Intranasal Perillyl Alcohol for Glioma Therapy: Molecular Mechanisms and Clinical Development




Abstract
1. Introduction
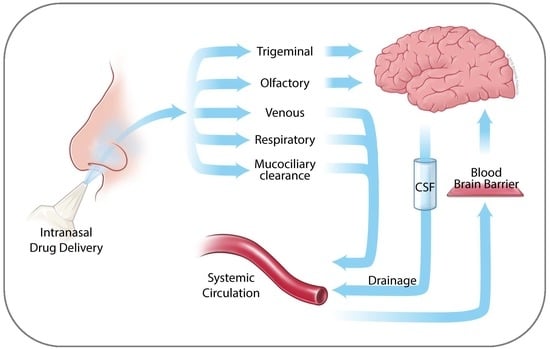
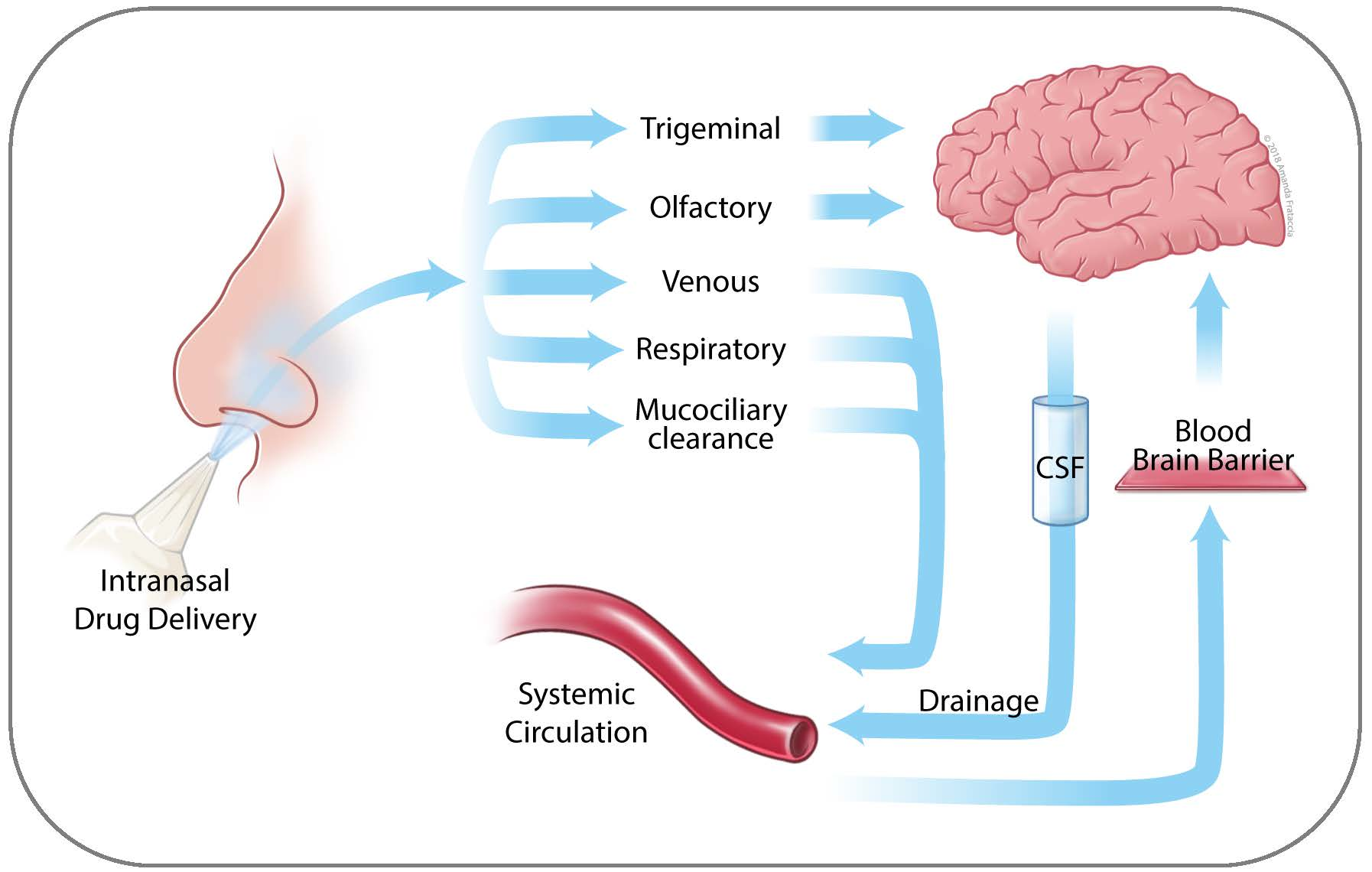
2. Anatomy of the Nasal Cavity
3. Nose to Brain Transport
4. Perillyl Alcohol
4.1. Molecular Mechanisms of POH Function
4.2. Role of Ras Pathway in POH Function
4.3. Clinical Testing of Oral POH
4.4. Intranasal POH
4.5. Intranasal POH in Combination
4.6. Exploration of Novel Therapeutic Applications of POH
4.6.1. POH as a Vehicle for Binary Drug Delivery
4.6.2. POH as a Covalent Modification of Established Drugs
5. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BBB | Blood-brain barrier |
| CHOP | CCAAT/Enhancer-binding protein homologous protein |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| ER | Endoplasmic reticulum |
| FPTase | Farnesyl-protein transferase |
| GB | Glioblastoma (multiforme) (see ref. [150] for details) |
| GGPTase | Geranylgeranyl-protein transferase |
| GRP78 | Glucose-regulated protein of 78 kDa |
| IC50 | Inhibitory concentration at 50% effect |
| IUPAC | International Union of Pure and Applied Chemistry |
| IN | Intranasal |
| JNK | c-Jun N-terminal kinase |
| KD | Ketogenic diet |
| MRI | Magnetic resonance imaging |
| MTD | Maximum tolerated dose |
| Na/K-ATPase | Sodium/potassium-adenosine triphosphatase |
| NEO100 | Clinical-grade, highly pure perillyl alcohol |
| POH | Perillyl alcohol |
| RT | Radiotherapy |
| TMZ | Temozolomide |
References
- American Cancer Society. Cancer Facts & Figures 2016; American Cancer Society: Atlanta, GA, USA, 2016. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Nayak, L.; Lee, E.Q.; Wen, P.Y. Epidemiology of brain metastases. Curr. Oncol. Rep. 2012, 14, 48–54. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Swanson, K.D.; Lok, E.; Wong, E.T. An Overview of Alternating Electric Fields Therapy (NovoTTF Therapy) for the Treatment of Malignant Glioma. Curr. Neurol. Neurosci. Rep. 2016, 16, 8. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Bernard-Arnoux, F.; Lamure, M.; Ducray, F.; Aulagner, G.; Honnorat, J.; Armoiry, X. The cost-effectiveness of tumor-treating fields therapy in patients with newly diagnosed glioblastoma. Neuro Oncol. 2016, 18, 1129–1136. [Google Scholar] [CrossRef]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neurooncol. 2012, 107, 359–364. [Google Scholar] [CrossRef]
- SEER, Surveillance, Epidemiology, and End Results (SEER) Program. Available online: www.seer.cancer.gov (accessed on 22 June 2018).
- Hofman, F.M.; Stathopoulos, A.; Kruse, C.A.; Chen, T.C.; Schijns, V.E. Immunotherapy of malignant gliomas using autologous and allogeneic tissue cells. Anticancer Agents Med. Chem. 2010, 10, 462–470. [Google Scholar] [CrossRef]
- Jackson, C.M.; Lim, M.; Drake, C.G. Immunotherapy for brain cancer: Recent progress and future promise. Clin. Cancer Res. 2014, 20, 3651–3659. [Google Scholar] [CrossRef]
- Tan, A.C.; Heimberger, A.B.; Khasraw, M. Immune Checkpoint Inhibitors in Gliomas. Curr. Oncol. Rep. 2017, 19, 23. [Google Scholar] [CrossRef]
- Kamath, S.D.; Kumthekar, P.U. Immune Checkpoint Inhibitors for the Treatment of Central Nervous System (CNS) Metastatic Disease. Front. Oncol. 2018, 8, 414. [Google Scholar] [CrossRef]
- Platten, M.; Bunse, L.; Riehl, D.; Bunse, T.; Ochs, K.; Wick, W. Vaccine Strategies in Gliomas. Curr. Treat. Opt. Neurol. 2018, 20, 11. [Google Scholar] [CrossRef]
- Schijns, V.; Pretto, C.; Strik, A.M.; Gloudemans-Rijkers, R.; Deviller, L.; Pierre, D.; Chung, J.; Dandekar, M.; Carrillo, J.A.; Kong, X.T.; et al. Therapeutic Immunization against Glioblastoma. Int. J. Mol. Sci. 2018, 19, 2540. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro Oncol. 2014, 16, 274–279. [Google Scholar] [CrossRef]
- Kong, Z.; Wang, Y.; Ma, W. Vaccination in the immunotherapy of glioblastoma. Hum. Vaccin Immunother. 2018, 14, 255–268. [Google Scholar] [CrossRef]
- Tivnan, A.; Heilinger, T.; Lavelle, E.C.; Prehn, J.H. Advances in immunotherapy for the treatment of glioblastoma. J. Neurooncol. 2017, 131, 1–9. [Google Scholar] [CrossRef]
- Weller, M.; Kaulich, K.; Hentschel, B.; Felsberg, J.; Gramatzki, D.; Pietsch, T.; Simon, M.; Westphal, M.; Schackert, G.; Tonn, J.C.; et al. Assessment and prognostic significance of the epidermal growth factor receptor vIII mutation in glioblastoma patients treated with concurrent and adjuvant temozolomide radiochemotherapy. Int. J. Cancer 2014, 134, 2437–2447. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Zhang, X.; Sharma, P.K.; Peter Goedegebuure, S.; Gillanders, W.E. Personalized cancer vaccines: Targeting the cancer mutanome. Vaccine 2017, 35, 1094–1100. [Google Scholar] [CrossRef]
- Tureci, O.; Vormehr, M.; Diken, M.; Kreiter, S.; Huber, C.; Sahin, U. Targeting the Heterogeneity of Cancer with Individualized Neoepitope Vaccines. Clin. Cancer Res. 2016, 22, 1885–1896. [Google Scholar] [CrossRef]
- Bota, D.A.; Alexandru-Abrams, D.; Pretto, C.; Hofman, F.M.; Chen, T.C.; Fu, B.; Carrillo, J.A.; Schijns, V.E.; Stathopoulos, A. Use of ERC-1671 Vaccine in a Patient with Recurrent Glioblastoma Multiforme after Progression during Bevacizumab Therapy: First Published Report. Perm. J. 2015, 19, 41–46. [Google Scholar] [CrossRef]
- Bota, D.A.; Chung, J.; Dandekar, M.; Carrillo, J.A.; Kong, X.T.; Fu, B.D.; Hsu, F.P.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C.; et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: Interim results and correlations with CD4(+) T-lymphocyte counts. CNS Oncol. 2018, 7, CNS22. [Google Scholar] [CrossRef]
- Schijns, V.E.; Pretto, C.; Devillers, L.; Pierre, D.; Hofman, F.M.; Chen, T.C.; Mespouille, P.; Hantos, P.; Glorieux, P.; Bota, D.A.; et al. First clinical results of a personalized immunotherapeutic vaccine against recurrent, incompletely resected, treatment-resistant glioblastoma multiforme (GBM) tumors, based on combined allo- and auto-immune tumor reactivity. Vaccine 2015, 33, 2690–2696. [Google Scholar] [CrossRef]
- Martinez-Garcia, M.; Alvarez-Linera, J.; Carrato, C.; Ley, L.; Luque, R.; Maldonado, X.; Martinez-Aguillo, M.; Navarro, L.M.; Vaz-Salgado, M.A.; Gil-Gil, M. SEOM clinical guidelines for diagnosis and treatment of glioblastoma (2017). Clin. Transl. Oncol. 2018, 20, 22–28. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Hopkins, K.; Tonn, J.C.; Stupp, R.; Falini, A.; Cohen-Jonathan-Moyal, E.; Frappaz, D.; Henriksson, R.; Balana, C.; et al. European Association for Neuro-Oncology Task Force on Malignant, G. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014, 15, e395–e403. [Google Scholar] [CrossRef]
- Muldoon, L.L.; Soussain, C.; Jahnke, K.; Johanson, C.; Siegal, T.; Smith, Q.R.; Hall, W.A.; Hynynen, K.; Senter, P.D.; Peereboom, D.M.; et al. Chemotherapy delivery issues in central nervous system malignancy: A reality check. J. Clin. Oncol. 2007, 25, 2295–2305. [Google Scholar] [CrossRef]
- Lockman, P.R.; Mittapalli, R.K.; Taskar, K.S.; Rudraraju, V.; Gril, B.; Bohn, K.A.; Adkins, C.E.; Roberts, A.; Thorsheim, H.R.; Gaasch, J.A.; et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res. 2010, 16, 5664–5678. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Zhang, R.D.; Price, J.E.; Fujimaki, T.; Bucana, C.D.; Fidler, I.J. Differential permeability of the blood-brain barrier in experimental brain metastases produced by human neoplasms implanted into nude mice. Am. J. Pathol. 1992, 141, 1115–1124. [Google Scholar]
- Joshi, S.; Meyers, P.M.; Ornstein, E. Intracarotid delivery of drugs: The potential and the pitfalls. Anesthesiology 2008, 109, 543–564. [Google Scholar] [CrossRef]
- Chen, T.C.; Da Fonseca, C.O.; Schonthal, A.H. Perillyl Alcohol and Its Drug-Conjugated Derivatives as Potential Novel Methods of Treating Brain Metastases. Int. J. Mol. Sci. 2016, 17, 1463. [Google Scholar] [CrossRef]
- Goodwin, J.T.; Clark, D.E. In silico predictions of blood-brain barrier penetration: Considerations to “keep in mind”. J. Pharmacol. Exp. Ther. 2005, 315, 477–483. [Google Scholar] [CrossRef]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef]
- Khan, A.R.; Liu, M.; Khan, M.W.; Zhai, G. Progress in brain targeting drug delivery system by nasal route. J. Control. Release 2017, 268, 364–389. [Google Scholar] [CrossRef]
- Dahl, R.; Mygind, N. Anatomy, physiology and function of the nasal cavities in health and disease. Adv. Drug Deliv. Rev. 1998, 29, 3–12. [Google Scholar]
- Harkema, J.R.; Carey, S.A.; Wagner, J.G. The nose revisited: A brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicol. Pathol. 2006, 34, 252–269. [Google Scholar] [CrossRef]
- Schipper, N.G.; Verhoef, J.C.; Merkus, F.W. The nasal mucociliary clearance: Relevance to nasal drug delivery. Pharm. Res. 1991, 8, 807–814. [Google Scholar] [CrossRef]
- Ganger, S.; Schindowski, K. Tailoring Formulations for Intranasal Nose-to-Brain Delivery: A Review on Architecture, Physico-Chemical Characteristics and Mucociliary Clearance of the Nasal Olfactory Mucosa. Pharmaceutics 2018, 10, 116. [Google Scholar] [CrossRef]
- Kimbell, J.S.; Segal, R.A.; Asgharian, B.; Wong, B.A.; Schroeter, J.D.; Southall, J.P.; Dickens, C.J.; Brace, G.; Miller, F.J. Characterization of deposition from nasal spray devices using a computational fluid dynamics model of the human nasal passages. J. Aerosol. Med. 2007, 20, 59–74. [Google Scholar] [CrossRef]
- Cauna, N. The fine structure of the arteriovenous anastomosis and its nerve supply in the human nasal respiratory mucosa. Anat. Rec. 1970, 168, 9–21. [Google Scholar] [CrossRef]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H., 2nd. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef]
- Djupesland, P.G.; Messina, J.C.; Mahmoud, R.A. The nasal approach to delivering treatment for brain diseases: An anatomic, physiologic, and delivery technology overview. Ther. Deliv. 2014, 5, 709–733. [Google Scholar] [CrossRef]
- Gizurarson, S. Anatomical and histological factors affecting intranasal drug and vaccine delivery. Curr. Drug Deliv. 2012, 9, 566–582. [Google Scholar] [CrossRef]
- Pardeshi, C.V.; Belgamwar, V.S. Direct nose to brain drug delivery via integrated nerve pathways bypassing the blood-brain barrier: An excellent platform for brain targeting. Expert Opin. Drug Deliv. 2013, 10, 957–972. [Google Scholar] [CrossRef]
- Samaridou, E.; Alonso, M.J. Nose-to-brain peptide delivery—The potential of nanotechnology. Bioorg. Med. Chem. 2018, 26, 2888–2905. [Google Scholar] [CrossRef]
- Turner, J.A.; Sears, J.M.; Loeser, J.D. Programmable intrathecal opioid delivery systems for chronic noncancer pain: A systematic review of effectiveness and complications. Clin. J. Pain 2007, 23, 180–195. [Google Scholar] [CrossRef]
- Joshi, S.; Ergin, A.; Wang, M.; Reif, R.; Zhang, J.; Bruce, J.N.; Bigio, I.J. Inconsistent blood brain barrier disruption by intraarterial mannitol in rabbits: Implications for chemotherapy. J. Neurooncol. 2011, 104, 11–19. [Google Scholar] [CrossRef]
- Bourganis, V.; Kammona, O.; Alexopoulos, A.; Kiparissides, C. Recent advances in carrier mediated nose-to-brain delivery of pharmaceutics. Eur. J. Pharm. Biopharm. 2018, 128, 337–362. [Google Scholar] [CrossRef]
- Pires, P.C.; Santos, A.O. Nanosystems in nose-to-brain drug delivery: A review of non-clinical brain targeting studies. J. Control. Release 2018, 270, 89–100. [Google Scholar] [CrossRef]
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Chougule, M.B.; Shoyele, S.A.; Alexander, A. Nose-to-brain drug delivery: An update on clinical challenges and progress towards approval of anti-Alzheimer drugs. J. Control. Release 2018, 281, 139–177. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Mills, J.J.; Chari, R.S.; Boyer, I.J.; Gould, M.N.; Jirtle, R.L. Induction of apoptosis in liver tumors by the monoterpene perillyl alcohol. Cancer Res. 1995, 55, 979–983. [Google Scholar]
- Stayrook, K.R.; McKinzie, J.H.; Barbhaiya, L.H.; Crowell, P.L. Effects of the antitumor agent perillyl alcohol on H-Ras vs. K-Ras farnesylation and signal transduction in pancreatic cells. Anticancer Res. 1998, 18, 823–828. [Google Scholar]
- Crowell, P.L.; Elson, C.E. Isoprenoids, Health and Disease. In Nutraceuticals and Functional Foods; Wildman, R.E.C., Ed.; CRC Press: Boca Raton, FL, USA, 2001; pp. 31–54. [Google Scholar]
- Hudes, G.R.; Szarka, C.E.; Adams, A.; Ranganathan, S.; McCauley, R.A.; Weiner, L.M.; Langer, C.J.; Litwin, S.; Yeslow, G.; Halberr, T.; et al. Phase I pharmacokinetic trial of perillyl alcohol (NSC 641066) in patients with refractory solid malignancies. Clin. Cancer Res. 2000, 6, 3071–3080. [Google Scholar]
- Ripple, G.H.; Gould, M.N.; Arzoomanian, R.Z.; Alberti, D.; Feierabend, C.; Simon, K.; Binger, K.; Tutsch, K.D.; Pomplun, M.; Wahamaki, A.; et al. Phase I clinical and pharmacokinetic study of perillyl alcohol administered four times a day. Clin. Cancer Res. 2000, 6, 390–396. [Google Scholar]
- Zhang, Z.; Chen, H.; Chan, K.K.; Budd, T.; Ganapathi, R. Gas chromatographic-mass spectrometric analysis of perillyl alcohol and metabolites in plasma. J. Chromatogr. B Biomed. Sci. Appl. 1999, 728, 85–95. [Google Scholar] [CrossRef]
- Koyama, M.; Sowa, Y.; Hitomi, T.; Iizumi, Y.; Watanabe, M.; Taniguchi, T.; Ichikawa, M.; Sakai, T. Perillyl alcohol causes G1 arrest through p15(INK4b) and p21(WAF1/Cip1) induction. Oncol. Rep. 2013, 29, 779–784. [Google Scholar] [CrossRef]
- Wiseman, D.A.; Werner, S.R.; Crowell, P.L. Cell cycle arrest by the isoprenoids perillyl alcohol, geraniol, and farnesol is mediated by p21(Cip1) and p27(Kip1) in human pancreatic adenocarcinoma cells. J. Pharmacol. Exp. Ther. 2007, 320, 1163–1170. [Google Scholar] [CrossRef]
- Yuri, T.; Danbara, N.; Tsujita-Kyutoku, M.; Kiyozuka, Y.; Senzaki, H.; Shikata, N.; Kanzaki, H.; Tsubura, A. Perillyl alcohol inhibits human breast cancer cell growth in vitro and in vivo. Breast Cancer Res. Treat. 2004, 84, 251–260. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Satomi, Y.; Ellis, M.J.; Haag, J.D.; Shi, W.; Sattler, C.A.; Gould, M.N. Activation of the transforming growth factor beta signaling pathway and induction of cytostasis and apoptosis in mammary carcinomas treated with the anticancer agent perillyl alcohol. Cancer Res. 1999, 59, 1917–1928. [Google Scholar]
- Bardon, S.; Picard, K.; Martel, P. Monoterpenes inhibit cell growth, cell cycle progression, and cyclin D1 gene expression in human breast cancer cell lines. Nutr. Cancer 1998, 32, 1–7. [Google Scholar] [CrossRef]
- Cerda, S.R.; Wilkinson, J.T.; Thorgeirsdottir, S.; Broitman, S.A. R-(+)-perillyl alcohol-induced cell cycle changes, altered actin cytoskeleton, and decreased ras and p34(cdc2) expression in colonic adenocarcinoma SW480 cells. J. Nutr. Biochem. 1999, 10, 19–30. [Google Scholar] [CrossRef]
- Fernandes, J.; da Fonseca, C.O.; Teixeira, A.; Gattass, C.R. Perillyl alcohol induces apoptosis in human glioblastoma multiforme cells. Oncol. Rep. 2005, 13, 943–947. [Google Scholar] [CrossRef]
- Shi, W.; Gould, M.N. Induction of cytostasis in mammary carcinoma cells treated with the anticancer agent perillyl alcohol. Carcinogenesis 2002, 23, 131–142. [Google Scholar] [CrossRef]
- Yeruva, L.; Pierre, K.J.; Elegbede, A.; Wang, R.C.; Carper, S.W. Perillyl alcohol and perillic acid induced cell cycle arrest and apoptosis in non small cell lung cancer cells. Cancer Lett. 2007, 257, 216–226. [Google Scholar] [CrossRef]
- Elegbede, J.A.; Flores, R.; Wang, R.C. Perillyl alcohol and perillaldehyde induced cell cycle arrest and cell death in BroTo and A549 cells cultured in vitro. Life Sci. 2003, 73, 2831–2840. [Google Scholar] [CrossRef]
- Satomi, Y.; Miyamoto, S.; Gould, M.N. Induction of AP-1 activity by perillyl alcohol in breast cancer cells. Carcinogenesis 1999, 20, 1957–1961. [Google Scholar] [CrossRef]
- Garcia, D.G.; de Castro-Faria-Neto, H.C.; da Silva, C.I.; de Souza e Souza, K.F.; Goncalves-de-Albuquerque, C.F.; Silva, A.R.; de Amorim, L.M.; Freire, A.S.; Santelli, R.E.; Diniz, L.P.; et al. Na/K-ATPase as a target for anticancer drugs: Studies with perillyl alcohol. Mol. Cancer 2015, 14, 105. [Google Scholar] [CrossRef]
- Cho, H.Y.; Wang, W.; Jhaveri, N.; Torres, S.; Tseng, J.; Leong, M.N.; Lee, D.J.; Goldkorn, A.; Xu, T.; Petasis, N.A.; et al. Perillyl alcohol for the treatment of temozolomide-resistant gliomas. Mol. Cancer Ther. 2012, 11, 2462–2472. [Google Scholar] [CrossRef]
- Sundin, T.; Peffley, D.M.; Gauthier, D.; Hentosh, P. The isoprenoid perillyl alcohol inhibits telomerase activity in prostate cancer cells. Biochimie 2012, 94, 2639–2648. [Google Scholar] [CrossRef]
- Sundin, T.; Peffley, D.M.; Hentosh, P. Disruption of an hTERT-mTOR-RAPTOR protein complex by a phytochemical perillyl alcohol and rapamycin. Mol. Cell. Biochem. 2013, 375, 97–104. [Google Scholar] [CrossRef]
- Peffley, D.M.; Sharma, C.; Hentosh, P.; Buechler, R.D. Perillyl alcohol and genistein differentially regulate PKB/Akt and 4E-BP1 phosphorylation as well as eIF4E/eIF4G interactions in human tumor cells. Arch. Biochem. Biophys. 2007, 465, 266–273. [Google Scholar] [CrossRef]
- Sundin, T.; Peffley, D.; Hentosh, P. eIF4E-Overexpression imparts perillyl alcohol and rapamycin-mediated regulation of telomerase reverse transcriptase. Exp. Cell Res. 2013, 319, 2103–2112. [Google Scholar] [CrossRef]
- Garcia, D.G.; Amorim, L.M.; de Castro Faria, M.V.; Freire, A.S.; Santelli, R.E.; Da Fonseca, C.O.; Quirico-Santos, T.; Burth, P. The anticancer drug perillyl alcohol is a Na/K-ATPase inhibitor. Mol. Cell. Biochem. 2010, 345, 29–34. [Google Scholar] [CrossRef]
- Ma, Y.; Bian, J.; Zhang, F. Inhibition of perillyl alcohol on cell invasion and migration depends on the Notch signaling pathway in hepatoma cells. Mol. Cell. Biochem. 2016, 411, 307–315. [Google Scholar] [CrossRef]
- Berchtold, C.M.; Chen, K.S.; Miyamoto, S.; Gould, M.N. Perillyl alcohol inhibits a calcium-dependent constitutive nuclear factor-kappaB pathway. Cancer Res. 2005, 65, 8558–8566. [Google Scholar] [CrossRef]
- Crowell, P.L.; Siar Ayoubi, A.; Burke, Y.D. Antitumorigenic effects of limonene and perillyl alcohol against pancreatic and breast cancer. Adv. Exp. Med. Biol. 1996, 401, 131–136. [Google Scholar]
- Jirtle, R.L.; Haag, J.D.; Ariazi, E.A.; Gould, M.N. Increased mannose 6-phosphate/insulin-like growth factor II receptor and transforming growth factor beta 1 levels during monoterpene-induced regression of mammary tumors. Cancer Res. 1993, 53, 3849–3852. [Google Scholar]
- Haag, J.D.; Gould, M.N. Mammary carcinoma regression induced by perillyl alcohol, a hydroxylated analog of limonene. Cancer Chemother. Pharmacol. 1994, 34, 477–483. [Google Scholar] [CrossRef]
- Stark, M.J.; Burke, Y.D.; McKinzie, J.H.; Ayoubi, A.S.; Crowell, P.L. Chemotherapy of pancreatic cancer with the monoterpene perillyl alcohol. Cancer Lett. 1995, 96, 15–21. [Google Scholar] [CrossRef]
- Ma, J.; Li, J.; Wang, K.S.; Mi, C.; Piao, L.X.; Xu, G.H.; Li, X.; Lee, J.J.; Jin, X. Perillyl alcohol efficiently scavenges activity of cellular ROS and inhibits the translational expression of hypoxia-inducible factor-1alpha via mTOR/4E-BP1 signaling pathways. Int. Immunopharmacol. 2016, 39, 1–9. [Google Scholar] [CrossRef]
- Teruszkin Balassiano, I.; Alves de Paulo, S.; Henriques Silva, N.; Curie Cabral, M.; Gibaldi, D.; Bozza, M.; Orlando da Fonseca, C.; Da Gloria da Costa Carvalho, M. Effects of perillyl alcohol in glial C6 cell line in vitro and anti-metastatic activity in chorioallantoic membrane model. Int. J. Mol. Med. 2002, 10, 785–788. [Google Scholar] [CrossRef]
- Afshordel, S.; Kern, B.; Clasohm, J.; Konig, H.; Priester, M.; Weissenberger, J.; Kogel, D.; Eckert, G.P. Lovastatin and perillyl alcohol inhibit glioma cell invasion, migration, and proliferation—Impact of Ras-/Rho-prenylation. Pharmacol. Res. 2015, 91, 69–77. [Google Scholar] [CrossRef]
- Mehta, S.; Lo Cascio, C. Developmentally regulated signaling pathways in glioma invasion. Cell. Mol. Life Sci. 2018, 75, 385–402. [Google Scholar] [CrossRef]
- Crowell, P.L.; Chang, R.R.; Ren, Z.B.; Elson, C.E.; Gould, M.N. Selective inhibition of isoprenylation of 21-26-kDa proteins by the anticarcinogen d-limonene and its metabolites. J. Biol. Chem. 1991, 266, 17679–17685. [Google Scholar]
- Crowell, P.L.; Ren, Z.; Lin, S.; Vedejs, E.; Gould, M.N. Structure-activity relationships among monoterpene inhibitors of protein isoprenylation and cell proliferation. Biochem. Pharmacol. 1994, 47, 1405–1415. [Google Scholar] [CrossRef]
- Gelb, M.H.; Tamanoi, F.; Yokoyama, K.; Ghomashchi, F.; Esson, K.; Gould, M.N. The inhibition of protein prenyltransferases by oxygenated metabolites of limonene and perillyl alcohol. Cancer Lett. 1995, 91, 169–175. [Google Scholar] [CrossRef]
- Ren, Z.; Elson, C.E.; Gould, M.N. Inhibition of type I and type II geranylgeranyl-protein transferases by the monoterpene perillyl alcohol in NIH3T3 cells. Biochem. Pharmacol. 1997, 54, 113–120. [Google Scholar] [CrossRef]
- Bassi, A.M.; Romano, P.; Mangini, S.; Colombo, M.; Canepa, C.; Nanni, G.; Casu, A. Protein and m-RNA expression of farnesyl-transferases, RhoA and RhoB in rat liver hepatocytes: Action of perillyl alcohol and vitamin A in vivo. J. Biomed. Sci. 2005, 12, 457–466. [Google Scholar] [CrossRef]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef]
- da Fonseca, C.O.; Linden, R.; Futuro, D.; Gattass, C.R.; Quirico-Santos, T. Ras pathway activation in gliomas: A strategic target for intranasal administration of perillyl alcohol. Arch. Immunol. Ther. Exp. 2008, 56, 267–276. [Google Scholar] [CrossRef]
- Gould, M.N. Prevention and therapy of mammary cancer by monoterpenes. J. Cell. Biochem. Suppl 1995, 22, 139–144. [Google Scholar] [CrossRef]
- Hardcastle, I.R.; Rowlands, M.G.; Barber, A.M.; Grimshaw, R.M.; Mohan, M.K.; Nutley, B.P.; Jarman, M. Inhibition of protein prenylation by metabolites of limonene. Biochem. Pharmacol. 1999, 57, 801–809. [Google Scholar] [CrossRef]
- Holstein, S.A.; Hohl, R.J. Monoterpene regulation of Ras and Ras-related protein expression. J. Lipid Res. 2003, 44, 1209–1215. [Google Scholar] [CrossRef]
- Ruch, R.J.; Sigler, K. Growth inhibition of rat liver epithelial tumor cells by monoterpenes does not involve Ras plasma membrane association. Carcinogenesis 1994, 15, 787–789. [Google Scholar] [CrossRef]
- Cerda, S.R.; Wilkinson, J.T.; Branch, S.K.; Broitman, S.A. Enhancement of sterol synthesis by the monoterpene perillyl alcohol is unaffected by competitive 3-hydroxy-3-methylglutaryl-CoA reductase inhibition. Lipids 1999, 34, 605–615. [Google Scholar] [CrossRef]
- Hohl, R.J.; Lewis, K. Differential effects of monoterpenes and lovastatin on RAS processing. J. Biol. Chem. 1995, 270, 17508–17512. [Google Scholar] [CrossRef]
- Karlson, J.; Borg-Karlson, A.K.; Unelius, R.; Shoshan, M.C.; Wilking, N.; Ringborg, U.; Linder, S. Inhibition of tumor cell growth by monoterpenes in vitro: Evidence of a Ras-independent mechanism of action. Anticancer Drugs 1996, 7, 422–429. [Google Scholar] [CrossRef]
- Clark, S.S.; Zhong, L.; Filiault, D.; Perman, S.; Ren, Z.; Gould, M.; Yang, X. Anti-leukemia effect of perillyl alcohol in Bcr/Abl-transformed cells indirectly inhibits signaling through Mek in a Ras- and Raf-independent fashion. Clin. Cancer Res. 2003, 9, 4494–4504. [Google Scholar]
- Lluria-Prevatt, M.; Morreale, J.; Gregus, J.; Alberts, D.S.; Kaper, F.; Giaccia, A.; Powell, M.B. Effects of perillyl alcohol on melanoma in the TPras mouse model. Cancer Epidemiol. Biomark. Prev. 2002, 11, 573–579. [Google Scholar]
- Chaudhary, S.C.; Alam, M.S.; Siddiqui, M.S.; Athar, M. Perillyl alcohol attenuates Ras-ERK signaling to inhibit murine skin inflammation and tumorigenesis. Chem. Biol. Interact. 2009, 179, 145–153. [Google Scholar] [CrossRef]
- Hohl, R.J. Monoterpenes as regulators of malignant cell proliferation. Adv. Exp. Med. Biol. 1996, 401, 137–146. [Google Scholar]
- Shojaei, S.; Kiumarsi, A.; Moghadam, A.R.; Alizadeh, J.; Marzban, H.; Ghavami, S. Perillyl Alcohol (Monoterpene Alcohol), Limonene. Enzymes 2014, 36, 7–32. [Google Scholar]
- Chen, T.C.; Fonseca, C.O.; Schönthal, A.H. Preclinical development and clinical use of perillyl alcohol for chemoprevention and cancer therapy. Am. J. Cancer Res. 2015, 5, 1580–1593. [Google Scholar]
- Murren, J.R.; Pizzorno, G.; DiStasio, S.A.; McKeon, A.; Peccerillo, K.; Gollerkari, A.; McMurray, W.; Burtness, B.A.; Rutherford, T.; Li, X.; et al. Phase I study of perillyl alcohol in patients with refractory malignancies. Cancer Biol. Ther. 2002, 1, 130–135. [Google Scholar] [CrossRef]
- Ripple, G.H.; Gould, M.N.; Stewart, J.A.; Tutsch, K.D.; Arzoomanian, R.Z.; Alberti, D.; Feierabend, C.; Pomplun, M.; Wilding, G.; Bailey, H.H. Phase I clinical trial of perillyl alcohol administered daily. Clin. Cancer Res. 1998, 4, 1159–1164. [Google Scholar]
- Azzoli, C.G.; Miller, V.A.; Ng, K.K.; Krug, L.M.; Spriggs, D.R.; Tong, W.P.; Riedel, E.R.; Kris, M.G. A phase I trial of perillyl alcohol in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2003, 51, 493–498. [Google Scholar]
- Bailey, H.H.; Attia, S.; Love, R.R.; Fass, T.; Chappell, R.; Tutsch, K.; Harris, L.; Jumonville, A.; Hansen, R.; Shapiro, G.R.; et al. Phase II trial of daily oral perillyl alcohol (NSC 641066) in treatment-refractory metastatic breast cancer. Cancer Chemother. Pharmacol. 2008, 62, 149–157. [Google Scholar] [CrossRef]
- Bailey, H.H.; Levy, D.; Harris, L.S.; Schink, J.C.; Foss, F.; Beatty, P.; Wadler, S. A phase II trial of daily perillyl alcohol in patients with advanced ovarian cancer: Eastern Cooperative Oncology Group Study E2E96. Gynecol. Oncol. 2002, 85, 464–468. [Google Scholar] [CrossRef]
- Liu, G.; Oettel, K.; Bailey, H.; Ummersen, L.V.; Tutsch, K.; Staab, M.J.; Horvath, D.; Alberti, D.; Arzoomanian, R.; Rezazadeh, H.; et al. Phase II trial of perillyl alcohol (NSC 641066) administered daily in patients with metastatic androgen independent prostate cancer. Investig. New Drugs 2003, 21, 367–372. [Google Scholar] [CrossRef]
- Meadows, S.M.; Mulkerin, D.; Berlin, J.; Bailey, H.; Kolesar, J.; Warren, D.; Thomas, J.P. Phase II trial of perillyl alcohol in patients with metastatic colorectal cancer. Int. J. Gastrointest. Cancer 2002, 32, 125–128. [Google Scholar] [CrossRef]
- Matos, J.M.; Schmidt, C.M.; Thomas, H.J.; Cummings, O.W.; Wiebke, E.A.; Madura, J.A.; Patrick, L.J., Sr.; Crowell, P.L. A pilot study of perillyl alcohol in pancreatic cancer. J. Surg. Res. 2008, 147, 194–199. [Google Scholar] [CrossRef]
- da Fonseca, C.O.; Masini, M.; Futuro, D.; Caetano, R.; Gattass, C.R.; Quirico-Santos, T. Anaplastic oligodendroglioma responding favorably to intranasal delivery of perillyl alcohol: A case report and literature review. Surg. Neurol. 2006, 66, 611–615. [Google Scholar] [CrossRef]
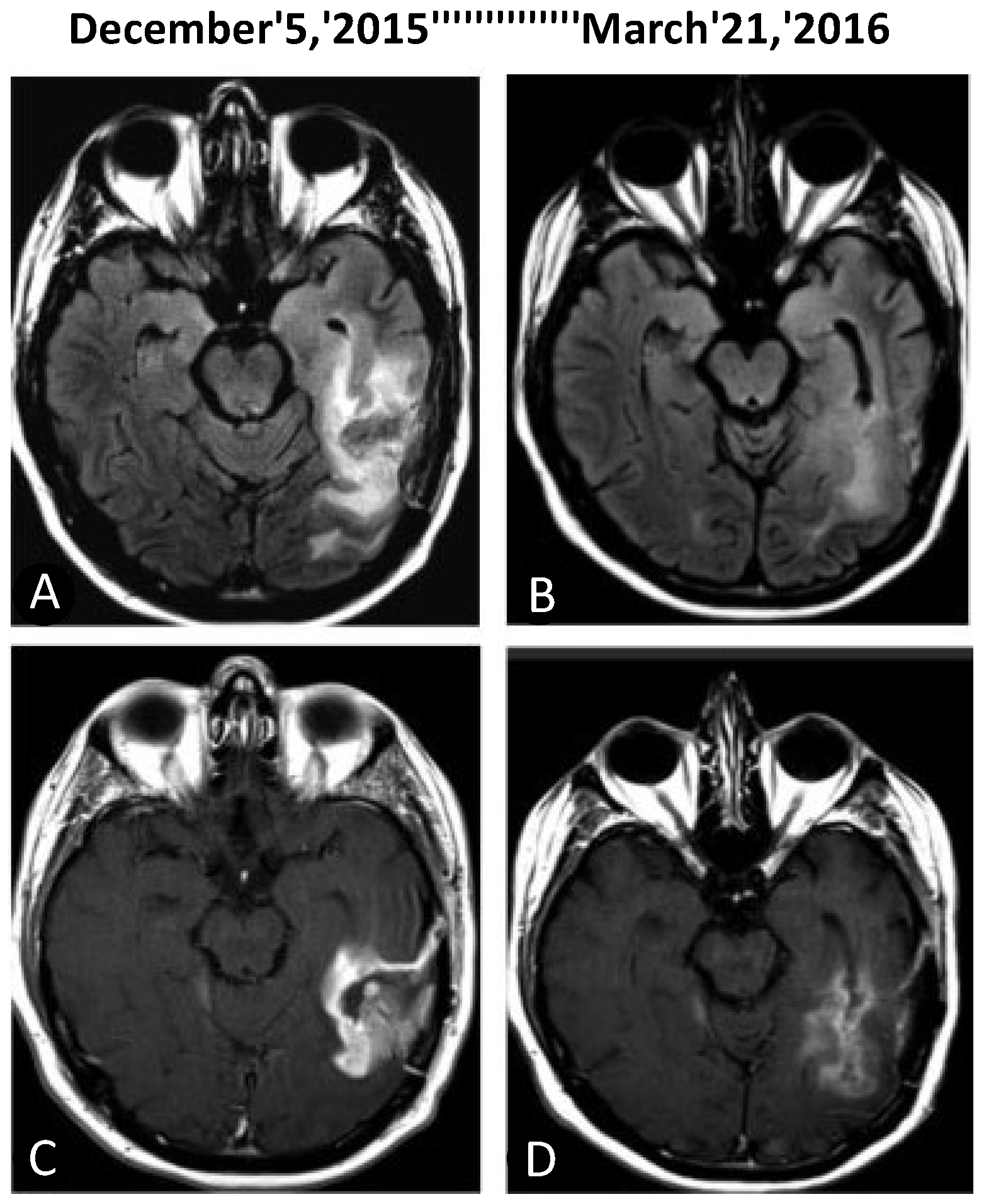
- da Fonseca, C.O.; Schwartsmann, G.; Fischer, J.; Nagel, J.; Futuro, D.; Quirico-Santos, T.; Gattass, C.R. Preliminary results from a phase I/II study of perillyl alcohol intranasal administration in adults with recurrent malignant gliomas. Surg. Neurol. 2008, 70, 259–266. [Google Scholar] [CrossRef]
- da Fonseca, C.O.; Simao, M.; Lins, I.R.; Caetano, R.O.; Futuro, D.; Quirico-Santos, T. Efficacy of monoterpene perillyl alcohol upon survival rate of patients with recurrent glioblastoma. J. Cancer Res. Clin. Oncol. 2011, 137, 287–293. [Google Scholar] [CrossRef]
- da Fonseca, C.O.; Teixeira, R.M.; Silva, J.C.; Fischer, J.D.S.D.G.; Meirelles, O.C.; Landeiro, J.A.; Quirico-Santos, T. Long-term outcome in patients with recurrent malignant glioma treated with Perillyl alcohol inhalation. Anticancer Res. 2013, 33, 5625–5631. [Google Scholar]
- Allen, B.G.; Bhatia, S.K.; Anderson, C.M.; Eichenberger-Gilmore, J.M.; Sibenaller, Z.A.; Mapuskar, K.A.; Schoenfeld, J.D.; Buatti, J.M.; Spitz, D.R.; Fath, M.A. Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism. Redox Biol. 2014, 2, 963–970. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Flores, R.; Poff, A.M.; D’Agostino, D.P.; Mukherjee, P. Metabolic therapy: A new paradigm for managing malignant brain cancer. Cancer Lett. 2015, 356 Pt A, 289–300. [Google Scholar] [CrossRef]
- Strowd, R.E., 3rd; Grossman, S.A. The Role of Glucose Modulation and Dietary Supplementation in Patients with Central Nervous System Tumors. Curr. Treat. Opt. Oncol. 2015, 16, 36. [Google Scholar] [CrossRef]
- Oliveira, C.L.P.; Mattingly, S.; Schirrmacher, R.; Sawyer, M.B.; Fine, E.J.; Prado, C.M. A Nutritional Perspective of Ketogenic Diet in Cancer: A Narrative Review. J. Acad. Nutr. Diet. 2018, 118, 668–688. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Santos, J.G.; Da Cruz, W.M.S.; Schönthal, A.H.; Salazar, M.D.; Fontes, C.A.P.; Quirico-Santos, T.; Da Fonseca, C.O. Patient with recurrent glioblastoma responding favorably to ketogenic diet combined with intranasal delivery of perillyl alcohol: A case report and Literature Review. Arq. Bras. Neurocir. 2017, 36, 194–199. [Google Scholar] [CrossRef]
- Santos, J.G.; Da Cruz, W.M.S.; Schonthal, A.H.; Salazar, M.D.; Fontes, C.A.P.; Quirico-Santos, T.; Da Fonseca, C.O. Efficacy of a ketogenic diet with concomitant intranasal perillyl alcohol as a novel strategy for the therapy of recurrent glioblastoma. Oncol. Lett. 2018, 15, 1263–1270. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signaling—From basic mechanisms to clinical applications. FEBS J. 2018. [Google Scholar] [CrossRef]
- Schönthal, A.H. Endoplasmic reticulum stress: Its role in disease and novel prospects for therapy. Scientifica 2012, 2012, 857516. [Google Scholar] [CrossRef]
- Schönthal, A.H. Targeting endoplasmic reticulum stress for cancer therapy. Front. Biosci. 2012, 4, 412–431. [Google Scholar] [CrossRef]
- Schönthal, A.H.; Chen, T.C.; Hofman, F.M.; Louie, S.G.; Petasis, N.A. Preclinical development of novel anti-tumor drugs targeting the endoplasmic reticulum stress response. Curr. Pharm. Des. 2011, 17, 2428–2438. [Google Scholar] [CrossRef]
- Peñaranda Fajardo, N.M.; Meijer, C.; Kruyt, F.A. The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem. Pharmacol. 2016, 118, 1–8. [Google Scholar] [CrossRef]
- Johnson, G.G.; White, M.C.; Grimaldi, M. Stressed to death: Targeting endoplasmic reticulum stress response induced apoptosis in gliomas. Curr. Pharm. Des. 2011, 17, 284–292. [Google Scholar] [CrossRef]
- Woolf, E.C.; Scheck, A.C. The ketogenic diet for the treatment of malignant glioma. J. Lipid Res. 2015, 56, 5–10. [Google Scholar] [CrossRef]
- Winter, S.F.; Loebel, F.; Dietrich, J. Role of ketogenic metabolic therapy in malignant glioma: A systematic review. Crit. Rev. Oncol. Hematol. 2017, 112, 41–58. [Google Scholar] [CrossRef]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef]
- Wang, W.; Swenson, S.; Cho, H.-Y.; Hofman, F.M.; Schönthal, A.H.; Chen, T.C. Efficient brain targeting and therapeutic intracranial activity of bortezomib through intranasal co-delivery with NEO100 in rodent glioblastoma models. J. Neurosurg. in press.
- Scott, K.; Hayden, P.J.; Will, A.; Wheatley, K.; Coyne, I. Bortezomib for the treatment of multiple myeloma. Cochrane Database Syst. Rev. 2016, 4, CD010816. [Google Scholar] [CrossRef]
- Hemeryck, A.; Geerts, R.; Monbaliu, J.; Hassler, S.; Verhaeghe, T.; Diels, L.; Verluyten, W.; van Beijsterveldt, L.; Mamidi, R.N.; Janssen, C.; et al. Tissue distribution and depletion kinetics of bortezomib and bortezomib-related radioactivity in male rats after single and repeated intravenous injection of 14 C-bortezomib. Cancer Chemother. Pharmacol. 2007, 60, 777–787. [Google Scholar] [CrossRef]
- Labussiere, M.; Pinel, S.; Delfortrie, S.; Plenat, F.; Chastagner, P. Proteasome inhibition by bortezomib does not translate into efficacy on two malignant glioma xenografts. Oncol. Rep. 2008, 20, 1283–1287. [Google Scholar]
- Wang, W.; Cho, H.Y.; Rosenstein-Sisson, R.; Marin Ramos, N.I.; Price, R.; Hurth, K.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C. Intratumoral delivery of bortezomib: Impact on survival in an intracranial glioma tumor model. J. Neurosurg. 2017, 128, 695–700. [Google Scholar] [CrossRef]
- Chen, T.C.; Cho, H.Y.; Wang, W.; Barath, M.; Sharma, N.; Hofman, F.M.; Schönthal, A.H. A novel temozolomide-perillyl alcohol conjugate exhibits superior activity against breast cancer cells in vitro and intracranial triple-negative tumor growth in vivo. Mol. Cancer Ther. 2014, 13, 1181–1193. [Google Scholar] [CrossRef]
- Chen, T.C.; Cho, H.Y.; Wang, W.; Nguyen, J.; Jhaveri, N.; Rosenstein-Sisson, R.; Hofman, F.M.; Schönthal, A.H. A novel temozolomide analog, NEO212, with enhanced activity against MGMT-positive melanoma in vitro and in vivo. Cancer Lett. 2015, 358, 144–151. [Google Scholar] [CrossRef]
- Chen, T.C.; Cho, H.Y.; Wang, W.; Wetzel, S.J.; Singh, A.; Nguyen, J.; Hofman, F.M.; Schönthal, A.H. Chemotherapeutic effect of a novel temozolomide analog on nasopharyngeal carcinoma in vitro and in vivo. J. Biomed. Sci. 2015, 22, 71–80. [Google Scholar] [CrossRef]
- Cho, H.Y.; Wang, W.; Jhaveri, N.; Lee, D.J.; Sharma, N.; Dubeau, L.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C. NEO212, Temozolomide Conjugated to Perillyl Alcohol, Is a Novel Drug for Effective Treatment of a Broad Range of Temozolomide-Resistant Gliomas. Mol. Cancer Ther. 2014, 13, 2004–2017. [Google Scholar] [CrossRef]
- Jhaveri, N.; Agasse, F.; Armstrong, D.; Peng, L.; Commins, D.; Wang, W.; Rosenstein-Sisson, R.; Vaikari, V.P.; Santiago, S.V.; Santos, T.; et al. A novel drug conjugate targeting proneural and mesenchymal subtypes of patient-derived glioma cancer stem cells. Cancer Lett. 2016, 371, 240–250. [Google Scholar] [CrossRef]
- Marin-Ramos, N.I.; Thein, T.Z.; Cho, H.Y.; Swenson, S.D.; Wang, W.; Schonthal, A.H.; Chen, T.C.; Hofman, F.M. NEO212 Inhibits Migration and Invasion of Glioma Stem Cells. Mol. Cancer Ther. 2018, 17, 625–637. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Temozolomide: Therapeutic limitations in the treatment of adult high-grade gliomas. Expert Rev. Neurother. 2010, 10, 1537–1544. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Phase (Duration) | Patients | Dosing | Daily Doses (mg/m2/day) | N | Published Results |
|---|---|---|---|---|---|
| Phase 2 (6-month, daily dosing) | Ovarian cancer | Oral (qid) | 4800 −6000 | 20 | [114] |
| Phase 2 (6-month, daily dosing) | Prostate cancer | Oral (qid) | 4800 | 15 | [115] |
| Phase 2 (6-month, daily dosing) | Breast cancer | Oral (qid) | 4800 −6000 | 14 | [113] |
| Phase 2 (6-month, daily dosing) | Colorectal cancer | Oral (qid) | 4800 −6400 | 27 | [116] |
| Phase 2 (15-day pilot study) | Pancreatic cancer | Oral (qid) | 4800 | 8 | [117] |
| Phase 2 (long-term daily dosing) | Brain cancer | Intranasal (qid) | 112 −271 | >250 | [118,119,120,121] |
| Phase 1/2a (NEO100) (long-term daily dosing) | Recurrent GB | Intranasal (qid) | 195 −586 | 25 (Phase 2) | N/A |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.C.; Da Fonseca, C.O.; Schönthal, A.H. Intranasal Perillyl Alcohol for Glioma Therapy: Molecular Mechanisms and Clinical Development. Int. J. Mol. Sci. 2018, 19, 3905. https://doi.org/10.3390/ijms19123905
Chen TC, Da Fonseca CO, Schönthal AH. Intranasal Perillyl Alcohol for Glioma Therapy: Molecular Mechanisms and Clinical Development. International Journal of Molecular Sciences. 2018; 19(12):3905. https://doi.org/10.3390/ijms19123905
Chicago/Turabian StyleChen, Thomas C., Clovis O. Da Fonseca, and Axel H. Schönthal. 2018. "Intranasal Perillyl Alcohol for Glioma Therapy: Molecular Mechanisms and Clinical Development" International Journal of Molecular Sciences 19, no. 12: 3905. https://doi.org/10.3390/ijms19123905
APA StyleChen, T. C., Da Fonseca, C. O., & Schönthal, A. H. (2018). Intranasal Perillyl Alcohol for Glioma Therapy: Molecular Mechanisms and Clinical Development. International Journal of Molecular Sciences, 19(12), 3905. https://doi.org/10.3390/ijms19123905