Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma

 , , ,
, , , 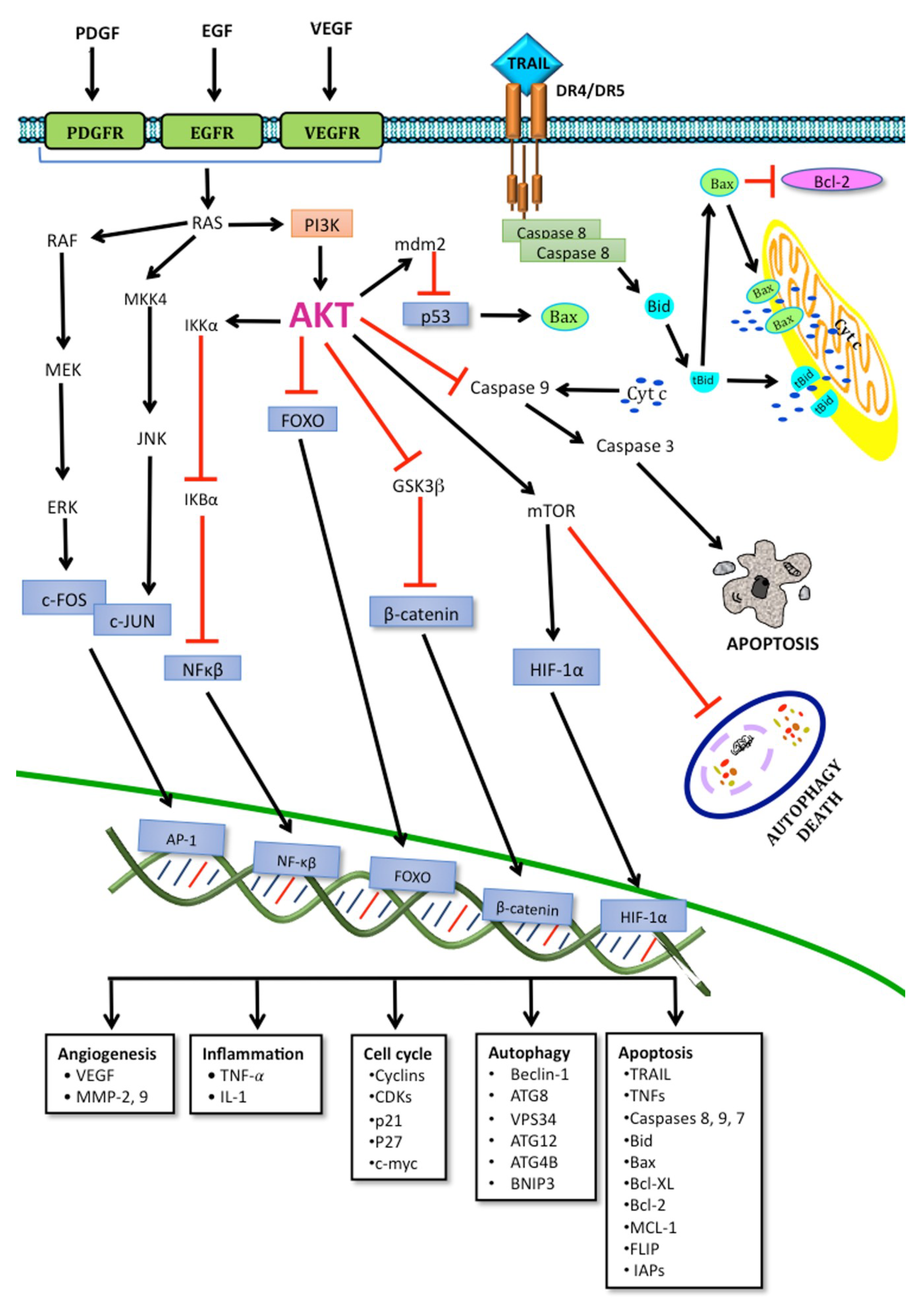{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Apoptosis
1.1.1. Apoptosis Pathways
1.1.2. Extrinsic pathway
1.1.3. Intrinsic Pathway
1.1.4. Apoptosis in Glioma
1.2. Autophagy
1.2.1. Molecular Mechanisms in Autophagy
1.2.2. Autophagy in Glioma
1.2.3. Autophagy as a Tumor Suppressor
1.2.4. Autophagy as a Tumor Promoter
1.3. Molecular Correlation between Apoptosis and Autophagy
1.4. Treatment Choices for Glioblastoma
Standard of Care
1.5. Small-Molecule Inhibitors
1.5.1. Erlotinib
1.5.2. Gefitinib
1.5.3. Imatinib
1.5.4. Sunitinib
1.5.5. Vandetanib
1.6. Targeting Downstream Intracellular Effector Molecules
1.6.1. The RAS/RAF/MAPK Pathway
Tipifarnib
Lonafarnib
1.6.2. Targeting PI3K/AKT/mTOR
Temsirolimus
Everolimus
Sirolimus
1.7. Bcl-2 Inhibitors
1.7.1. ABT-737
1.7.2. Gossypol
1.7.3. Berberine
1.8. TRAIL/TRAILR Pathway Activators
1.8.1. Recombinant TRAIL, sTRAIL, and Anti-DR4 and -5 Antibodies
1.8.2. Taxol
1.8.3. TIC10/ONC201
2. Conclusions
Funding
Conflicts of Interest
Abbreviations
| 3 MA | methyladenine |
| Activating factor-1 | Apaf-1 |
| Activating transcription factor 4 | ATF4 |
| Activator of transcription-3 | STAT3 |
| Adenosine triphosphate | ATP |
| AMBRA-1 | Activating Molecule in Beclin 1-Regulated Autophagy |
| AMP-activated kinase | AMPK |
| Apoptosis-inducing factor | AIF |
| Autophagy-related genes | ATG |
| Ataxia telangiectasia mutated protein | ATM |
| ATP-binding cassette (ABC) transporters | ABCG |
| Bax-interacting factor 1 | Bif-1 |
| B cell lymphoma 2 | Bcl-2 |
| B cell lymphoma-extra-large | Bcl-xL |
| BH3-type proteins in the Bcl-2 family | BNIP3 |
| Binding Protein Homology Protein | CHOP |
| Ca2+/calmodulin-dependent kinase kinase | CaMKKβ |
| Calcium channel, voltage-dependent gamma subunit 4 | CACNG4 |
| Calcineurin-dependent 1 | NFATC1 |
| Caspase recruitment domain | CARD |
| Caveolin-1 | Cav-1 |
| Central nervous system | CNS |
| c-Jun N-terminal kinase | JNK |
| Coat protein complex II | COPII |
| Colony-stimulating factor-1 | CSF1R |
| C vacuolar protein | C-VPS |
| Cytochrome c | cyt c |
| Death effector domain | DED |
| Death Domain | DD |
| Death-inducing signaling complex | DISC |
| Diffuse Intrinsic Pontine Gliomas | DIPG |
| DNA damage-regulated autophagy modulator | DRAM |
| Elongation factor-2 | elF2α |
| Elongation factor-2 kinase | eEF2 kinase |
| Epidermal growth factor receptor | EGFR |
| EGFR-targeted diphtheria toxin | DT-EGF |
| Extracellular matrix | ECM |
| Farnesyltransferase inhibitors | FTIs |
| Fas-associated death domain | FADD |
| Fas ligand | Fas-L |
| Fas receptor | FasR |
| Fibroblast growth factor receptor 4 | FGFR4 |
| FK-binding protein-12 | FKBP-12 |
| Fms-like tyrosine kinase-3 | FLT3 |
| Focal adhesion kinase | FAK |
| Food and Drug Administration | FDA |
| G protein β-subunit-like protein | GβL |
| Glioblastoma multiforme | GBM |
| Glioma Stem Cells | GSC |
| Glioma stem/progenitor cells | GSPCs |
| Heat shock cognate 71 kDa protein | Hsc70 |
| Heat shock 27-kD protein 1 | HSPB1 |
| Heat shock 70-kD protein 1B | HSPA1B |
| High-mobility group box protein 1 | HMGB1 |
| Human multidrug resistance protein 3 | MRP3 |
| Inhibitor of apoptosis | IAP |
| Inositol 1,4,5-triphosphate receptor | IP3R |
| Lysosomal-associate membrane protein 2A receptor | LAMP2A |
| Methylguanine-O6-methyltransferase | MGMT |
| Mitogen-activated protein kinase | MAPK |
| Monocarboxylate transporter-4 | MCT4 |
| Neurotrophic tyrosine kinase receptor type-1 | NTRK1 |
| Nitrogen reactive species | NOS |
| Overall survival | OS |
| Paxillin | PXN |
| Phagophore assembly site | PAS |
| Phosphatidylinositol 3-phosphate | PI3P |
| Phosphatidylinositol-4,5-bisphosphate | PIP2 |
| Phosphatidylinositol-3,4,5-trisphosphate | PIP3 |
| Phosphatidylethanolamine | PE |
| Phospholipase C-γ1 | PLC-γ1 |
| Platelet-derived growth factor receptor | PDGR |
| Proline-rich AKT substrate of 40 kDa | PRAS40 |
| Progression-free survival | PFS |
| Protein endoplasmic reticulum kinase | PERK |
| RAS-related C3 botulinum toxin substrate 1 | RAC1 |
| Reactive oxygen species | ROS |
| Ribosomal S6 kinase 1 | RSK1 |
| Second Mitochondria-derived Activator of Caspases | Smac |
| Direct IAP-Binding protein with Low PI | DIABLO |
| Serine/threonine kinases phosphoinositide-dependent kinase 1 | PDK1 |
| Stem cell-factor | Kit |
| Smoothened homolog | SMO |
| Target of rapamycin complex 1 | TORC1 |
| Temozolomide | TMZ |
| Tyrosine-kinase inhibitors | TKI |
| Transcription factor 7-like 1 | TCF7L1 |
| Transforming growth factor beta 3 | TGFβ3 |
| Transforming growth factor-β-activating kinase 1 | TAK1 |
| Tensin homolog on chromosome ten | PTEN |
| Toll-like receptor 4 | TLR4 |
| Transport protein particle III | TRAPPIII |
| Tumor Necrosis Factor receptors | TNF |
| Tumor Necrosis Factor receptors | TNFR |
| Tumor necrosis factor-related apoptosis-inducing ligand | TRAIL |
| Unc-51-Like Kinase ½ | ULK1/ULK2 |
| UV irradiation resistance-associated tumor suppressor gene | UVRAG |
| Vascular endothelial growth factor receptor | VEGFR |
References
- DeAngelis, L.M. Brain tumors. N. Engl. J. Med. 2001, 344, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Crocetti, E.; Trama, A.; Stiller, C.; Caldarella, A.; Soffietti, R.; Jaal, J.; Weber, D.C.; Ricardi, U.; Slowinski, J.; Brandes, A.; et al. Epidemiology of glial and non-glial brain tumours in Europe. Eur. J. Cancer 2012, 48, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Budke, M.; Isla-Guerrero, A.; Perez-Lopez, C.; Perez-Alvarez, M.; Garcia-Grande, A.; Bello, M.J.; Rey, J. A comparative study of the treatment of high grade gliomas. Rev. Neurol. 2003, 37, 912–916. [Google Scholar] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Grobben, B.; De Deyn, P.P.; Slegers, H. Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion. Cell Tissue Res. 2002, 310, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Mischel, P.S.; Cloughesy, T.F. Targeted molecular therapy of GBM. Brain Pathol. 2003, 13, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.M.; Amankulor, N.M.; Pitter, K.; Helmy, K.; Squatrito, M.; Holland, E.C. Astrocyte-specific expression patterns associated with the PDGF-induced glioma microenvironment. PLoS ONE 2012, 7, e32453. [Google Scholar] [CrossRef] [PubMed]
- Machein, M.R.; Plate, K.H. VEGF in brain tumors. J. Neuro-Oncol. 2000, 50, 109–120. [Google Scholar] [CrossRef]
- Feldkamp, M.M.; Lau, N.; Guha, A. Signal transduction pathways and their relevance in human astrocytomas. J. Neuro-Oncol. 1997, 35, 223–248. [Google Scholar] [CrossRef]
- Chakravarti, A.; Delaney, M.A.; Noll, E.; Black, P.M.; Loeffler, J.S.; Muzikansky, A.; Dyson, N.J. Prognostic and pathologic significance of quantitative protein expression profiling in human gliomas. Clin. Cancer Res. 2001, 7, 2387–2395. [Google Scholar] [PubMed]
- Kornienko, A.; Mathieu, V.; Rastogi, S.K.; Lefranc, F.; Kiss, R. Therapeutic agents triggering nonapoptotic cancer cell death. J. Med. Chem. 2013, 56, 4823–4839. [Google Scholar] [CrossRef] [PubMed]
- Djedid, R.; Tomasi, O.; Haidara, A.; Rynkowski, M.; Lefranc, F. Glioblastoma treatment in 2010. Rev. Med. Brux. 2009, 30, 496–505. [Google Scholar] [PubMed]
- Trejo-Solis, C.; Jimenez-Farfan, D.; Rodriguez-Enriquez, S.; Fernandez-Valverde, F.; Cruz-Salgado, A.; Ruiz-Azuara, L.; Sotelo, J. Copper compound induces autophagy and apoptosis of glioma cells by reactive oxygen species and JNK activation. BMC Cancer 2012, 12, 156. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.; Bajpai, V.K.; Sahasrabuddhe, A.A.; Kumar, A.; Sinha, R.A.; Behari, S.; Godbole, M.M. Inhibition of N-(4-hydroxyphenyl)retinamide-induced autophagy at a lower dose enhances cell death in malignant glioma cells. Carcinogenesis 2008, 29, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Jo, G.H.; Bogler, O.; Chwae, Y.J.; Yoo, H.; Lee, S.H.; Park, J.B.; Kim, Y.J.; Kim, J.H.; Gwak, H.S. Radiation-induced autophagy contributes to cell death and induces apoptosis partly in malignant glioma cells. Cancer Res. Treat. 2015, 47, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Thayyullathil, F.; Rahman, A.; Pallichankandy, S.; Patel, M.; Galadari, S. ROS-dependent prostate apoptosis response-4 (Par-4) up-regulation and ceramide generation are the prime signaling events associated with curcumin-induced autophagic cell death in human malignant glioma. FEBS Open Bio 2014, 4, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Keshmiri-Neghab, H.; Goliaei, B.; Nikoofar, A. Gossypol enhances radiation induced autophagy in glioblastoma multiforme. Gen. Physiol. Biophys. 2014, 33, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Garcia, N.; Pedraza-Chaverri, J.; Mares-Samano, J.J.; Orozco-Ibarra, M.; Cruz-Salgado, A.; Jimenez-Anguiano, A.; Sotelo, J.; Trejo-Solis, C. Antiapoptotic Effects of EGb 761. ECAM 2013, 2013, 495703. [Google Scholar] [CrossRef] [PubMed]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Hunter, A.M.; LaCasse, E.C.; Korneluk, R.G. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007, 12, 1543–1568. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S. Molecular steps of cell suicide: An insight into immune senescence. J. Clin. Immunol. 2000, 20, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Nijhawan, D.; Honarpour, N.; Wang, X. Apoptosis in neural development and disease. Ann. Rev. Neurosci. 2000, 23, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Orlinick, J.R.; Vaishnaw, A.K.; Elkon, K.B. Structure and function of Fas/Fas ligand. Int. Rev. Immunol. 1999, 18, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Cahuzac, N.; Baum, W.; Kirkin, V.; Conchonaud, F.; Wawrezinieck, L.; Marguet, D.; Janssen, O.; Zornig, M.; Hueber, A.O. Fas ligand is localized to membrane rafts, where it displays increased cell death-inducing activity. Blood 2006, 107, 2384–2391. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Fukunaga, R.; Brannan, C.I.; Itoh, N.; Yonehara, S.; Copeland, N.G.; Jenkins, N.A.; Nagata, S. The cDNA structure, expression, and chromosomal assignment of the mouse Fas antigen. J. Immunol. 1992, 148, 1274–1279. [Google Scholar] [PubMed]
- Bechmann, I.; Mor, G.; Nilsen, J.; Eliza, M.; Nitsch, R.; Naftolin, F. FasL (CD95L, Apo1L) is expressed in the normal rat and human brain: Evidence for the existence of an immunological brain barrier. Glia 1999, 27, 62–74. [Google Scholar] [CrossRef]
- Choi, C.; Park, J.Y.; Lee, J.; Lim, J.H.; Shin, E.C.; Ahn, Y.S.; Kim, C.H.; Kim, S.J.; Kim, J.D.; Choi, I.S.; et al. Fas ligand and Fas are expressed constitutively in human astrocytes and the expression increases with IL-1, IL-6, TNF-alpha, or IFN-gamma. J. Immunol. 1999, 162, 1889–1895. [Google Scholar] [PubMed]
- MacEwan, D.J. TNF ligands and receptors—A matter of life and death. Br. J. Pharmacol. 2002, 135, 855–875. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.; Landau, A.M.; Desbarats, J. Fas-beyond death: A regenerative role for Fas in the nervous system. Apoptosis 2003, 8, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C.; Kischkel, F.C.; Krammer, P.H.; Peter, M.E. Analysis of the CD95 (APO-1/Fas) death-inducing signaling complex by high-resolution two-dimensional gel electrophoresis. Methods Enzymol. 2000, 322, 363–373. [Google Scholar] [PubMed]
- Zhang, J.; Zhang, D.; Hua, Z. FADD and its phosphorylation. IUBMB Life 2004, 56, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Angelats, M.; Cidlowski, J.A. Molecular evidence for the nuclear localization of FADD. Cell Death Differ. 2003, 10, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Forbes-Hernandez, T.Y.; Giampieri, F.; Gasparrini, M.; Mazzoni, L.; Quiles, J.L.; Alvarez-Suarez, J.M.; Battino, M. The effects of bioactive compounds from plant foods on mitochondrial function: A focus on apoptotic mechanisms. Food Chem. Toxicol. 2014, 68, 154–182. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Deedigan, L.; Jencz, S.; Mehrabadi, Y.; Houlden, L.; Albarenque, S.M.; Zwacka, R.M. Caspase-10: A molecular switch from cell-autonomous apoptosis to communal cell death in response to chemotherapeutic drug treatment. Cell Death Differ. 2018, 25, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Chen, G.S.; Lin, P.Y.; Pan, I.H.; Wang, S.T.; Lin, S.H.; Yu, H.S.; Lin, C.C. Tazarotene induces apoptosis in human basal cell carcinoma via activation of caspase-8/t-Bid and the reactive oxygen species-dependent mitochondrial pathway. DNA Cell Biol. 2014, 33, 652–666. [Google Scholar] [CrossRef] [PubMed]
- Sastry, P.S.; Rao, K.S. Apoptosis and the nervous system. J. Neurochem. 2000, 74, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ashe, P.C.; Berry, M.D. Apoptotic signaling cascades. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 199–214. [Google Scholar] [CrossRef]
- Zimmermann, K.C.; Bonzon, C.; Green, D.R. The machinery of programmed cell death. Pharmacol. Ther. 2001, 92, 57–70. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Hockenbery, D.M. Targeting mitochondria for cancer therapy. Environ. Mol. Mutagen. 2010, 51, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Moroni, M.C.; Hickman, E.S.; Lazzerini Denchi, E.; Caprara, G.; Colli, E.; Cecconi, F.; Muller, H.; Helin, K. Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol. 2001, 3, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Angosto, M. Bases Moleculares de la Apoptosis. Anal. Real Acad. Nac. Farm. 2003, 69, 29. [Google Scholar]
- Verhagen, A.M.; Silke, J.; Ekert, P.G.; Pakusch, M.; Kaufmann, H.; Connolly, L.M.; Day, C.L.; Tikoo, A.; Burke, R.; Wrobel, C.; et al. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J. Biol. Chem. 2002, 277, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.-D. Cell Cycle Arrest and Apoptosis; Springer: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Wu, G.S.; Burns, T.F.; McDonald, E.R., 3rd; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.D.; Zhou, J.Y.; Muschel, R.; et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 1997, 17, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Sinha, S.C.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.D.; Xavier, J.M.; Steer, C.J.; Rodrigues, C.M. The role of p53 in apoptosis. Discov. Med. 2010, 9, 145–152. [Google Scholar] [PubMed]
- Altin, S.E.; Schulze, P.C. p53-upregulated modulator of apoptosis (PUMA): A novel proapoptotic molecule in the failing heart. Circulation 2011, 124, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Strozyk, E.; Kulms, D. The role of AKT/mTOR pathway in stress response to UV-irradiation: Implication in skin carcinogenesis by regulation of apoptosis, autophagy and senescence. Int. J. Mol. Sci. 2013, 14, 15260–15285. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, J.K.; Vahakangas, K.H.; Helppi, H.M.; Bloigu, R.; Paakko, P.; Turpeenniemi-Hujanen, T. Specific TP53 mutations predict aggressive phenotype in head and neck squamous cell carcinoma: A retrospective archival study. Head Neck Oncol. 2011, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, S.; Se-Kwon, K. Cell survival and apoptosis signaling as therapeutic target for cancer: Marine bioactive compounds. Int. J. Mol. Sci. 2013, 14, 2334–2354. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, F.; Sunayama, J.; Mori, Y.; Hattori, S.; Shimizu, S.; Tsujimoto, Y.; Yoshioka, K.; Masuyama, N.; Gotoh, Y. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 2004, 23, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Stegh, A.H.; Kim, H.; Bachoo, R.M.; Forloney, K.L.; Zhang, J.; Schulze, H.; Park, K.; Hannon, G.J.; Yuan, J.; Louis, D.N.; et al. Bcl2L12 inhibits post-mitochondrial apoptosis signaling in glioblastoma. Genes Dev. 2007, 21, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Wagner, S.; Kerkau, S.; Dichgans, J.; Tonn, J.C.; Weller, M. BCL-2 promotes migration and invasiveness of human glioma cells. FEBS Lett. 1998, 440, 419–424. [Google Scholar] [CrossRef]
- Wick, W.; Wild-Bode, C.; Frank, B.; Weller, M. BCL-2-induced glioma cell invasiveness depends on furin-like proteases. J. Neurochem. 2004, 91, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, D.; Sharma, B.S.; Gupta, S.K.; Kaul, D.; Vasishta, R.K.; Khosla, V.K. Expression of Bcl2 proto-oncogene in primary tumors of the central nervous system. Neurol. India 2002, 50, 290–294. [Google Scholar] [PubMed]
- Steinbach, J.P.; Weller, M. Apoptosis in Gliomas: Molecular Mechanisms and Therapeutic Implications. J. Neuro-Oncol. 2004, 70, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Strik, H.; Deininger, M.; Streffer, J.; Grote, E.; Wickboldt, J.; Dichgans, J.; Weller, M.; Meyermann, R. BCL-2 family protein expression in initial and recurrent glioblastomas: Modulation by radiochemotherapy. J. Neurol. Neurosurg. Psychiatry 1999, 67, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Ruano, Y.; Mollejo, M.; Camacho, F.I.; Rodriguez de Lope, A.; Fiano, C.; Ribalta, T.; Martinez, P.; Hernandez-Moneo, J.L.; Melendez, B. Identification of survival-related genes of the phosphatidylinositol 3’-kinase signaling pathway in glioblastoma multiforme. Cancer 2008, 112, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Cartron, P.F.; Loussouarn, D.; Campone, M.; Martin, S.A.; Vallette, F.M. Prognostic impact of the expression/phosphorylation of the BH3-only proteins of the BCL-2 family in glioblastoma multiforme. Cell Death Dis. 2012, 3, e421. [Google Scholar] [CrossRef] [PubMed]
- Blahovcova, E.; Richterova, R.; Kolarovszki, B.; Dobrota, D.; Racay, P.; Hatok, J. Apoptosis-related gene expression in tumor tissue samples obtained from patients diagnosed with glioblastoma multiforme. Int. J. Mol. Med. 2015, 36, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.C.; Weyhenmeyer, B.; Schmid, J.; Kilbride, S.M.; Rehm, M.; Huber, H.J.; Senft, C.; Weissenberger, J.; Seifert, V.; Dunst, M.; et al. Activation of executioner caspases is a predictor of progression-free survival in glioblastoma patients: A systems medicine approach. Cell Death Dis. 2013, 4, e629. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Li, T.; Ma, D.Z.; Ji, Y.X.; Zhi, H. Overexpression of FADD and Caspase-8 inhibits proliferation and promotes apoptosis of human glioblastoma cells. Biomed. Pharmacother. 2017, 93, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Saggioro, F.P.; Neder, L.; Stavale, J.N.; Paixao-Becker, A.N.; Malheiros, S.M.; Soares, F.A.; Pittella, J.E.; Matias, C.C.; Colli, B.O.; Carlotti, C.G., Jr.; et al. Fas, FasL, and cleaved caspases 8 and 3 in glioblastomas: A tissue microarray-based study. Pathol. Res. Pract. 2014, 210, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ashley, D.M.; Riffkin, C.D.; Muscat, A.M.; Knight, M.J.; Kaye, A.H.; Novak, U.; Hawkins, C.J. Caspase 8 is absent or low in many ex vivo gliomas. Cancer 2005, 104, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Knight, M.J.; Riffkin, C.D.; Muscat, A.M.; Ashley, D.M.; Hawkins, C.J. Analysis of FasL and TRAIL induced apoptosis pathways in glioma cells. Oncogene 2001, 20, 5789–5798. [Google Scholar] [CrossRef] [PubMed]
- Kuijlen, J.M.; Mooij, J.J.; Platteel, I.; Hoving, E.W.; van der Graaf, W.T.; Span, M.M.; Hollema, H.; den Dunnen, W.F. TRAIL-receptor expression is an independent prognostic factor for survival in patients with a primary glioblastoma multiforme. J. Neuro-Oncol. 2006, 78, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Elias, A.; Siegelin, M.D.; Steinmuller, A.; von Deimling, A.; Lass, U.; Korn, B.; Mueller, W. Epigenetic silencing of death receptor 4 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in gliomas. Clin. Cancer Res. 2009, 15, 5457–5465. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, B.; Glaser, T.; Naumann, U.; Kugler, S.; Isenmann, S.; Bahr, M.; Korneluk, R.; Liston, P.; Weller, M. Expression and biological activity of X-linked inhibitor of apoptosis (XIAP) in human malignant glioma. Cell Death Differ. 1999, 6, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Cloonan, S.M.; Choi, A.M. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Karp, C.; Strohecker, A.M.; Guo, Y.; Mathew, R. Role of autophagy in suppression of inflammation and cancer. Curr. Opin. Cell Biol. 2010, 22, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, G.; Neufeld, T.P. Autophagy: A forty-year search for a missing membrane source. PLoS Biol. 2006, 4, e36. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.M.; Chen, L.H.; Chen, M.T.; Ma, H.I.; Su, T.L.; Hsieh, P.C.; Chien, C.S.; Jiang, B.H.; Chen, Y.C.; Lin, Y.H.; et al. Targeting autophagy enhances BO-1051-induced apoptosis in human malignant glioma cells. Cancer Chemother. Pharmacol. 2012, 69, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, N.; Klionsky, D.J.; Reggiori, F. Genetic aberrations in macroautophagy genes leading to diseases. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, F.; Afrin, S.; Forbes-Hernandez, T.Y.; Gasparrini, M.; Cianciosi, D.; Reboredo-Rodriguez, P.; Varela-Lopez, A.; Quiles, J.L.; Battino, M. Autophagy in Human Health and Disease: Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. BioEssays 2018, 40, e1800008. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Akioka, M.; Kondo-Kakuta, C.; Yamamoto, H.; Ohsumi, Y. Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J. Cell Sci. 2013, 126, 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Farre, J.C.; Subramani, S. Mechanistic insights into selective autophagy pathways: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2016, 17, 537–552. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.H.; Yip, C.K.; Shi, Y.; Chait, B.T.; Wang, Q.J. Beclin 1-Vps34 Complex Architecture: Understanding the Nuts and Bolts of Therapeutic Targets. Front. Biol. 2015, 10, 398–426. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Klionsky, D.J. Physiological functions of Atg6/Beclin 1: A unique autophagy-related protein. Cell Res. 2007, 17, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [PubMed]
- Law, F.; Seo, J.H.; Wang, Z.; DeLeon, J.L.; Bolis, Y.; Brown, A.; Zong, W.X.; Du, G.; Rocheleau, C.E. The VPS34 PI3K negatively regulates RAB-5 during endosome maturation. J. Cell Sci. 2017, 130, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Tooze, S.A.; Reggiori, F. The puzzling origin of the autophagosomal membrane. F1000 Biol. Rep. 2011, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; McEwan, D.G.; Dikic, I. The LC3 interactome at a glance. J. Cell Sci. 2014, 127, 3–9. [Google Scholar] [CrossRef]
- Monastyrska, I.; Rieter, E.; Klionsky, D.J.; Reggiori, F. Multiple roles of the cytoskeleton in autophagy. Biol. Rev. Camb. Philos. Soc. 2009, 84, 431–448. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.; Isaacs, A.M. The role of ESCRT proteins in fusion events involving lysosomes, endosomes and autophagosomes. Biochem. Soc. Trans. 2010, 38, 1469–1473. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Platta, H.W.; Abrahamsen, H.; Thoresen, S.B.; Stenmark, H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem. J. 2012, 441, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Lin, Y.C.; Chen, Y.H.; Chen, C.M.; Pang, L.Y.; Chen, H.A.; Wu, P.R.; Lin, M.Y.; Jiang, S.T.; Tsai, T.F.; et al. Cul3-KLHL20 Ubiquitin Ligase Governs the Turnover of ULK1 and VPS34 Complexes to Control Autophagy Termination. Mol. Cell 2016, 61, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK and autophagy get connected. EMBO J. 2011, 30, 634–635. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.L. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Bai, H.M.; Chen, L.; Li, B.; Lu, Y.C. Reduced expression of LC3B-II and Beclin 1 in glioblastoma multiforme indicates a down-regulated autophagic capacity that relates to the progression of astrocytic tumors. J. Clin. Neurosci. 2010, 17, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Pirtoli, L.; Cevenini, G.; Tini, P.; Vannini, M.; Oliveri, G.; Marsili, S.; Mourmouras, V.; Rubino, G.; Miracco, C. The prognostic role of Beclin 1 protein expression in high-grade gliomas. Autophagy 2009, 5, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Patric, I.R.; Patil, V.; Shwetha, S.D.; Hegde, A.S.; Chandramouli, B.A.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Methylation silencing of ULK2, an autophagy gene, is essential for astrocyte transformation and tumor growth. J. Biol. Chem. 2014, 289, 22306–22318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tian, H.; Miao, Y.; Feng, X.; Li, Y.; Wang, H.; Song, X. Upregulation of p72 Enhances Malignant Migration and Invasion of Glioma Cells by Repressing Beclin1 Expression. Biochem. Biokhimiia 2016, 81, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Kondo, Y.; Aldape, K.; Yamamoto, A.; Iwado, E.; Yokoyama, T.; Hollingsworth, E.F.; Kobayashi, R.; Hess, K.; Shinojima, N.; et al. Monitoring autophagy in glioblastoma with antibody against isoform B of human microtubule-associated protein 1 light chain 3. Autophagy 2008, 4, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, T.; Hu, J.; Zhang, R.; Rao, Y.; Wang, S.; Chen, R.; McLendon, R.E.; Friedman, A.H.; Keir, S.T.; et al. miR-33a promotes glioma-initiating cell self-renewal via PKA and NOTCH pathways. J. Clin. Investig. 2014, 124, 4489–4502. [Google Scholar] [CrossRef] [PubMed]
- Jennewein, L.; Ronellenfitsch, M.W.; Antonietti, P.; Ilina, E.I.; Jung, J.; Stadel, D.; Flohr, L.M.; Zinke, J.; von Renesse, J.; Drott, U.; et al. Diagnostic and clinical relevance of the autophago-lysosomal network in human gliomas. Oncotarget 2016, 7, 20016–20032. [Google Scholar] [CrossRef] [PubMed]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.O.; Pratheeshkumar, P.; Roy, R.V.; Hitron, J.A.; Wang, L.; Zhang, Z.; Shi, X. Nrf2/p62 signaling in apoptosis resistance and its role in cadmium-induced carcinogenesis. J. Biol. Chem. 2014, 289, 28660–28675. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Oh, E.; Yoo, J.Y.; Choi, K.S.; Yoon, M.J.; Yun, C.O. Adenovirus expressing dual c-Met-specific shRNA exhibits potent antitumor effect through autophagic cell death accompanied by senescence-like phenotypes in glioblastoma cells. Oncotarget 2015, 6, 4051–4065. [Google Scholar] [CrossRef] [PubMed]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [PubMed]
- Lepine, S.; Allegood, J.C.; Edmonds, Y.; Milstien, S.; Spiegel, S. Autophagy induced by deficiency of sphingosine-1-phosphate phosphohydrolase 1 is switched to apoptosis by calpain-mediated autophagy-related gene 5 (Atg5) cleavage. J. Biol. Chem. 2011, 286, 44380–44390. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Qi, Q.; Hua, X.; Li, X.; Zhang, W.; Sun, H.; Li, S.; Wang, X.; Li, B. Beclin 1, an autophagy-related gene, augments apoptosis in U87 glioblastoma cells. Oncol. Rep. 2014, 31, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Kaza, N.; Kohli, L.; Roth, K.A. Autophagy in brain tumors: A new target for therapeutic intervention. Brain Pathol. 2012, 22, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.T.; Sun, G.H.; Cha, T.L.; Kao, C.C.; Chang, S.Y.; Kuo, S.C.; Way, T.D. CSC-3436 switched tamoxifen-induced autophagy to apoptosis through the inhibition of AMPK/mTOR pathway. J. Biomed. Sci. 2016, 23, 60. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Du, J.; Hua, S.; Zhang, H.; Gu, C.; Wang, J.; Yang, L.; Huang, J.; Yu, J.; Liu, F. Suppression of autophagy augments the radiosensitizing effects of STAT3 inhibition on human glioma cells. Exp. Cell Res. 2015, 330, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, W.; Wang, C.; Leng, X.; Lian, S.; Feng, J.; Li, J.; Wang, H. Inhibition of autophagy enhances apoptosis induced by proteasome inhibitor bortezomib in human glioblastoma U87 and U251 cells. Mol. Cell. Biochem. 2014, 385, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Chiavarina, B.; Pavlides, S.; Whitaker-Menezes, D.; Tsirigos, A.; Witkiewicz, A.; Lin, Z.; Balliet, R.; Howell, A.; et al. Understanding the "lethal" drivers of tumor-stroma co-evolution: Emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Biol. Ther. 2010, 10, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, M.P.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NF-κB activation in the tumor stromal microenvironment. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cardena, G.; Martasek, P.; Masters, B.S.; Skidd, P.M.; Couet, J.; Li, S.; Lisanti, M.P.; Sessa, W.C. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J. Biol. Chem. 1997, 272, 25437–25440. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial "lactate shuttle" in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [PubMed]
- Regina, A.; Jodoin, J.; Khoueir, P.; Rolland, Y.; Berthelet, F.; Moumdjian, R.; Fenart, L.; Cecchelli, R.; Demeule, M.; Beliveau, R. Down-regulation of caveolin-1 in glioma vasculature: Modulation by radiotherapy. J. Neurosci. Res. 2004, 75, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Goncalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro-Oncology 2013, 15, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J. Detachment-induced autophagy during anoikis and lumen formation in epithelial acini. Autophagy 2008, 4, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Avivar-Valderas, A.; Salas, E.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Nagi, C.; Debnath, J.; Aguirre-Ghiso, J.A. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell. Biol. 2011, 31, 3616–3629. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Vial, D.; McKeown-Longo, P.J. Role of EGFR expression levels in the regulation of integrin function by EGF. Mol. Carcinogen. 2016, 55, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Peart, T.; Ramos Valdes, Y.; Correa, R.J.; Fazio, E.; Bertrand, M.; McGee, J.; Prefontaine, M.; Sugimoto, A.; DiMattia, G.E.; Shepherd, T.G. Intact LKB1 activity is required for survival of dormant ovarian cancer spheroids. Oncotarget 2015, 6, 22424–22438. [Google Scholar] [CrossRef] [PubMed]
- Magnus, N.; Garnier, D.; Meehan, B.; McGraw, S.; Lee, T.H.; Caron, M.; Bourque, G.; Milsom, C.; Jabado, N.; Trasler, J.; et al. Tissue factor expression provokes escape from tumor dormancy and leads to genomic alterations. Proc. Natl. Acad. Sci. USA 2014, 111, 3544–3549. [Google Scholar] [CrossRef] [PubMed]
- Magnus, N.; Garnier, D.; Rak, J. Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood 2010, 116, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Daido, S.; Kanzawa, T.; Kondo, S.; Kondo, Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 2005, 26, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Levin-Salomon, V.; Bialik, S.; Kimchi, A. DAP-kinase and autophagy. Apoptosis 2014, 19, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Strappazzon, F.; Vietri-Rudan, M.; Campello, S.; Nazio, F.; Florenzano, F.; Fimia, G.M.; Piacentini, M.; Levine, B.; Cecconi, F. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 2011, 30, 1195–1208. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Ishaq, M.; Singh, S.K. Caspase-mediated crosstalk between autophagy and apoptosis: Mutual adjustment or matter of dominance. J. Cancer Res. Ther. 2015, 11, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.; Lane, J.D. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 2009, 122, 2554–2566. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, V.; Wirawan, E.; Romagnoli, A.; Ciccosanti, F.; Lisi, G.; Lippens, S.; Cecconi, F.; Fimia, G.M.; Vandenabeele, P.; Corazzari, M.; et al. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. 2012, 19, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Zhao, W.; Zeng, R.X. Autophagic degradation of caspase-8 protects U87MG cells against H2O2-induced oxidative stress. APJCP 2013, 14, 4095–4099. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Cao, L.; Kang, R.; Yang, M.; Wang, Z.; Peng, Y.; Tan, Y.; Liu, L.; Xie, M.; Zhao, Y.; et al. UV irradiation resistance-associated gene suppresses apoptosis by interfering with BAX activation. EMBO Rep. 2011, 12, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, Y.; Long, M.; Luo, T.; Bian, J.; Liu, X.; Gu, J.; Zou, H.; Song, R.; Wang, Y.; et al. Beclin-1-mediated Autophagy Protects Against Cadmium-activated Apoptosis via the Fas/FasL Pathway in Primary Rat Proximal Tubular Cell Culture. Sci. Rep. 2017, 7, 977. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hou, W.; Goldstein, L.A.; Lu, C.; Stolz, D.B.; Yin, X.M.; Rabinowich, H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J. Biol. Chem. 2008, 283, 19665–19677. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, J.; Moore, F.; Rao, A.; Barclay, W.W.; Thomas, L.R.; Grant, K.W.; Cramer, S.D.; Thorburn, A. Selective inactivation of a Fas-associated death domain protein (FADD)-dependent apoptosis and autophagy pathway in immortal epithelial cells. Mol. Biol. Cell 2005, 16, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Martin, G.; Hoyer-Hansen, M.; Garcia-Garcia, C.; Fumarola, C.; Farkas, T.; Lopez-Rivas, A.; Jaattela, M. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009, 28, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Park, K.J.; Lee, S.H.; Kim, T.I.; Lee, H.W.; Lee, C.H.; Kim, E.H.; Jang, J.Y.; Choi, K.S.; Kwon, M.H.; Kim, Y.S. A human scFv antibody against TRAIL receptor 2 induces autophagic cell death in both TRAIL-sensitive and TRAIL-resistant cancer cells. Cancer Res. 2007, 67, 7327–7334. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.; Bond, E.E.; Levine, A.J.; Bond, G.L. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov. 2008, 7, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Kakudo, Y.; Takahashi, S.; Sakamoto, Y.; Kato, S.; Ishioka, C. Overexpression of DRAM enhances p53-dependent apoptosis. Cancer Med. 2013, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Laforge, M.; Limou, S.; Harper, F.; Casartelli, N.; Rodrigues, V.; Silvestre, R.; Haloui, H.; Zagury, J.F.; Senik, A.; Estaquier, J. DRAM triggers lysosomal membrane permeabilization and cell death in CD4(+) T cells infected with HIV. PLoS Pathog. 2013, 9, e1003328. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Shi, Y.; Guo, X.H.; Ouyang, Y.B.; Wang, S.S.; Liu, D.J.; Wang, A.N.; Li, N.; Chen, D.X. Phosphorylated AKT inhibits the apoptosis induced by DRAM-mediated mitophagy in hepatocellular carcinoma by preventing the translocation of DRAM to mitochondria. Cell Death Dis. 2014, 5, e1078. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Qi, L.; Wu, J.C.; Qin, Z.H. DRAM1 regulates autophagy flux through lysosomes. PLoS ONE 2013, 8, e63245. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Karuman, P.; Gozani, O.; Odze, R.D.; Zhou, X.C.; Zhu, H.; Shaw, R.; Brien, T.P.; Bozzuto, C.D.; Ooi, D.; Cantley, L.C.; et al. The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol. Cell 2001, 7, 1307–1319. [Google Scholar] [CrossRef]
- Gao, W.; Shen, Z.; Shang, L.; Wang, X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011, 18, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Fox, C.J.; Mu, J.; Solt, L.; Xu, A.; Cinalli, R.M.; Birnbaum, M.J.; Lindsten, T.; Thompson, C.B. The PP2A-associated protein alpha4 is an essential inhibitor of apoptosis. Science 2004, 306, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Ro, S.H.; Semple, I.A.; Park, H.; Park, H.; Park, H.W.; Kim, M.; Kim, J.S.; Lee, J.H. Sestrin2 promotes Unc-51-like kinase 1 mediated phosphorylation of p62/sequestosome-1. FEBS J. 2014, 281, 3816–3827. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Carmo, A.; Carvalheiro, H.; Crespo, I.; Nunes, I.; Lopes, M.C. Effect of temozolomide on the U-118 glioma cell line. Oncol. Lett. 2011, 2, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- De Salvo, M.; Maresca, G.; D’Agnano, I.; Marchese, R.; Stigliano, A.; Gagliassi, R.; Brunetti, E.; Raza, G.H.; De Paula, U.; Bucci, B. Temozolomide induced c-Myc-mediated apoptosis via Akt signalling in MGMT expressing glioblastoma cells. Int. J. Radiat. Biol. 2011, 87, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Wurstle, S.; Schneider, F.; Ringel, F.; Gempt, J.; Lammer, F.; Delbridge, C.; Wu, W.; Schlegel, J. Temozolomide induces autophagy in primary and established glioblastoma cells in an EGFR independent manner. Oncol. Lett. 2017, 14, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Zhang, L.; Wu, J.; Zhou, L.; Ren, Y.J.; Yang, W.Q.; Ming, Z.J.; Chen, B.; Wang, J.; Zhang, Y.; et al. Inhibition of elongation factor-2 kinase augments the antitumor activity of Temozolomide against glioma. PLoS ONE 2013, 8, e81345. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Batista, L.F.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.B.; Wang, Z.; Shu, F.; Jin, Y.H.; Liu, H.Y.; Wang, Q.J.; Yang, Y. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J. Biol. Chem. 2010, 285, 40461–40471. [Google Scholar] [CrossRef] [PubMed]
- Ramis, G.; Thomas-Moya, E.; Fernandez de Mattos, S.; Rodriguez, J.; Villalonga, P. EGFR inhibition in glioma cells modulates Rho signaling to inhibit cell motility and invasion and cooperates with temozolomide to reduce cell growth. PLoS ONE 2012, 7, e38770. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.R.; Rath, P.; Oyinlade, O.; Lopez, H.; Mughal, S.; Xia, S.; Li, Y.; Kaur, H.; Zhou, X.; Ahmed, A.K.; et al. Crizotinib and erlotinib inhibits growth of c-Met(+)/EGFRvIII(+) primary human glioblastoma xenografts. Clin. Neurol. Neurosurg. 2018, 171, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Karpel-Massler, G.; Westhoff, M.A.; Kast, R.E.; Dwucet, A.; Karpel-Massler, S.; Nonnenmacher, L.; Siegelin, M.D.; Wirtz, C.R.; Debatin, K.M.; Halatsch, M.E. Simultaneous Interference with HER1/EGFR and RAC1 Signaling Drives Cytostasis and Suppression of Survivin in Human Glioma Cells in Vitro. Neurochem. Res. 2017, 42, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Shingu, T.; Holmes, L.; Henry, V.; Wang, Q.; Latha, K.; Gururaj, A.E.; Gibson, L.A.; Doucette, T.; Lang, F.F.; Rao, G.; et al. Suppression of RAF/MEK or PI3K synergizes cytotoxicity of receptor tyrosine kinase inhibitors in glioma tumor-initiating cells. J. Trans. Med. 2016, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Eimer, S.; Belaud-Rotureau, M.A.; Airiau, K.; Jeanneteau, M.; Laharanne, E.; Veron, N.; Vital, A.; Loiseau, H.; Merlio, J.P.; Belloc, F. Autophagy inhibition cooperates with erlotinib to induce glioblastoma cell death. Cancer Biol. Ther. 2011, 11, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Kesavabhotla, K.; Schlaff, C.D.; Shin, B.; Mubita, L.; Kaplan, R.; Tsiouris, A.J.; Pannullo, S.C.; Christos, P.; Lavi, E.; Scheff, R.; et al. Phase I/II study of oral erlotinib for treatment of relapsed/refractory glioblastoma multiforme and anaplastic astrocytoma. J. Exp. Ther. Oncol. 2012, 10, 71–81. [Google Scholar] [PubMed]
- Van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Giglio, P.; Hu, J.; Groves, M.; Merrell, R.; Conrad, C.; Phuphanich, S.; Puduvalli, V.K.; Loghin, M.; Paleologos, N.; et al. A phase II study of bevacizumab and erlotinib after radiation and temozolomide in MGMT unmethylated GBM patients. J. Neuro-Oncol. 2016, 126, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.L.; Molinaro, A.M.; Phillips, J.J.; Butowski, N.A.; Chang, S.M.; Perry, A.; Costello, J.F.; DeSilva, A.A.; Rabbitt, J.E.; Prados, M.D. A single-institution phase II trial of radiation, temozolomide, erlotinib, and bevacizumab for initial treatment of glioblastoma. Neuro-Oncology 2014, 16, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Qaddoumi, I.; Kocak, M.; Pai Panandiker, A.S.; Armstrong, G.T.; Wetmore, C.; Crawford, J.R.; Lin, T.; Boyett, J.M.; Kun, L.E.; Boop, F.A.; et al. Phase II Trial of Erlotinib during and after Radiotherapy in Children with Newly Diagnosed High-Grade Gliomas. Front. Oncol. 2014, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Norden, A.D.; Cloughesy, T.F.; Robins, H.I.; Lieberman, F.S.; Gilbert, M.R.; Mehta, M.P.; et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro-Oncology 2014, 16, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Halatsch, M.E.; Low, S.; Mursch, K.; Hielscher, T.; Schmidt, U.; Unterberg, A.; Vougioukas, V.I.; Feuerhake, F. Candidate genes for sensitivity and resistance of human glioblastoma multiforme cell lines to erlotinib. Laboratory investigation. J. Neurosurg. 2009, 111, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Shen, C.C.; Su, H.L.; Chen, C.J. Gefitinib induces apoptosis in human glioma cells by targeting Bad phosphorylation. J. Neuro-Oncol. 2011, 105, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Guillamo, J.S.; de Bouard, S.; Valable, S.; Marteau, L.; Leuraud, P.; Marie, Y.; Poupon, M.F.; Parienti, J.J.; Raymond, E.; Peschanski, M. Molecular mechanisms underlying effects of epidermal growth factor receptor inhibition on invasion, proliferation, and angiogenesis in experimental glioma. Clin. Cancer Res. 2009, 15, 3697–3704. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Kuan, Y.H.; Ou, Y.C.; Li, J.R.; Wu, C.C.; Pan, P.H.; Chen, W.Y.; Huang, H.Y.; Chen, C.J. Autophagy contributes to gefitinib-induced glioma cell growth inhibition. Exp. Cell Res. 2014, 327, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, Y.; Zhang, L.; Ren, X.; Huber-Keener, K.J.; Liu, X.; Zhou, L.; Liao, J.; Keihack, H.; Yan, L.; et al. MK-2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Mol. Cancer Ther. 2012, 11, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Li, J.R.; Wu, C.C.; Ou, Y.C.; Chen, W.Y.; Kuan, Y.H.; Wang, W.Y.; Chen, C.J. Valproic acid sensitizes human glioma cells to gefitinib-induced autophagy. IUBMB Life 2015, 67, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Wang, M.; Robins, H.I.; Lautenschlaeger, T.; Curran, W.J.; Brachman, D.G.; Schultz, C.J.; Choucair, A.; Dolled-Filhart, M.; Christiansen, J.; et al. RTOG 0211: A phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Doherty, L.; Gigas, D.C.; Kesari, S.; Drappatz, J.; Kim, R.; Zimmerman, J.; Ostrowsky, L.; Wen, P.Y. Pilot study of the combination of EGFR and mTOR inhibitors in recurrent malignant gliomas. Neurology 2006, 67, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Bady, P.; Kamoshima, Y.; Kouwenhoven, M.C.; Delorenzi, M.; Lambiv, W.L.; Hamou, M.F.; Matter, M.S.; Koch, A.; et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib—A phase II trial. Mol. Cancer Ther. 2011, 10, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Uhm, J.H.; Ballman, K.V.; Wu, W.; Giannini, C.; Krauss, J.C.; Buckner, J.C.; James, C.D.; Scheithauer, B.W.; Behrens, R.J.; Flynn, P.J.; et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Hu, J.; Gomez, G.G.; Taylor, T.; Villa, G.R.; Pizzo, D.; VandenBerg, S.R.; Thorne, A.H.; Chen, C.C.; Mischel, P.S.; et al. A urokinase receptor-Bim signaling axis emerges during EGFR inhibitor resistance in mutant EGFR glioblastoma. Cancer Res. 2015, 75, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Ranza, E.; Mazzini, G.; Facoetti, A.; Nano, R. In-vitro effects of the tyrosine kinase inhibitor imatinib on glioblastoma cell proliferation. J. Neuro-Oncol. 2010, 96, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Kilic, T.; Alberta, J.A.; Zdunek, P.R.; Acar, M.; Iannarelli, P.; O’Reilly, T.; Buchdunger, E.; Black, P.M.; Stiles, C.D. Intracranial inhibition of platelet-derived growth factor-mediated glioblastoma cell growth by an orally active kinase inhibitor of the 2-phenylaminopyrimidine class. Cancer Res. 2000, 60, 5143–5150. [Google Scholar] [PubMed]
- Yang, L.; Xu, Z.Y.; Chen, X.H.; Wang, K.W.; Li, G.F.; Chen, Z.L. Effect of imatinib at different concentrations on rat C6 glioma cell apoptosis and cell cycle. J. South. Med. Univ. 2010, 30, 1089–1091. [Google Scholar]
- Shingu, T.; Fujiwara, K.; Bogler, O.; Akiyama, Y.; Moritake, K.; Shinojima, N.; Tamada, Y.; Yokoyama, T.; Kondo, S. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. Int. J. Cancer 2009, 124, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Bilir, A.; Erguven, M.; Oktem, G.; Ozdemir, A.; Uslu, A.; Aktas, E.; Bonavida, B. Potentiation of cytotoxicity by combination of imatinib and chlorimipramine in glioma. Int. J. Oncol. 2008, 32, 829–839. [Google Scholar] [PubMed]
- Erguven, M.; Yazihan, N.; Aktas, E.; Sabanci, A.; Li, C.J.; Oktem, G.; Bilir, A. Carvedilol in glioma treatment alone and with imatinib in vitro. Int. J. Oncol. 2010, 36, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Sathornsumetee, S.; Rich, J.N.; Quinn, J.A.; Lagattuta, T.F.; Egorin, M.J.; Gururangan, S.; McLendon, R.; et al. Safety and pharmacokinetics of dose-intensive imatinib mesylate plus temozolomide: Phase 1 trial in adults with malignant glioma. Neuro-Oncology 2008, 10, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Yung, W.K.; Lamborn, K.R.; Dahia, P.L.; Wang, Y.; Peng, B.; Abrey, L.E.; Raizer, J.; Cloughesy, T.F.; Fink, K.; et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clin. Cancer Res. 2006, 12, 4899–4907. [Google Scholar] [CrossRef] [PubMed]
- Leis, J.F.; Stepan, D.E.; Curtin, P.T.; Ford, J.M.; Peng, B.; Schubach, S.; Druker, B.J.; Maziarz, R.T. Central nervous system failure in patients with chronic myelogenous leukemia lymphoid blast crisis and Philadelphia chromosome positive acute lymphoblastic leukemia treated with imatinib (STI-571). Leuk. Lymphoma 2004, 45, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Le Coutre, P.; Kreuzer, K.A.; Pursche, S.; Bonin, M.; Leopold, T.; Baskaynak, G.; Dorken, B.; Ehninger, G.; Ottmann, O.; Jenke, A.; et al. Pharmacokinetics and cellular uptake of imatinib and its main metabolite CGP74588. Cancer Chemother. Pharmacol. 2004, 53, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Neville, K.; Parise, R.A.; Thompson, P.; Aleksic, A.; Egorin, M.J.; Balis, F.M.; McGuffey, L.; McCully, C.; Berg, S.L.; Blaney, S.M. Plasma and cerebrospinal fluid pharmacokinetics of imatinib after administration to nonhuman primates. Clin. Cancer Res. 2004, 10, 2525–2529. [Google Scholar] [CrossRef] [PubMed]
- Razis, E.; Selviaridis, P.; Labropoulos, S.; Norris, J.L.; Zhu, M.J.; Song, D.D.; Kalebic, T.; Torrens, M.; Kalogera-Fountzila, A.; Karkavelas, G.; et al. Phase II study of neoadjuvant imatinib in glioblastoma: Evaluation of clinical and molecular effects of the treatment. Clin. Cancer Res. 2009, 15, 6258–6266. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.; Lindh, M.B.; Jarvius, M.; Puputti, M.; Nister, M.; Nupponen, N.N.; Paulus, W.; Soderberg, O.; Dresemann, G.; von Deimling, A.; et al. Prognostic but not predictive role of platelet-derived growth factor receptors in patients with recurrent glioblastoma. Int. J. Cancer 2011, 128, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Evans, I.M.; Li, N.; Sidlauskas, K.; Paliashvili, K.; Lockwood, N.; Barrett, A.; Brandner, S.; Zachary, I.C.; Frankel, P. Imatinib and Nilotinib increase glioblastoma cell invasion via Abl-independent stimulation of p130Cas and FAK signalling. Sci. Rep. 2016, 6, 27378. [Google Scholar] [CrossRef] [PubMed]
- Moeckel, S.; Meyer, K.; Leukel, P.; Heudorfer, F.; Seliger, C.; Stangl, C.; Bogdahn, U.; Proescholdt, M.; Brawanski, A.; Vollmann-Zwerenz, A.; et al. Response-predictive gene expression profiling of glioma progenitor cells in vitro. PLoS ONE 2014, 9, e108632. [Google Scholar] [CrossRef] [PubMed]
- De Bouard, S.; Herlin, P.; Christensen, J.G.; Lemoisson, E.; Gauduchon, P.; Raymond, E.; Guillamo, J.S. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro-Oncology 2007, 9, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, A.K.; Shouman, S.; El-Demerdash, E.; Elgendy, M.; Abdel-Naim, A.B. Chloroquine synergizes sunitinib cytotoxicity via modulating autophagic, apoptotic and angiogenic machineries. Chem.-Biol. Interact. 2014, 217, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Loilome, W.; Siu, I.M.; Tyler, B.; Gallia, G.L.; Riggins, G.J. Evaluation of tyrosine kinase inhibitor combinations for glioblastoma therapy. PLoS ONE 2012, 7, e44372. [Google Scholar] [CrossRef] [PubMed]
- Czabanka, M.; Bruenner, J.; Parmaksiz, G.; Broggini, T.; Topalovic, M.; Bayerl, S.H.; Auf, G.; Kremenetskaia, I.; Nieminen, M.; Jabouille, A.; et al. Combined temozolomide and sunitinib treatment leads to better tumour control but increased vascular resistance in O6-methylguanine methyltransferase-methylated gliomas. Eur. J. Cancer 2013, 49, 2243–2252. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Vredenburgh, J.J.; Coan, A.; Desjardins, A.; Peters, K.B.; Gururangan, S.; Sathornsumetee, S.; Rich, J.N.; Herndon, J.E.; Friedman, H.S. Phase I study of sunitinib and irinotecan for patients with recurrent malignant glioma. J. Neuro-Oncol. 2011, 105, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; Smith, P.; Sul, J.; Salgado, C.; Iwamoto, F.M.; Shih, J.H.; Fine, H.A. Continuous daily sunitinib for recurrent glioblastoma. J. Neuro-Oncol. 2013, 111, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Balana, C.; Gil, M.J.; Perez, P.; Reynes, G.; Gallego, O.; Ribalta, T.; Capellades, J.; Gonzalez, S.; Verger, E. Sunitinib administered prior to radiotherapy in patients with non-resectable glioblastoma: Results of a phase II study. Target. Oncol. 2014, 9, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Oberoi, R.K.; Mittapalli, R.K.; Elmquist, W.F. Pharmacokinetic assessment of efflux transport in sunitinib distribution to the brain. J. Pharmacol. Exp. Ther. 2013, 347, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Lv, H.; Mazloom, A.R.; Xu, H.; Ma’ayan, A.; Gallo, J.M. Activation of alternate prosurvival pathways accounts for acquired sunitinib resistance in U87MG glioma xenografts. J. Pharmacol. Exp. Ther. 2012, 343, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, M.; Johansson, M.; Andersson, U.; Bergh, A.; Bergenheim, A.T.; Henriksson, R. The tyrosine kinase inhibitor ZD6474 inhibits tumour growth in an intracerebral rat glioma model. Br. J. Cancer 2004, 91, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Yiin, J.J.; Hu, B.; Schornack, P.A.; Sengar, R.S.; Liu, K.W.; Feng, H.; Lieberman, F.S.; Chiou, S.H.; Sarkaria, J.N.; Wiener, E.C.; et al. ZD6474, a multitargeted inhibitor for receptor tyrosine kinases, suppresses growth of gliomas expressing an epidermal growth factor receptor mutant, EGFRvIII, in the brain. Mol. Cancer Ther. 2010, 9, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zheng, H.; Ruan, J.; Fang, W.; Li, A.; Tian, G.; Niu, X.; Luo, S.; Zhao, P. Autophagy inhibition induces enhanced proapoptotic effects of ZD6474 in glioblastoma. Br. J. Cancer 2013, 109, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, M.; Johansson, M.; Bergstrom, P.; Bergenheim, A.T.; Henriksson, R. Effects of the VEGFR inhibitor ZD6474 in combination with radiotherapy and temozolomide in an orthotopic glioma model. J. Neuro-Oncol. 2008, 88, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Drappatz, J.; Norden, A.D.; Wong, E.T.; Doherty, L.M.; Lafrankie, D.C.; Ciampa, A.; Kesari, S.; Sceppa, C.; Gerard, M.; Phan, P.; et al. Phase I study of vandetanib with radiotherapy and temozolomide for newly diagnosed glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; McNeill, K.A.; Sul, J.; Iwamoto, F.M.; Shih, J.; Fine, H.A. A phase I/II trial of vandetanib for patients with recurrent malignant glioma. Neuro-Oncology 2012, 14, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q.; Kaley, T.J.; Duda, D.G.; Schiff, D.; Lassman, A.B.; Wong, E.T.; Mikkelsen, T.; Purow, B.W.; Muzikansky, A.; Ancukiewicz, M.; et al. A Multicenter, Phase II, Randomized, Noncomparative Clinical Trial of Radiation and Temozolomide with or without Vandetanib in Newly Diagnosed Glioblastoma Patients. Clin. Cancer Res. 2015, 21, 3610–3618. [Google Scholar] [CrossRef] [PubMed]
- Pham, K.; Luo, D.; Siemann, D.W.; Law, B.K.; Reynolds, B.A.; Hothi, P.; Foltz, G.; Harrison, J.K. VEGFR inhibitors upregulate CXCR4 in VEGF receptor-expressing glioblastoma in a TGFbetaR signaling-dependent manner. Cancer Lett. 2015, 360, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Arbab, A.S. Activation of alternative pathways of angiogenesis and involvement of stem cells following anti-angiogenesis treatment in glioma. Histol. Histopathol. 2012, 27, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Delmas, C.; End, D.; Rochaix, P.; Favre, G.; Toulas, C.; Cohen-Jonathan, E. The farnesyltransferase inhibitor R115777 reduces hypoxia and matrix metalloproteinase 2 expression in human glioma xenograft. Clin. Cancer Res. 2003, 9, 6062–6068. [Google Scholar] [PubMed]
- Wang, C.C.; Liao, Y.P.; Mischel, P.S.; Iwamoto, K.S.; Cacalano, N.A.; McBride, W.H. HDJ-2 as a target for radiosensitization of glioblastoma multiforme cells by the farnesyltransferase inhibitor R115777 and the role of the p53/p21 pathway. Cancer Res. 2006, 66, 6756–6762. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Kuhn, J.; Robins, H.I.; Abrey, L.; Wen, P.; Fink, K.; Lieberman, F.S.; Mehta, M.; Chang, S.; Yung, A.; et al. Phase I trial of tipifarnib in patients with recurrent malignant glioma taking enzyme-inducing antiepileptic drugs: A North American Brain Tumor Consortium Study. J. Clin. Oncol. 2005, 23, 6647–6656. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.; Mikkelsen, T.; Lesser, G.; Grossman, S.; Ye, X.; Desideri, S.; Fisher, J.; Wright, J.; New Approaches to Brain Tumor Therapy, C.N.S.C. Phase II preradiation R115777 (tipifarnib) in newly diagnosed GBM with residual enhancing disease. Neuro-Oncology 2008, 10, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Ducassou, A.; Uro-Coste, E.; Verrelle, P.; Filleron, T.; Benouaich-Amiel, A.; Lubrano, V.; Sol, J.C.; Delisle, M.B.; Favre, G.; Ken, S.; et al. alphavbeta3 Integrin and Fibroblast growth factor receptor 1 (FGFR1): Prognostic factors in a phase I-II clinical trial associating continuous administration of Tipifarnib with radiotherapy for patients with newly diagnosed glioblastoma. Eur. J. Cancer 2013, 49, 2161–2169. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Wen, P.Y.; Robins, H.I.; Chang, S.M.; Groves, M.D.; Fink, K.L.; Junck, L.; Schiff, D.; Abrey, L.; Gilbert, M.R.; et al. Phase II trial of tipifarnib in patients with recurrent malignant glioma either receiving or not receiving enzyme-inducing antiepileptic drugs: A North American Brain Tumor Consortium Study. J. Clin. Oncol. 2006, 24, 3651–3656. [Google Scholar] [CrossRef] [PubMed]
- Feldkamp, M.M.; Lau, N.; Roncari, L.; Guha, A. Isotype-specific Ras.GTP-levels predict the efficacy of farnesyl transferase inhibitors against human astrocytomas regardless of Ras mutational status. Cancer Res. 2001, 61, 4425–4431. [Google Scholar] [PubMed]
- Glass, T.L.; Liu, T.J.; Yung, W.K. Inhibition of cell growth in human glioblastoma cell lines by farnesyltransferase inhibitor SCH66336. Neuro-Oncology 2000, 2, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Chaponis, D.; Barnes, J.W.; Dellagatta, J.L.; Kesari, S.; Fast, E.; Sauvageot, C.; Panagrahy, D.; Greene, E.R.; Ramakrishna, N.; Wen, P.Y.; et al. Lonafarnib (SCH66336) improves the activity of temozolomide and radiation for orthotopic malignant gliomas. J. Neuro-Oncol. 2011, 104, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Chen, B.; Su, C.H.; Zhao, R.; Xu, Z.X.; Sun, L.; Lee, M.H.; Yeung, S.C. Autophagy induced by farnesyltransferase inhibitors in cancer cells. Cancer Biol. Ther. 2008, 7, 1679–1684. [Google Scholar] [CrossRef] [PubMed]
- Yust-Katz, S.; Liu, D.; Yuan, Y.; Liu, V.; Kang, S.; Groves, M.; Puduvalli, V.; Levin, V.; Conrad, C.; Colman, H.; et al. Phase 1/1b study of lonafarnib and temozolomide in patients with recurrent or temozolomide refractory glioblastoma. Cancer 2013, 119, 2747–2753. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.M.; Wen, P.; Cloughesy, T.; Greenberg, H.; Schiff, D.; Conrad, C.; Fink, K.; Robins, H.I.; De Angelis, L.; Raizer, J.; et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Investig. New Drugs 2005, 23, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Uhrbom, L.; Nerio, E.; Holland, E.C. Dissecting tumor maintenance requirements using bioluminescence imaging of cell proliferation in a mouse glioma model. Nat. Med. 2004, 10, 1257–1260. [Google Scholar] [CrossRef] [PubMed]
- Pitter, K.L.; Galban, C.J.; Galban, S.; Tehrani, O.S.; Li, F.; Charles, N.; Bradbury, M.S.; Becher, O.J.; Chenevert, T.L.; Rehemtulla, A.; et al. Perifosine and CCI 779 co-operate to induce cell death and decrease proliferation in PTEN-intact and PTEN-deficient PDGF-driven murine glioblastoma. PLoS ONE 2011, 6, e14545. [Google Scholar] [CrossRef]
- Tsoli, M.; Liu, J.; Franshaw, L.; Shen, H.; Cheng, C.; Jung, M.; Joshi, S.; Ehteda, A.; Khan, A.; Montero-Carcabosso, A.; et al. Dual targeting of mitochondrial function and mTOR pathway as a therapeutic strategy for diffuse intrinsic pontine glioma. Oncotarget 2018, 9, 7541–7556. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Mammalian target of rapamycin inhibitors, temsirolimus and torin 1, attenuate stemness-associated properties and expression of mesenchymal markers promoted by phorbol-myristate-acetate and oncostatin-M in glioblastoma cells. Tumour Biol. 2017, 39, 1010428317695921. [Google Scholar] [CrossRef] [PubMed]
- Lassen, U.; Sorensen, M.; Gaziel, T.B.; Hasselbalch, B.; Poulsen, H.S. Phase II study of bevacizumab and temsirolimus combination therapy for recurrent glioblastoma multiforme. AntiCancer Res. 2013, 33, 1657–1660. [Google Scholar] [PubMed]
- Lee, E.Q.; Kuhn, J.; Lamborn, K.R.; Abrey, L.; DeAngelis, L.M.; Lieberman, F.; Robins, H.I.; Chang, S.M.; Yung, W.K.; Drappatz, J.; et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro-Oncology 2012, 14, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; van den Bent, M.J.; Taphoorn, M.J.; Steuve, J.; Brandes, A.A.; Hamou, M.F.; Wick, A.; et al. Phase II Study of Radiotherapy and Temsirolimus versus Radiochemotherapy with Temozolomide in Patients with Newly Diagnosed Glioblastoma without MGMT Promoter Hypermethylation (EORTC 26082). Clin. Cancer Res. 2016, 22, 4797–4806. [Google Scholar] [CrossRef] [PubMed]
- Josset, E.; Burckel, H.; Noel, G.; Bischoff, P. The mTOR inhibitor RAD001 potentiates autophagic cell death induced by temozolomide in a glioblastoma cell line. AntiCancer Res. 2013, 33, 1845–1851. [Google Scholar] [PubMed]
- Venkatesh, H.S.; Chaumeil, M.M.; Ward, C.S.; Haas-Kogan, D.A.; James, C.D.; Ronen, S.M. Reduced phosphocholine and hyperpolarized lactate provide magnetic resonance biomarkers of PI3K/Akt/mTOR inhibition in glioblastoma. Neuro-Oncology 2012, 14, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Olmez, I.; Brenneman, B.; Xiao, A.; Serbulea, V.; Benamar, M.; Zhang, Y.; Manigat, L.; Abbas, T.; Lee, J.; Nakano, I.; et al. Combined CDK4/6 and mTOR Inhibition Is Synergistic against Glioblastoma via Multiple Mechanisms. Clin. Cancer Res. 2017, 23, 6958–6968. [Google Scholar] [CrossRef] [PubMed]
- Minocha, M.; Khurana, V.; Qin, B.; Pal, D.; Mitra, A.K. Co-administration strategy to enhance brain accumulation of vandetanib by modulating P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp1/Abcg2) mediated efflux with m-TOR inhibitors. Int. J. Pharm. 2012, 434, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Goudar, R.K.; Shi, Q.; Hjelmeland, M.D.; Keir, S.T.; McLendon, R.E.; Wikstrand, C.J.; Reese, E.D.; Conrad, C.A.; Traxler, P.; Lane, H.A.; et al. Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol. Cancer Ther. 2005, 4, 101–112. [Google Scholar] [PubMed]
- Alonso, M.M.; Jiang, H.; Yokoyama, T.; Xu, J.; Bekele, N.B.; Lang, F.F.; Kondo, S.; Gomez-Manzano, C.; Fueyo, J. Delta-24-RGD in combination with RAD001 induces enhanced anti-glioma effect via autophagic cell death. Mol. Ther. 2008, 16, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Wendland, M.; Dipetrillo, T.A.; Corn, B.W.; Mehta, M.P. RTOG 0913: A phase 1 study of daily everolimus (RAD001) in combination with radiation therapy and temozolomide in patients with newly diagnosed glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Shih, K.C.; Shepard, G.C.; Tillinghast, G.W.; Brinker, B.T.; Spigel, D.R. Phase II study of concurrent radiation therapy, temozolomide, and bevacizumab followed by bevacizumab/everolimus as first-line treatment for patients with glioblastoma. Clin. Adv. Hematol. Oncol. 2012, 10, 240–246. [Google Scholar] [PubMed]
- Kreisl, T.N.; Lassman, A.B.; Mischel, P.S.; Rosen, N.; Scher, H.I.; Teruya-Feldstein, J.; Shaffer, D.; Lis, E.; Abrey, L.E. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM). J. Neuro-Oncol. 2009, 92, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Arcella, A.; Biagioni, F.; Antonietta Oliva, M.; Bucci, D.; Frati, A.; Esposito, V.; Cantore, G.; Giangaspero, F.; Fornai, F. Rapamycin inhibits the growth of glioblastoma. Brain Res. 2013, 1495, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Lu, K.V.; Zhu, S.; Dia, E.Q.; Vivanco, I.; Shackleford, G.M.; Cavenee, W.K.; Mellinghoff, I.K.; Cloughesy, T.F.; Sawyers, C.L.; et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006, 66, 7864–7869. [Google Scholar] [CrossRef] [PubMed]
- Iwamaru, A.; Kondo, Y.; Iwado, E.; Aoki, H.; Fujiwara, K.; Yokoyama, T.; Mills, G.B.; Kondo, S. Silencing mammalian target of rapamycin signaling by small interfering RNA enhances rapamycin-induced autophagy in malignant glioma cells. Oncogene 2007, 26, 1840–1851. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005, 65, 3336–3346. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.Z.; Long, L.M.; Ji, W.J.; Liang, Z.Q. Rapamycin induces differentiation of glioma stem/progenitor cells by activating autophagy. Chin. J. Cancer 2011, 30, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Iwado, E.; Kondo, Y.; Aoki, H.; Hayashi, Y.; Georgescu, M.M.; Sawaya, R.; Hess, K.R.; Mills, G.B.; Kawamura, H.; et al. Autophagy-inducing agents augment the antitumor effect of telerase-selve oncolytic adenovirus OBP-405 on glioblastoma cells. Gene Ther. 2008, 15, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Lee, Y.S.; Lee, M.; Lee, H.J.; Lee, B.W.; Lee, H.J.; Lee, K.E.; Jung, M.K.; et al. Radioresistant cancer cells can be conditioned to enter senescence by mTOR inhibition. Cancer Res. 2013, 73, 4267–4277. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.P.C.; Kuo, J.S.; Chiang, H.C.; Wang, H.E.; Wang, Y.S.; Huang, C.C.; Huang, Y.C.; Chi, M.S.; Mehta, M.P.; Chi, K.H. Temozolomide, sirolimus and chloroquine is a new therapeutic combination that synergizes to disrupt lysosomal function and cholesterol homeostasis in GBM cells. Oncotarget 2018, 9, 6883–6896. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Gururangan, S.; Friedman, A.H.; Herndon, J.E., 2nd; Marcello, J.; Norfleet, J.A.; McLendon, R.E.; Sampson, J.H.; et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J. Neuro-Oncol. 2010, 96, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Chheda, M.G.; Wen, P.Y.; Hochberg, F.H.; Chi, A.S.; Drappatz, J.; Eichler, A.F.; Yang, D.; Beroukhim, R.; Norden, A.D.; Gerstner, E.R.; et al. Vandetanib plus sirolimus in adults with recurrent glioblastoma: Results of a phase I and dose expansion cohort study. J. Neuro-Oncol. 2015, 121, 627–634. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Hockenbery, D.; Nunez, G.; Milliman, C.; Schreiber, R.D.; Korsmeyer, S.J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990, 348, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Tagscherer, K.E.; Fassl, A.; Campos, B.; Farhadi, M.; Kraemer, A.; Bock, B.C.; Macher-Goeppinger, S.; Radlwimmer, B.; Wiestler, O.D.; Herold-Mende, C.; et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene 2008, 27, 6646–6656. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, D.R.; Jane, E.P.; Pollack, I.F. Cucurbitacin-I inhibits Aurora kinase A, Aurora kinase B and survivin, induces defects in cell cycle progression and promotes ABT-737-induced cell death in a caspase-independent manner in malignant human glioma cells. Cancer Biol. Ther. 2015, 16, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Jane, E.P.; Premkumar, D.R.; Cavaleri, J.M.; Sutera, P.A.; Rajasekar, T.; Pollack, I.F. Dinaciclib, a Cyclin-Dependent Kinase Inhibitor Promotes Proteasomal Degradation of Mcl-1 and Enhances ABT-737-Mediated Cell Death in Malignant Human Glioma Cell Lines. J. Pharmacol. Exp. Ther. 2016, 356, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Kiprianova, I.; Remy, J.; Milosch, N.; Mohrenz, I.V.; Seifert, V.; Aigner, A.; Kogel, D. Sorafenib Sensitizes Glioma Cells to the BH3 Mimetic ABT-737 by Targeting MCL1 in a STAT3-Dependent Manner. Neoplasia 2015, 17, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Jane, E.P.; Premkumar, D.R.; DiDomenico, J.D.; Hu, B.; Cheng, S.Y.; Pollack, I.F. YM-155 potentiates the effect of ABT-737 in malignant human glioma cells via survivin and Mcl-1 downregulation in an EGFR-dependent context. Mol. Cancer Ther. 2013, 12, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Cristofanon, S.; Fulda, S. ABT-737 promotes tBid mitochondrial accumulation to enhance TRAIL-induced apoptosis in glioblastoma cells. Cell Death Dis. 2012, 3, e432. [Google Scholar] [CrossRef] [PubMed]
- Gratas, C.; Sery, Q.; Rabe, M.; Oliver, L.; Vallette, F.M. Bak and Mcl-1 are essential for Temozolomide induced cell death in human glioma. Oncotarget 2014, 5, 2428–2435. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.C.; Loh, J.K.; Li, Y.Y.; Huang, W.S.; Chou, C.H.; Cheng, J.T.; Wang, Y.T.; Lieu, A.S.; Howng, S.L.; Hong, Y.R.; et al. Bcl2L12 with a BH3-like domain in regulating apoptosis and TMZ-induced autophagy: A prospective combination of ABT-737 and TMZ for treating glioma. Int. J. Oncol. 2015, 46, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Voss, V.; Senft, C.; Lang, V.; Ronellenfitsch, M.W.; Steinbach, J.P.; Seifert, V.; Kogel, D. The pan-Bcl-2 inhibitor (-)-gossypol triggers autophagic cell death in malignant glioma. Mol. Cancer Res. 2010, 8, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Coyle, T.; Levante, S.; Shetler, M.; Winfield, J. In vitro and in vivo cytotoxicity of gossypol against central nervous system tumor cell lines. J. Neuro-Oncol. 1994, 19, 25–35. [Google Scholar] [CrossRef]
- Jang, J.H.; Surh, Y.J. Potentiation of cellular antioxidant capacity by Bcl-2: Implications for its antiapoptotic function. Biochem. Pharmacol. 2003, 66, 1371–1379. [Google Scholar] [CrossRef]
- Antonietti, P.; Linder, B.; Hehlgans, S.; Mildenberger, I.C.; Burger, M.C.; Fulda, S.; Steinbach, J.P.; Gessler, F.; Rodel, F.; Mittelbronn, M.; et al. Interference with the HSF1/HSP70/BAG3 Pathway Primes Glioma Cells to Matrix Detachment and BH3 Mimetic-Induced Apoptosis. Mol. Cancer Ther. 2017, 16, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Boiani, M.; Daniel, C.; Liu, X.; Hogarty, M.D.; Marnett, L.J. The stress protein BAG3 stabilizes Mcl-1 protein and promotes survival of cancer cells and resistance to antagonist ABT-737. J. Biol. Chem. 2013, 288, 6980–6990. [Google Scholar] [CrossRef] [PubMed]
- Festa, M.; Del Valle, L.; Khalili, K.; Franco, R.; Scognamiglio, G.; Graziano, V.; De Laurenzi, V.; Turco, M.C.; Rosati, A. BAG3 protein is overexpressed in human glioblastoma and is a potential target for therapy. Am. J. Pathol. 2011, 178, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Jarzabek, M.A.; Amberger-Murphy, V.; Callanan, J.J.; Gao, C.; Zagozdzon, A.M.; Shiels, L.; Wang, J.; Ligon, K.L.; Rich, B.E.; Dicker, P.; et al. Interrogation of gossypol therapy in glioblastoma implementing cell line and patient-derived tumour models. Br. J. Cancer 2014, 111, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Bushunow, P.; Reidenberg, M.M.; Wasenko, J.; Winfield, J.; Lorenzo, B.; Lemke, S.; Himpler, B.; Corona, R.; Coyle, T. Gossypol treatment of recurrent adult malignant gliomas. J. Neuro-Oncol. 1999, 43, 79–86. [Google Scholar] [CrossRef]
- Imanshahidi, M.; Hosseinzadeh, H. Pharmacological and therapeutic effects of Berberis vulgaris and its active constituent, berberine. Phytother. Res. 2008, 22, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Eom, K.S.; Kim, H.J.; So, H.S.; Park, R.; Kim, T.Y. Berberine-induced apoptosis in human glioblastoma T98G cells is mediated by endoplasmic reticulum stress accompanying reactive oxygen species and mitochondrial dysfunction. Biol. Pharm. Bull. 2010, 33, 1644–1649. [Google Scholar] [CrossRef] [PubMed]
- Eom, K.S.; Hong, J.M.; Youn, M.J.; So, H.S.; Park, R.; Kim, J.M.; Kim, T.Y. Berberine induces G1 arrest and apoptosis in human glioblastoma T98G cells through mitochondrial/caspases pathway. Biol. Pharm. Bull. 2008, 31, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Lai, K.C.; Yang, J.S.; Liao, C.L.; Hsia, T.C.; Chen, G.W.; Lin, J.J.; Lin, H.J.; Chiu, T.H.; Tang, Y.J.; et al. Involvement of reactive oxygen species and caspase-dependent pathway in berberine-induced cell cycle arrest and apoptosis in C6 rat glioma cells. Int. J. Oncol. 2009, 34, 1681–1690. [Google Scholar] [PubMed]
- Lin, T.H.; Kuo, H.C.; Chou, F.P.; Lu, F.J. Berberine enhances inhibition of glioma tumor cell migration and invasiveness mediated by arsenic trioxide. BMC Cancer 2008, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qi, Q.; Feng, Z.; Zhang, X.; Huang, B.; Chen, A.; Prestegarden, L.; Li, X.; Wang, J. Berberine induces autophagy in glioblastoma by targeting the AMPK/mTOR/ULK1-pathway. Oncotarget 2016, 7, 66944–66958. [Google Scholar] [CrossRef] [PubMed]
- Mariani, S.M.; Krammer, P.H. Differential regulation of TRAIL and CD95 ligand in transformed cells of the T and B lymphocyte lineage. Eur. J. Immunol. 1998, 28, 973–982. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Jane, E.P.; Premkumar, D.R.; Pollack, I.F. Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-κB signaling pathway. Mol. Cancer Ther. 2011, 10, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Fan, L.; Pang, Z.; Ren, J.; Ren, Y.; Li, J.; Chen, J.; Wen, Z.; Jiang, X. TRAIL and doxorubicin combination enhances anti-glioblastoma effect based on passive tumor targeting of liposomes. J. Control. Release 2011, 154, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Bagci-Onder, T.; Agarwal, A.; Flusberg, D.; Wanningen, S.; Sorger, P.; Shah, K. Real-time imaging of the dynamics of death receptors and therapeutics that overcome TRAIL resistance in tumors. Oncogene 2013, 32, 2818–2827. [Google Scholar] [CrossRef] [PubMed]
- Bago, J.R.; Alfonso-Pecchio, A.; Okolie, O.; Dumitru, R.; Rinkenbaugh, A.; Baldwin, A.S.; Miller, C.R.; Magness, S.T.; Hingtgen, S.D. Therapeutically engineered induced neural stem cells are tumour-homing and inhibit progression of glioblastoma. Nat. Commun. 2016, 7, 10593. [Google Scholar] [CrossRef] [PubMed]
- Hingtgen, S.; Ren, X.; Terwilliger, E.; Classon, M.; Weissleder, R.; Shah, K. Targeting multiple pathways in gliomas with stem cell and viral delivered S-TRAIL and Temozolomide. Mol. Cancer Ther. 2008, 7, 3575–3585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, X.; Hu, S.; Zheng, M.; Zhang, J.; Zhao, J.; Zhang, X.; Yan, B.; Jia, L.; Zhao, J.; et al. Identification of miRNA-7 by genome-wide analysis as a critical sensitizer for TRAIL-induced apoptosis in glioblastoma cells. Nucleic Acids Res. 2017, 45, 5930–5944. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Zhao, W.; Song, L.; Ying, W.; Guo, X. Pro-apoptotic effect of TRAIL-transfected endothelial progenitor cells on glioma cells. Oncol. Lett. 2018, 15, 5004–5012. [Google Scholar] [CrossRef] [PubMed]
- Bremer, E.; Samplonius, D.F.; van Genne, L.; Dijkstra, M.H.; Kroesen, B.J.; de Leij, L.F.; Helfrich, W. Simultaneous inhibition of epidermal growth factor receptor (EGFR) signaling and enhanced activation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor-mediated apoptosis induction by an scFv:sTRAIL fusion protein with specificity for human EGFR. J. Biol. Chem. 2005, 280, 10025–10033. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Ni, C.W.; Lin, Y.Z.; Yin, L.; Jiang, C.B.; Lv, C.T.; Le, Y.; Lang, Y.; Zhao, C.Y.; Yang, K.; et al. Targeted induction of apoptosis in glioblastoma multiforme cells by an MRP3-specific TRAIL fusion protein in vitro. Tumour Biol. 2014, 35, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Vermot-Desroches, C.; Sergent, E.; Bonnin, B.; Wijdenes, J. Characterization of monoclonal antibodies directed against trail or trail receptors. Cell. Immunol. 2005, 236, 86–91. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Banik, N.L.; Ray, S.K. Molecular Mechanisms of Taxol for Induction of Cell Death in Glioblastomas. Glioblastoma 2010, 283–298. [Google Scholar]
- George, J.; Banik, N.L.; Ray, S.K. Combination of taxol and Bcl-2 siRNA induces apoptosis in human glioblastoma cells and inhibits invasion, angiogenesis and tumour growth. J. Cell. Mol. Med. 2009, 13, 4205–4218. [Google Scholar] [CrossRef] [PubMed]
- Unterkircher, T.; Cristofanon, S.; Vellanki, S.H.; Nonnenmacher, L.; Karpel-Massler, G.; Wirtz, C.R.; Debatin, K.M.; Fulda, S. Bortezomib primes glioblastoma, including glioblastoma stem cells, for TRAIL by increasing tBid stability and mitochondrial apoptosis. Clin. Cancer Res. 2011, 17, 4019–4030. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Meyer, E.; Debatin, K.M. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene 2002, 21, 2283–2294. [Google Scholar] [CrossRef] [PubMed]
- Burton, T.R.; Henson, E.S.; Azad, M.B.; Brown, M.; Eisenstat, D.D.; Gibson, S.B. BNIP3 acts as transcriptional repressor of death receptor-5 expression and prevents TRAIL-induced cell death in gliomas. Cell Death Dis. 2013, 4, e587. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Krigsfeld, G.; Mayes, P.A.; Patel, L.; Dicker, D.T.; Patel, A.S.; Dolloff, N.G.; Messaris, E.; Scata, K.A.; Wang, W.; et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci. Transl. Med. 2013, 5, 171ra117. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.S.; He, J.; Liu, H.; Lee, H.C.; Wang, H.; Ishizawa, J.; Allen, J.E.; Andreeff, M.; Orlowski, R.Z.; Davis, R.E.; et al. The Imipridone ONC201 Induces Apoptosis and Overcomes Chemotherapy Resistance by Up-Regulation of Bim in Multiple Myeloma. Neoplasia 2017, 19, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.L.; Van den Heuvel, A.P.; Allen, J.E.; Prabhu, V.V.; Dicker, D.T.; El-Deiry, W.S. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases. Sci. Signal. 2016, 9, ra18. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Zhang, X.; Hu, C.H.; Langridge, T.; Tarapore, R.S.; Allen, J.E.; Oster, W.; Duvic, M. ONC201 selectively induces apoptosis in cutaneous T-cell lymphoma cells via activating pro-apoptotic integrated stress response and inactivating JAK/STAT and NF-κB pathways. Oncotarget 2017, 8, 61761–61776. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Kline, C.L.; Prabhu, V.V.; Wagner, J.; Ishizawa, J.; Madhukar, N.; Lev, A.; Baumeister, M.; Zhou, L.; Lulla, A.; et al. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget 2016, 7, 74380–74392. [Google Scholar] [CrossRef] [PubMed]
- Karpel-Massler, G.; Ba, M.; Shu, C.; Halatsch, M.E.; Westhoff, M.A.; Bruce, J.N.; Canoll, P.; Siegelin, M.D. TIC10/ONC201 synergizes with Bcl-2/Bcl-xL inhibition in glioblastoma by suppression of Mcl-1 and its binding partners in vitro and in vivo. Oncotarget 2015, 6, 36456–36471. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trejo-Solís, C.; Serrano-Garcia, N.; Escamilla-Ramírez, Á.; Castillo-Rodríguez, R.A.; Jimenez-Farfan, D.; Palencia, G.; Calvillo, M.; Alvarez-Lemus, M.A.; Flores-Nájera, A.; Cruz-Salgado, A.; et al. Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma. Int. J. Mol. Sci. 2018, 19, 3773. https://doi.org/10.3390/ijms19123773
Trejo-Solís C, Serrano-Garcia N, Escamilla-Ramírez Á, Castillo-Rodríguez RA, Jimenez-Farfan D, Palencia G, Calvillo M, Alvarez-Lemus MA, Flores-Nájera A, Cruz-Salgado A, et al. Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma. International Journal of Molecular Sciences. 2018; 19(12):3773. https://doi.org/10.3390/ijms19123773
Chicago/Turabian StyleTrejo-Solís, Cristina, Norma Serrano-Garcia, Ángel Escamilla-Ramírez, Rosa A. Castillo-Rodríguez, Dolores Jimenez-Farfan, Guadalupe Palencia, Minerva Calvillo, Mayra A. Alvarez-Lemus, Athenea Flores-Nájera, Arturo Cruz-Salgado, and et al. 2018. "Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma" International Journal of Molecular Sciences 19, no. 12: 3773. https://doi.org/10.3390/ijms19123773
APA StyleTrejo-Solís, C., Serrano-Garcia, N., Escamilla-Ramírez, Á., Castillo-Rodríguez, R. A., Jimenez-Farfan, D., Palencia, G., Calvillo, M., Alvarez-Lemus, M. A., Flores-Nájera, A., Cruz-Salgado, A., & Sotelo, J. (2018). Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma. International Journal of Molecular Sciences, 19(12), 3773. https://doi.org/10.3390/ijms19123773