Genomics: New Light on Alzheimer’s Disease Research


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
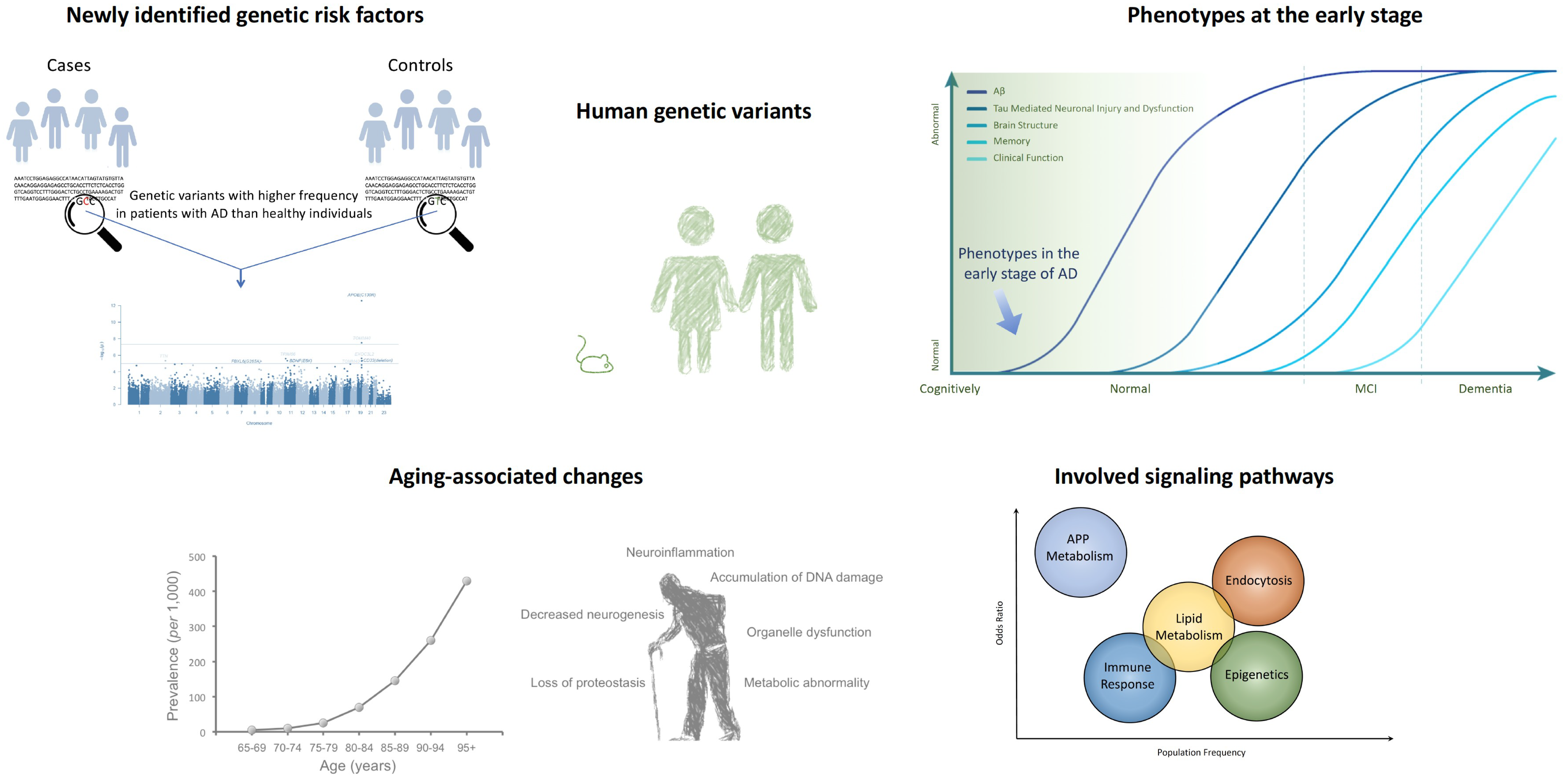
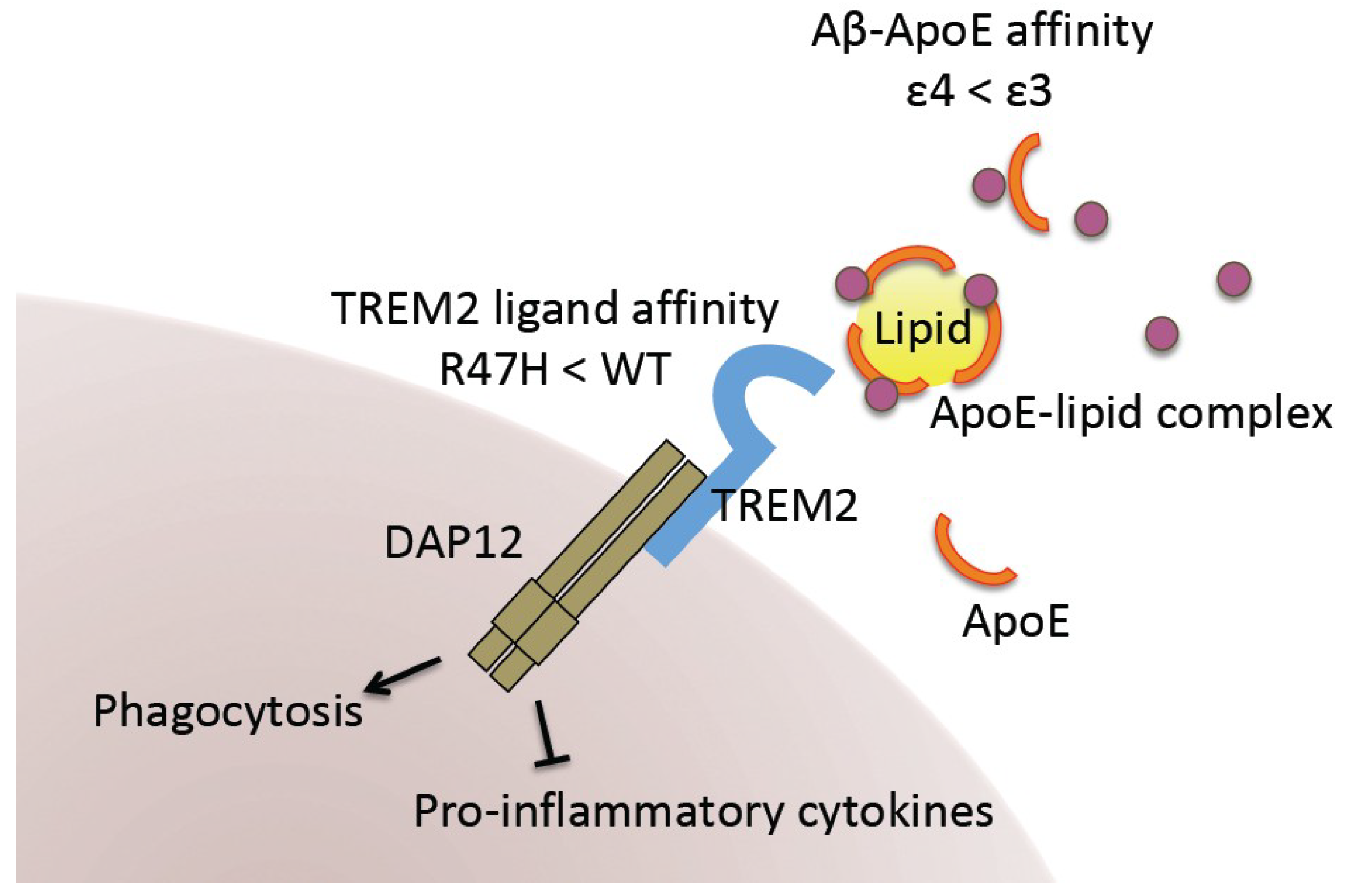
2. GWAS Shed New Light on AD
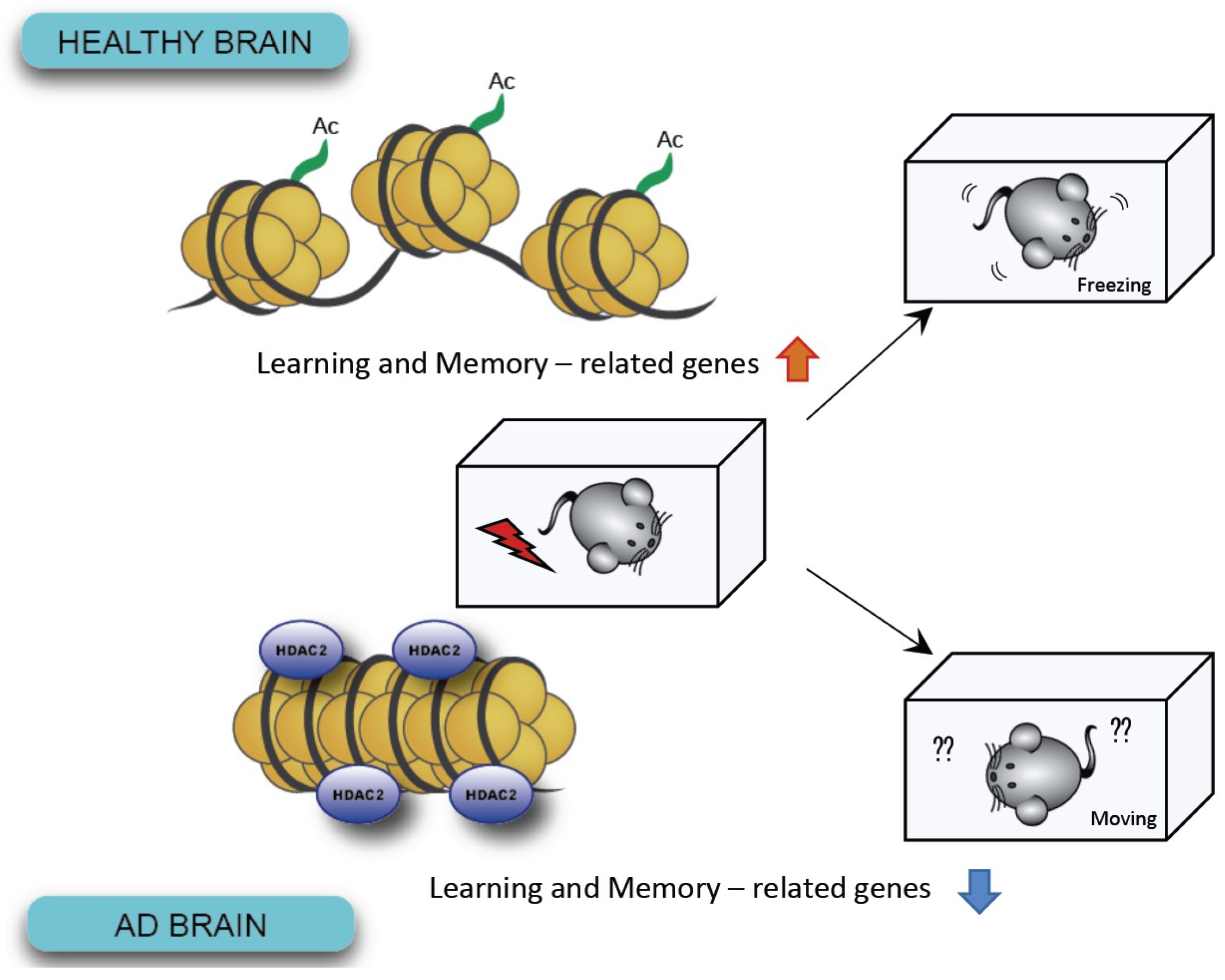
3. Epigenetic Modification: Changes in Gene Expression without Genomic Alteration
4. Aging is still One of the Greatest Risk Factors for AD
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MAPT | Microtubule associated protein tau |
| APOE | Apolipoprotein E |
| CLU | Clusterin |
| CR1 | Complement C3b/C4b receptor 1 |
| BIN1 | Bridging integrator 1 |
| PICALM | Phosphatidylinositol binding clathrin assembly protein |
| CD33 | CD33 molecule |
| ABCA7 | ATP binding cassette subfamily A member 7 |
| CD2AP | CD2 associated protein |
| EPHA1 | EPH receptor A1 |
| MS4 | Membrane spanning 4-domains |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| H3K18/23 | Histone H3 lysine 18/23 |
| H3K4me3 | Tri-methylation on histone H3 lysine 4 |
| H3K27ac | Acetylation on histone H3 lysine 27 |
| H4K16ac | Acetylation on histone H4 lysine 16 |
| NMDA | N-methyl-d-aspartate |
References
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 2018, 14, 367–429. [Google Scholar]
- Prince, M.J.; Comas-Herrera, A.; Knapp, M.; Guerchet, M.M.; Karagiannidou, M. World Alzheimer Report 2016—Improving Healthcare for People Living with Dementia: Coverage, Quality and Costs Now and in the Future; Alzheimer’s Disease International: London, UK, 2016. [Google Scholar]
- Maurer, K.; Volk, S.; Gerbaldo, H. Auguste D and Alzheimer’s disease. Lancet 1997, 349, 1546–1549. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef] [PubMed]
- St George-Hyslop, P.H.; Tanzi, R.E.; Polinsky, R.J.; Haines, J.L.; Nee, L.; Watkins, P.C.; Myers, R.H.; Feldman, R.G.; Pollen, D.; Drachman, D. The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science 1987, 235, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. Thirty years of Alzheimer’s disease genetics: The implications of systematic meta-analyses. Nat. Rev. Neurosci. 2008, 9, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Van Leuven, F. Single and multiple transgenic mice as models for Alzheimer’s disease. Prog. Neurobiol. 2000, 61, 305–312. [Google Scholar] [CrossRef]
- Lee, E.B.; Leng, L.Z.; Zhang, B.; Kwong, L.; Trojanowski, J.Q.; Abel, T.; Lee, V.M.-Y. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J. Biol. Chem. 2006, 281, 4292–4299. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.-Y.; Li, B.; Peng, D.; Wang, C.-Y.; Wang, G.-Y.; Li, P.; Le, Y.-Y.; Wang, J.M.; Ye, G.; Chen, J.-H. A novel monoclonal antibody against the N-terminus of Aβ1-42 reduces plaques and improves cognition in a mouse model of Alzheimer’s disease. PLoS ONE 2017, 12, e0180076. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, J.R.M.; Marques, D.F.F.; Baptista, S.J.; Pereira, C.M.F.; Moreira, P.I.; Dinis, T.C.P.; Santos, A.E.; Salvador, J.A.R. Highlights in BACE1 Inhibitors for Alzheimer’s Disease Treatment. Front Chem. 2018, 6, 178. [Google Scholar] [CrossRef] [PubMed]
- Ridler, C. Alzheimer disease: BACE1 inhibitors block new Aβ plaque formation. Nat. Rev. Neurol. 2018, 14, 126. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. γ-Secretase inhibitors and modulators for Alzheimer’s disease. J. Neurochem. 2012, 120 (Suppl. 1), 89–98. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Plattner, F.; Angelo, M.; Giese, K.P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 2006, 281, 25457–25465. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Avila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol. Neurosci. 2014, 7, 46. [Google Scholar] [PubMed]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Giusti-Rodríguez, P.; Zhou, Y.; Rudenko, A.; Cho, S.; Ota, K.T.; Park, C.; Patzke, H.; Madabhushi, R.; Pan, L.; et al. Activity-dependent p25 generation regulates synaptic plasticity and Aβ-induced cognitive impairment. Cell 2014, 157, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kritskiy, O.; Watson, L.A.; Barker, S.J.; Dey, D.; Raja, W.K.; Lin, Y.-T.; Ko, T.; Cho, S.; Penney, J.; et al. Inhibition of p25/Cdk5 Attenuates Tauopathy in Mouse and iPSC Models of Frontotemporal Dementia. J. Neurosci. 2017, 37, 9917–9924. [Google Scholar] [CrossRef] [PubMed]
- Mazanetz, M.P.; Fischer, P.M. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 2007, 6, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Kondadi, A.K.; Wang, S.; Montagner, S.; Kladt, N.; Korwitz, A.; Martinelli, P.; Herholz, D.; Baker, M.J.; Schauss, A.C.; Langer, T.; et al. Loss of the m-AAA protease subunit AFG₃L₂ causes mitochondrial transport defects and tau hyperphosphorylation. EMBO J. 2014, 33, 1011–1026. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 2011, 70, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Espuny-Camacho, I.; Arranz, A.M.; Fiers, M.; Snellinx, A.; Ando, K.; Munck, S.; Bonnefont, J.; Lambot, L.; Corthout, N.; Omodho, L.; et al. Hallmarks of Alzheimer’s Disease in Stem-Cell-Derived Human Neurons Transplanted into Mouse Brain. Neuron 2017, 93, 1066–1081. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.P.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.-E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.-T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.-H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet. Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Onos, K.D.; Sukoff Rizzo, S.J.; Howell, G.R.; Sasner, M. Toward more predictive genetic mouse models of Alzheimer’s disease. Brain Res. Bull. 2016, 122, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Ittner, L.M. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat. Rev. Neurosci. 2008, 9, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.S.; Loy, E.Y.; Pawitan, Y.; Chia, K.S. The pursuit of genome-wide association studies: Where are we now? J. Hum. Genet. 2010, 55, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S. The genetics of Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.-S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Brás, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Pimenova, A.A.; Marcora, E.; Goate, A.M. A Tale of Two Genes: Microglial Apoe and Trem2. Immunity 2017, 47, 398–400. [Google Scholar] [CrossRef] [PubMed]
- Ignatius, M.J.; Gebicke-Härter, P.J.; Skene, J.H.; Schilling, J.W.; Weisgraber, K.H.; Mahley, R.W.; Shooter, E.M. Expression of apolipoprotein E during nerve degeneration and regeneration. Proc. Natl. Acad. Sci. USA 1986, 83, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Boyles, J.K.; Pitas, R.E.; Wilson, E.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Investig. 1985, 76, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Pitas, R.E.; Boyles, J.K.; Lee, S.H.; Foss, D.; Mahley, R.W. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta 1987, 917, 148–161. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Namba, Y.; Tomonaga, M.; Kawasaki, H.; Otomo, E.; Ikeda, K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991, 541, 163–166. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.M.; Weisgraber, K.H. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J. Biol. Chem. 1996, 271, 19053–19057. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Painter, M.M.; Bu, G.; Kanekiyo, T. Apolipoprotein E as a Therapeutic Target in Alzheimer’s Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs 2016, 30, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M. TREMs in the immune system and beyond. Nat. Rev. Immunol. 2003, 3, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; Chen, T.T.; Tchao, N.K.; Seaman, W.E. Pattern recognition by TREM-2: Binding of anionic ligands. J. Immunol. 2003, 171, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Rochford, C.D.P.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, I.R.; Colonna, M. Activating and inhibitory functions of DAP12. Nat. Rev. Immunol. 2007, 7, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, I.R.; Gilfillan, S.; Cella, M.; Aoshi, T.; Miller, M.; Piccio, L.; Hernandez, M.; Colonna, M. Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 2006, 177, 3520–3524. [Google Scholar] [CrossRef] [PubMed]
- Hao, K.; Di Narzo, A.F.; Ho, L.; Luo, W.; Li, S.; Chen, R.; Li, T.; Dubner, L.; Pasinetti, G.M. Shared genetic etiology underlying Alzheimer’s disease and type 2 diabetes. Mol. Aspects Med. 2015, 43–44, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Annese, A.; Manzari, C.; Lionetti, C.; Picardi, E.; Horner, D.S.; Chiara, M.; Caratozzolo, M.F.; Tullo, A.; Fosso, B.; Pesole, G.; et al. Whole transcriptome profiling of Late-Onset Alzheimer’s Disease patients provides insights into the molecular changes involved in the disease. Sci. Rep. 2018, 8, 4282. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-C.; Tan, L.; Wang, H.-F.; Jiang, T.; Cao, L.; Wang, C.; Wang, J.; Tan, C.-C.; Meng, X.-F.; Yu, J.-T. Rate of early onset Alzheimer’s disease: A systematic review and meta-analysis. Ann. Transl. Med. 2015, 3, 38. [Google Scholar] [PubMed]
- Gong, C.-X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi-Targets for Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mut, J.V.; Gräff, J. Epigenetic Alterations in Alzheimer’s Disease. Front Behav. Neurosci. 2015, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Bradley-Whitman, M.A.; Lovell, M.A. Epigenetic changes in the progression of Alzheimer’s disease. Mech. Ageing Dev. 2013, 134, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Rotllant, D.; Maes, T. Understanding Epigenetic Alterations in Alzheimer’s and Parkinson’s Disease: Towards Targeted Biomarkers and Therapies. Curr. Pharm. Des. 2017, 23, 839–857. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sananbenesi, F.; Wang, X.; Dobbin, M.; Tsai, L.-H. Recovery of learning and memory is associated with chromatin remodelling. Nature 2007, 447, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Schrag, M.; Crofton, A.; Trivedi, R.; Vinters, H.; Kirsch, W. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 2012, 12, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.-S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.-H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Rei, D.; Guan, J.-S.; Wang, W.-Y.; Seo, J.; Hennig, K.M.; Nieland, T.J.F.; Fass, D.M.; Kao, P.F.; Kahn, M.; et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 2012, 483, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Cheng, J.; Penney, J.; Gao, F.; Rueda, R.; Wang, J.; Yamakawa, S.; Kritskiy, O.; Gjoneska, E.; Tsai, L.-H. The Transcription Factor Sp3 Cooperates with HDAC2 to Regulate Synaptic Function and Plasticity in Neurons. Cell Rep. 2017, 20, 1319–1334. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Schroeder, F.A.; Wey, H.-Y.; Borra, R.; Wagner, F.F.; Reis, S.; Kim, S.W.; Holson, E.B.; Haggarty, S.J.; Hooker, J.M. In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J. Med. Chem. 2014, 57, 7999–8009. [Google Scholar] [CrossRef] [PubMed]
- Wey, H.-Y.; Wang, C.; Schroeder, F.A.; Logan, J.; Price, J.C.; Hooker, J.M. Kinetic Analysis and Quantification of [11C]Martinostat for in Vivo HDAC Imaging of the Brain. ACS Chem. Neurosci. 2015, 6, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, M.M.; Madabhushi, R.; Pan, L.; Chen, Y.; Kim, D.; Gao, J.; Ahanonu, B.; Pao, P.-C.; Qiu, Y.; Zhao, Y.; et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat. Neurosci. 2013, 16, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Bardai, F.H.; Price, V.; Zaayman, M.; Wang, L.; D’Mello, S.R. Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J. Biol. Chem. 2012, 287, 35444–35453. [Google Scholar] [CrossRef] [PubMed]
- Gjoneska, E.; Pfenning, A.R.; Mathys, H.; Quon, G.; Kundaje, A.; Tsai, L.-H.; Kellis, M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 2015, 518, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Xin, S.-H.; Tan, L.; Cao, X.; Yu, J.-T.; Tan, L. Clearance of Amyloid Beta and Tau in Alzheimer’s Disease: From Mechanisms to Therapy. Neurotox Res. 2018, 34, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Jevtic, S.; Sengar, A.S.; Salter, M.W.; McLaurin, J. The role of the immune system in Alzheimer disease: Etiology and treatment. Ageing Res. Rev. 2017, 40, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Kim-Ha, J.; Kim, Y.-J. Age-related epigenetic regulation in the brain and its role in neuronal diseases. BMB Rep. 2016, 49, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Dean, B.; Thomas, E.A. Disease- and age-related changes in histone acetylation at gene promoters in psychiatric disorders. Transl. Psychiatry 2011, 1, e64. [Google Scholar] [CrossRef] [PubMed]
- Nativio, R.; Donahue, G.; Berson, A.; Lan, Y.; Amlie-Wolf, A.; Tuzer, F.; Toledo, J.B.; Gosai, S.J.; Gregory, B.D.; Torres, C.; et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, J.-T.; Tan, M.-S.; Jiang, T.; Tan, L. Epigenetic mechanisms in Alzheimer’s disease: Implications for pathogenesis and therapy. Ageing Res. Rev. 2013, 12, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Brody, H. Organization of the cerebral cortex. III. A study of aging in the human cerebral cortex. J. Comp. Neurol. 1955, 102, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.J. Neuronal loss, neurofibrillary tangles and granulovacuolar degeneration in the hippocampus with ageing and dementia. A quantitative study. Acta Neuropathol. 1977, 37, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Brizzee, K.R.; Ordy, J.M.; Bartus, R.T. Localization of cellular changes within multimodal sensory regions in aged monkey brain: Possible implications for age-related cognitive loss. NBA 1980, 1, 45–52. [Google Scholar] [CrossRef]
- West, M.J.; Coleman, P.D.; Flood, D.G.; Troncoso, J.C. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 1994, 344, 769–772. [Google Scholar] [CrossRef]
- Gazzaley, A.H.; Thakker, M.M.; Hof, P.R.; Morrison, J.H. Preserved number of entorhinal cortex layer II neurons in aged macaque monkeys. NBA 1997, 18, 549–553. [Google Scholar] [CrossRef]
- Rapp, P.R.; Gallagher, M. Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc. Natl. Acad. Sci. USA 1996, 93, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Woodruff-Pak, D.S.; Foy, M.R.; Akopian, G.G.; Lee, K.H.; Zach, J.; Nguyen, K.P.T.; Comalli, D.M.; Kennard, J.A.; Agelan, A.; Thompson, R.F. Differential effects and rates of normal aging in cerebellum and hippocampus. PNAS 2010, 107, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.; Schliemann, T.; Sørensen, J.C.; Zimmer, J.; West, M.J. Memory impaired aged rats: No loss of principal hippocampal and subicular neurons. NBA 1996, 17, 143–147. [Google Scholar] [CrossRef]
- Burke, S.N.; Barnes, C.A. Neural plasticity in the ageing brain. Nat. Rev. Neurosci. 2006, 7, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Dickstein, D.L.; Weaver, C.M.; Luebke, J.I.; Hof, P.R. Dendritic spine changes associated with normal aging. Neuroscience 2013, 251, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.; Sethares, C. The effects of age on the cells in layer 1 of primate cerebral cortex. Cereb. Cortex 2002, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Page, T.L.; Einstein, M.; Duan, H.; He, Y.; Flores, T.; Rolshud, D.; Erwin, J.M.; Wearne, S.L.; Morrison, J.H.; Hof, P.R. Morphological alterations in neurons forming corticocortical projections in the neocortex of aged Patas monkeys. Neurosci. Lett. 2002, 317, 37–41. [Google Scholar] [CrossRef]
- Dumitriu, D.; Hao, J.; Hara, Y.; Kaufmann, J.; Janssen, W.G.M.; Lou, W.; Rapp, P.R.; Morrison, J.H. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J. Neurosci. 2010, 30, 7507–7515. [Google Scholar] [CrossRef] [PubMed]
- Cesa, R.; Scelfo, B.; Strata, P. Activity-dependent presynaptic and postsynaptic structural plasticity in the mature cerebellum. J. Neurosci. 2007, 27, 4603–4611. [Google Scholar] [CrossRef] [PubMed]
- Tessier, C.R.; Broadie, K. Activity-dependent modulation of neural circuit synaptic connectivity. Front Mol. Neurosci. 2009, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, Y.; Tanaka, T.; Tominaga-Yoshino, K.; Ogura, A. Persistent synapse loss induced by repetitive LTD in developing rat hippocampal neurons. PLoS ONE 2010, 5, e10390. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.S.; Arons, A.; Mitchell, T.I.; Pignatelli, M.; Ryan, T.J.; Tonegawa, S. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature 2016, 531, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Ridge, P.G.; Kauwe, J.S.K. Mitochondria and Alzheimer’s Disease: The Role of Mitochondrial Genetic Variation. Curr. Genet. Med. Rep. 2018, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Rinne, J.O.; Tsui, W.H.; Berti, V.; Li, Y.; Wang, H.; Murray, J.; Scheinin, N.; Någren, K.; Williams, S.; et al. Increased fibrillar amyloid-{beta} burden in normal individuals with a family history of late-onset Alzheimer’s. PNAS 2010, 107, 5949–5954. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, Y.J.; Kim, Y.H.; Bhalla, M.; Lee, S.B.; Seo, J. Genomics: New Light on Alzheimer’s Disease Research. Int. J. Mol. Sci. 2018, 19, 3771. https://doi.org/10.3390/ijms19123771
Jung YJ, Kim YH, Bhalla M, Lee SB, Seo J. Genomics: New Light on Alzheimer’s Disease Research. International Journal of Molecular Sciences. 2018; 19(12):3771. https://doi.org/10.3390/ijms19123771
Chicago/Turabian StyleJung, Yeong Ju, Yoon Ha Kim, Mridula Bhalla, Sung Bae Lee, and Jinsoo Seo. 2018. "Genomics: New Light on Alzheimer’s Disease Research" International Journal of Molecular Sciences 19, no. 12: 3771. https://doi.org/10.3390/ijms19123771
APA StyleJung, Y. J., Kim, Y. H., Bhalla, M., Lee, S. B., & Seo, J. (2018). Genomics: New Light on Alzheimer’s Disease Research. International Journal of Molecular Sciences, 19(12), 3771. https://doi.org/10.3390/ijms19123771