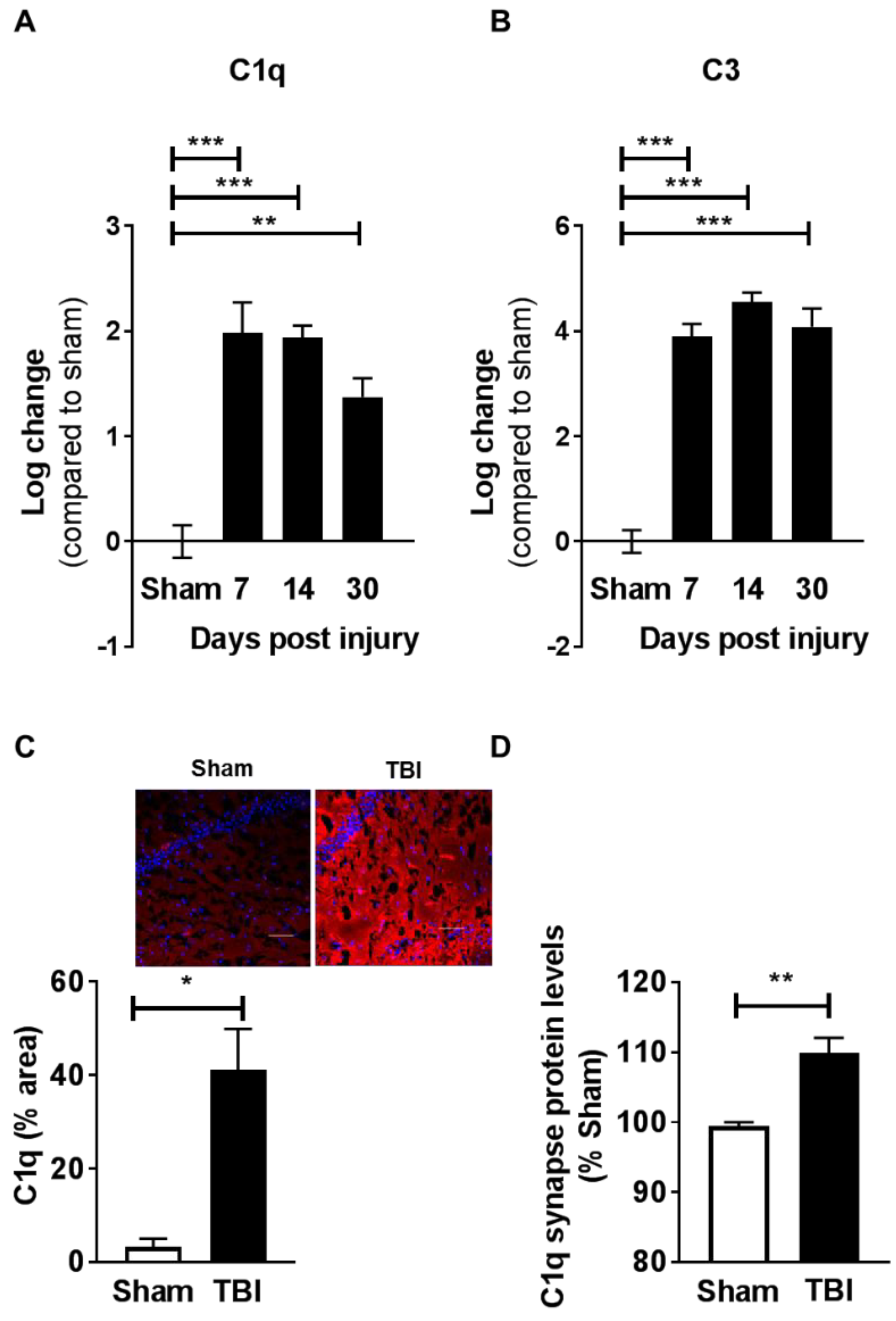
2.4. Contusion Injury Induces Robust Complement Initiation in the Aged Brain
2.5. Contusive Injury Results in Chronic Synapse Loss in the Aged Brain
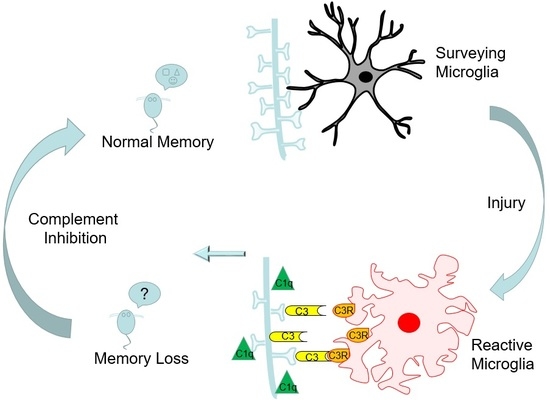
2.6. Complement Blockade Prevents Memory Deficits in Aged Animals after Contusion Injury
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Trauma Surgery
4.3. In Vivo MR Acquisitions
4.4. MR Data Analysis
4.5. Tissue Collection
4.6. qPCR Analysis
4.7. Microglia Isolation and Phagocytic Assay
4.8. Immunohistochemistry Analysis
4.9. FLOW Synaptocytometry
4.10. C1q Antibody Administration
4.11. Novel Object Recognition
4.12. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Senathi-Raja, D.; Ponsford, J.; Schonberger, M. Impact of age on long-term cognitive function after traumatic brain injury. Neuropsychology 2010, 24, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Spitz, G.; Ponsford, J.L.; Rudzki, D.; Maller, J.J. Association between cognitive performance and functional outcome following traumatic brain injury: A longitudinal multilevel examination. Neuropsychology 2012, 26, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; McCormick, W.C.; Kagan, S.H. Traumatic brain injury in older adults: Epidemiology, outcomes, and future implications. J. Am. Geriatr. Soc. 2006, 54, 1590–1595. [Google Scholar] [CrossRef] [PubMed]
- Himanen, L.; Portin, R.; Isoniemi, H.; Helenius, H.; Kurki, T.; Tenovuo, O. Longitudinal cognitive changes in traumatic brain injury: A 30-year follow-up study. Neurology 2006, 66, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Hukkelhoven, C.W.; Steyerberg, E.W.; Rampen, A.J.; Farace, E.; Habbema, J.D.; Marshall, L.F.; Murray, G.D.; Maas, A.I. Patient age and outcome following severe traumatic brain injury: An analysis of 5600 patients. J. Neurosurg. 2003, 99, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Schonberger, M.; Ponsford, J.; Reutens, D.; Beare, R.; O’Sullivan, R. The Relationship between age, injury severity, and MRI findings after traumatic brain injury. J. Neurotrauma 2009, 26, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.; Krukowski, K.; Morganti, J.M.; Riparip, L.K.; Rosi, S. Persistent Infiltration and Impaired Response of Peripherally-Derived Monocytes after Traumatic Brain Injury in the Aged Brain. Int. J. Mol. Sci. 2018, 19, 1616. [Google Scholar] [CrossRef] [PubMed]
- Morganti, J.M.; Jopson, T.D.; Liu, S.; Riparip, L.K.; Guandique, C.K.; Gupta, N.; Ferguson, A.R.; Rosi, S. CCR2 Antagonism Alters Brain Macrophage Polarization and Ameliorates Cognitive Dysfunction Induced by Traumatic Brain Injury. J. Neurosci. 2015, 35, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Morganti, J.M.; Riparip, L.K.; Chou, A.; Liu, S.; Gupta, N.; Rosi, S. Age exacerbates the CCR2/5-mediated neuroinflammatory response to traumatic brain injury. J. Neuroinflamm. 2016, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Stoica, B.A.; Sabirzhanov, B.; Burns, M.P.; Faden, A.I.; Loane, D.J. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol. Aging 2013, 34, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.A.; Moss, L.D.; Lee, J.Y.; Tajiri, N.; Acosta, S.; Hudson, C.; Parag, S.; Cooper, D.R.; Borlongan, C.V.; Bickford, P.C. Long noncoding RNA MALAT1 in exosomes drives regenerative function and modulates inflammation-linked networks following traumatic brain injury. J. Neuroinflamm. 2018, 15, 204. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lin, R.; Nguyen, H.; Grant Liska, M.; Lippert, T.; Kaneko, Y.; Borlongan, C.V. Histopathological and Behavioral Assessments of Aging Effects on Stem Cell Transplants in an Experimental Traumatic Brain Injury. Methods Mol. Biol. 2018. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.; Dodart, J.C.; et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J. Neurosci. 2015, 35, 13029–13042. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.E.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [PubMed]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.H.; Madison, D.V.; Mateos, J.M.; Fraser, D.A.; Lovelett, E.A.; Coutellier, L.; Kim, L.; Tsai, H.H.; Huang, E.J.; Rowitch, D.H.; et al. A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 2013, 33, 13460–13474. [Google Scholar] [CrossRef] [PubMed]
- Lighthall, J.W. Controlled cortical impact: A new experimental brain injury model. J. Neurotrauma 1988, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Guglielmetti, C.; Chou, A.; Krukowski, K.; Najac, C.; Feng, X.; Riparip, L.K.; Rosi, S.; Chaumeil, M.M. In vivo metabolic imaging of Traumatic Brain Injury. Sci. Rep. 2017, 7, 17525. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Prieto, G.A.; Snigdha, S.; Baglietto-Vargas, D.; Smith, E.D.; Berchtold, N.C.; Tong, L.; Ajami, D.; LaFerla, F.M.; Rebek, J., Jr.; Cotman, C.W. Synapse-specific IL-1 receptor subunit reconfiguration augments vulnerability to IL-1beta in the aged hippocampus. Proc. Natl. Acad. Sci. USA 2015, 112, E5078–E5087. [Google Scholar] [CrossRef] [PubMed]
- Snigdha, S.; Prieto, G.A.; Petrosyan, A.; Loertscher, B.M.; Dieskau, A.P.; Overman, L.E.; Cotman, C.W. H3K9me3 Inhibition Improves Memory, Promotes Spine Formation, and Increases BDNF Levels in the Aged Hippocampus. J. Neurosci. 2016, 36, 3611–3622. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, K.; Grue, K.; Frias, E.S.; Pietrykowski, J.; Jones, T.; Nelson, G.; Rosi, S. Female Mice are Protected from Space Radiation-Induced Maladaptive Responses. Brain Behav. Immun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Onyszchuk, G.; He, Y.Y.; Berman, N.E.; Brooks, W.M. Detrimental effects of aging on outcome from traumatic brain injury: A behavioral, magnetic resonance imaging, and histological study in mice. J. Neurotrauma 2008, 25, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Timaru-Kast, R.; Luh, C.; Gotthardt, P.; Huang, C.; Schafer, M.K.; Engelhard, K.; Thal, S.C. Influence of age on brain edema formation, secondary brain damage and inflammatory response after brain trauma in mice. PLoS ONE 2012, 7, e43829. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Niemi, E.C.; Wang, S.H.; Lee, C.C.; Bingham, D.; Zhang, J.; Cozen, M.L.; Charo, I.; Huang, E.J.; Liu, J.; et al. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J. Neurotrauma 2014, 31, 1677–1688. [Google Scholar] [CrossRef] [PubMed]
- Norris, G.T.; Smirnov, I.; Filiano, A.J.; Shadowen, H.M.; Cody, K.R.; Thompson, J.A.; Harris, T.H.; Gaultier, A.; Overall, C.C.; Kipnis, J. Neuronal integrity and complement control synaptic material clearance by microglia after CNS injury. J. Exp. Med. 2018, 215, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.I.; Zhou, J.; Botto, M.; Tenner, A.J. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2004, 24, 6457–6465. [Google Scholar] [CrossRef] [PubMed]
- Alawieh, A.; Langley, E.F.; Weber, S.; Adkins, D.; Tomlinson, S. Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kossmann, T.; Stahel, P.F.; Morganti-Kossmann, M.C.; Jones, J.L.; Barnum, S.R. Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J. Neuroimmunol. 1997, 73, 63–69. [Google Scholar] [CrossRef]
- Stahel, P.F.; Morganti-Kossmann, M.C.; Perez, D.; Redaelli, C.; Gloor, B.; Trentz, O.; Kossmann, T. Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. J. Neurotrauma 2001, 18, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.; Krukowski, K.; Jopson, T.; Zhu, P.J.; Costa-Mattioli, M.; Walter, P.; Rosi, S. Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury. Proc. Natl. Acad. Sci. USA 2017, 114, E6420–E6426. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jopson, T.D.; Paladini, M.S.; Liu, S.; West, B.L.; Gupta, N.; Rosi, S. Colony-stimulating factor 1 receptor blockade prevents fractionated whole-brain irradiation-induced memory deficits. J. Neuroinflamm. 2016, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, K.; Feng, X.; Paladini, M.S.; Chou, A.; Sacramento, K.; Grue, K.; Riparip, L.K.; Jones, T.; Campbell-Beachler, M.; Nelson, G.; et al. Temporary microglia-depletion after cosmic radiation modifies phagocytic activity and prevents cognitive deficits. Sci. Rep. 2018, 8, 7857. [Google Scholar] [CrossRef] [PubMed]
- Mumby, D.G.; Gaskin, S.; Glenn, M.J.; Schramek, T.E.; Lehmann, H. Hippocampal damage and exploratory preferences in rats: Memory for objects, places, and contexts. Learn. Mem. 2002, 9, 49–57. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krukowski, K.; Chou, A.; Feng, X.; Tiret, B.; Paladini, M.-S.; Riparip, L.-K.; Chaumeil, M.M.; Lemere, C.; Rosi, S. Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits. Int. J. Mol. Sci. 2018, 19, 3753. https://doi.org/10.3390/ijms19123753
Krukowski K, Chou A, Feng X, Tiret B, Paladini M-S, Riparip L-K, Chaumeil MM, Lemere C, Rosi S. Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits. International Journal of Molecular Sciences. 2018; 19(12):3753. https://doi.org/10.3390/ijms19123753
Chicago/Turabian StyleKrukowski, Karen, Austin Chou, Xi Feng, Brice Tiret, Maria-Serena Paladini, Lara-Kirstie Riparip, Myriam M. Chaumeil, Cynthia Lemere, and Susanna Rosi. 2018. "Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits" International Journal of Molecular Sciences 19, no. 12: 3753. https://doi.org/10.3390/ijms19123753
APA StyleKrukowski, K., Chou, A., Feng, X., Tiret, B., Paladini, M.-S., Riparip, L.-K., Chaumeil, M. M., Lemere, C., & Rosi, S. (2018). Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits. International Journal of Molecular Sciences, 19(12), 3753. https://doi.org/10.3390/ijms19123753