Hydrogen Sulfide: A Therapeutic Option in Systemic Sclerosis



Abstract
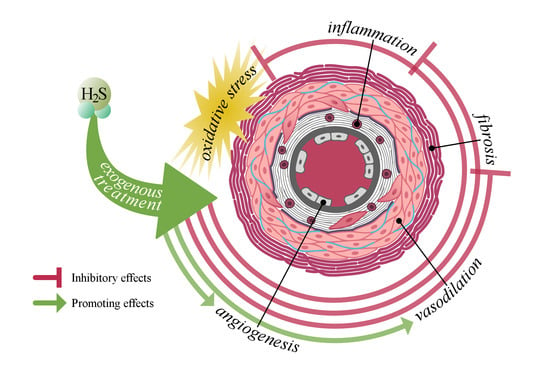
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
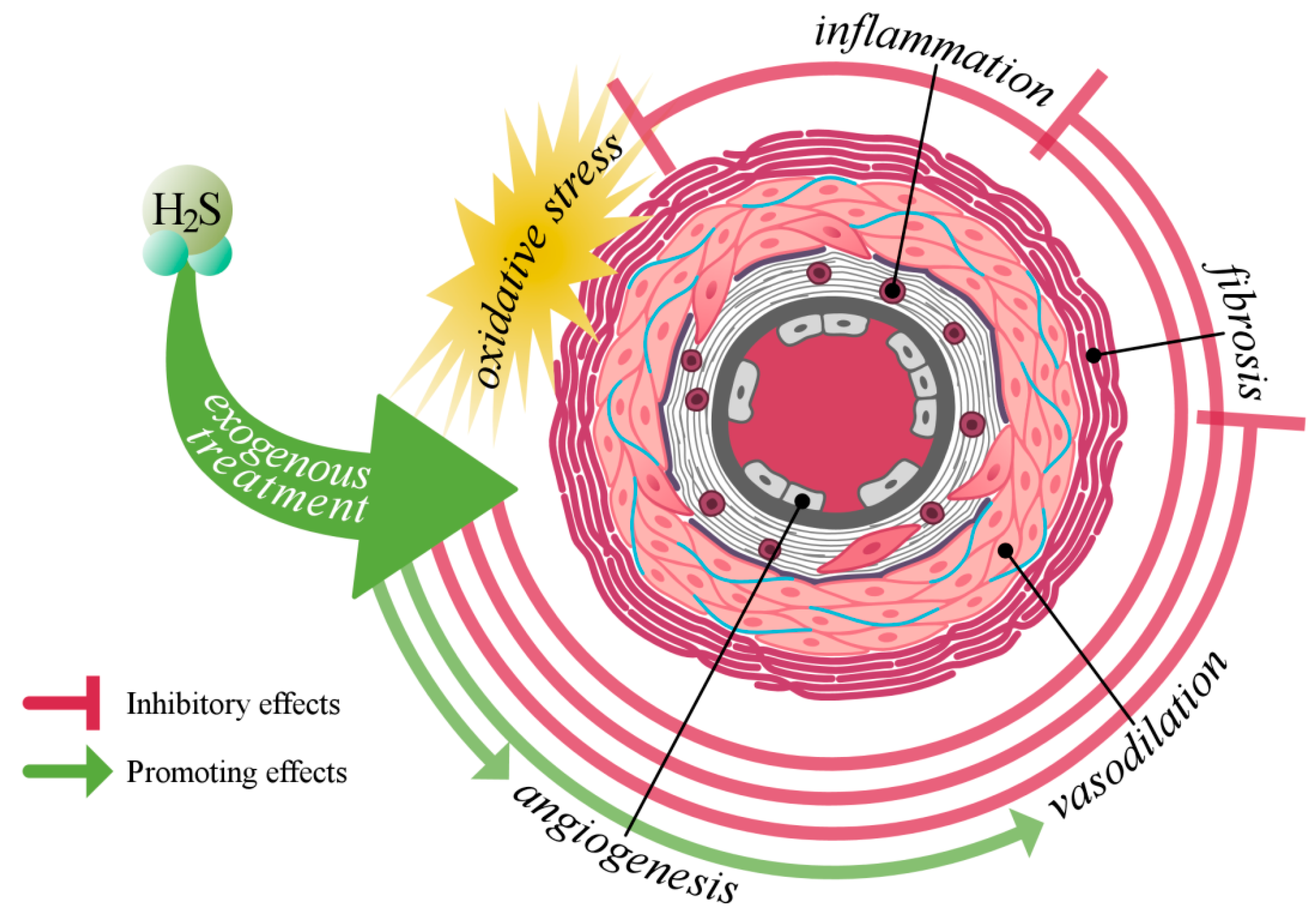
1. Introduction
2. Role of Nitric Oxide and Carbon Monoxide in Systemic Sclerosis
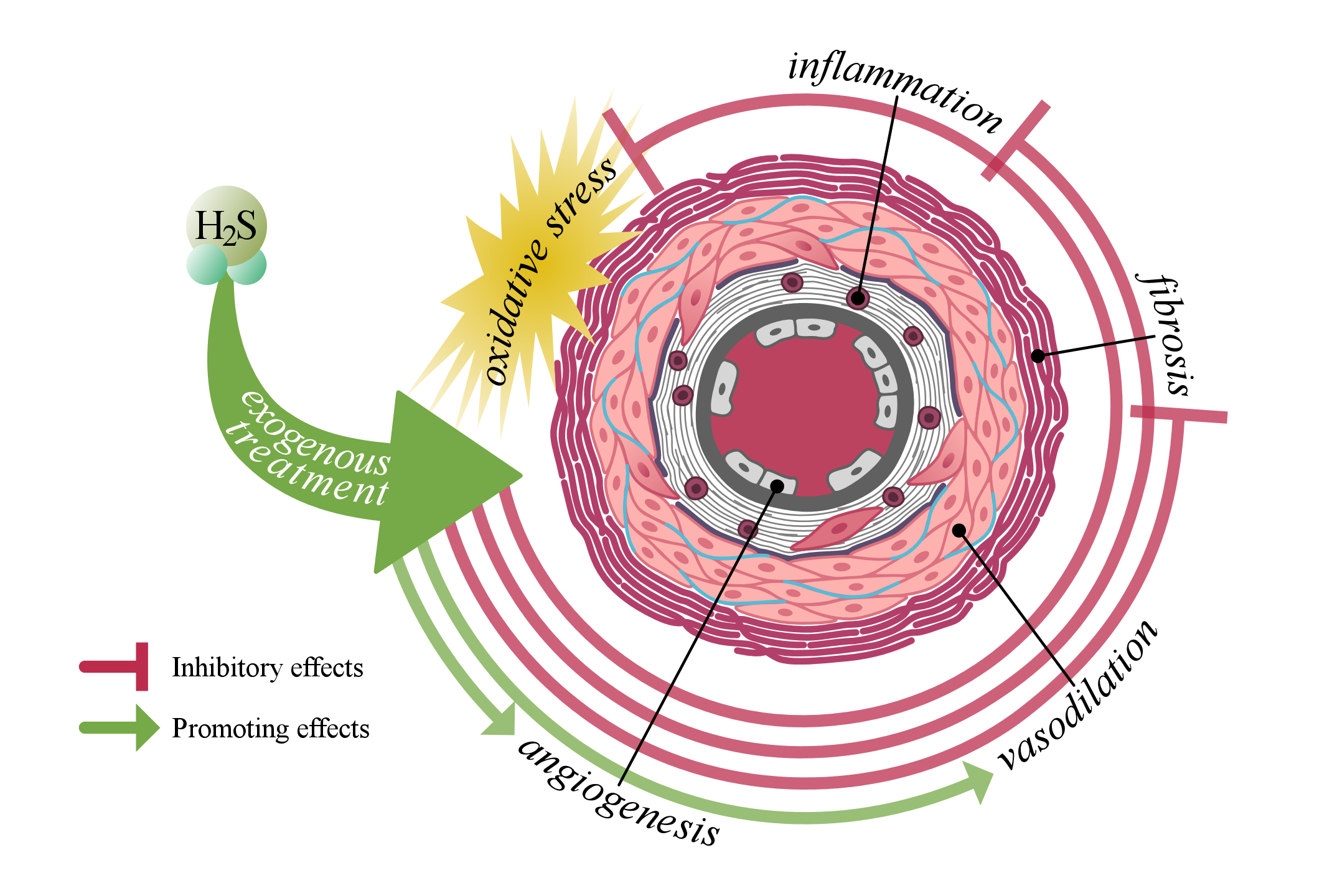
3. The Role of Hydrogen Sulfide in Vascular Biology
3.1. Production of H2S
3.2. Vascular Biology
3.2.1. Vascular Tone Regulation
3.2.2. Cell Proliferation and Angiogenesis
3.2.3. Antioxidant Effect of H2S
4. The Potential Role of H2S in the Development of Systemic Sclerosis-Related Vasculopathy
4.1. Link between H2S and Vascular Injury
4.2. Link between H2S and Inflammation
4.3. Link between H2S and Fibrosis
5. Hydrogen Sulfide as a Therapeutic Option
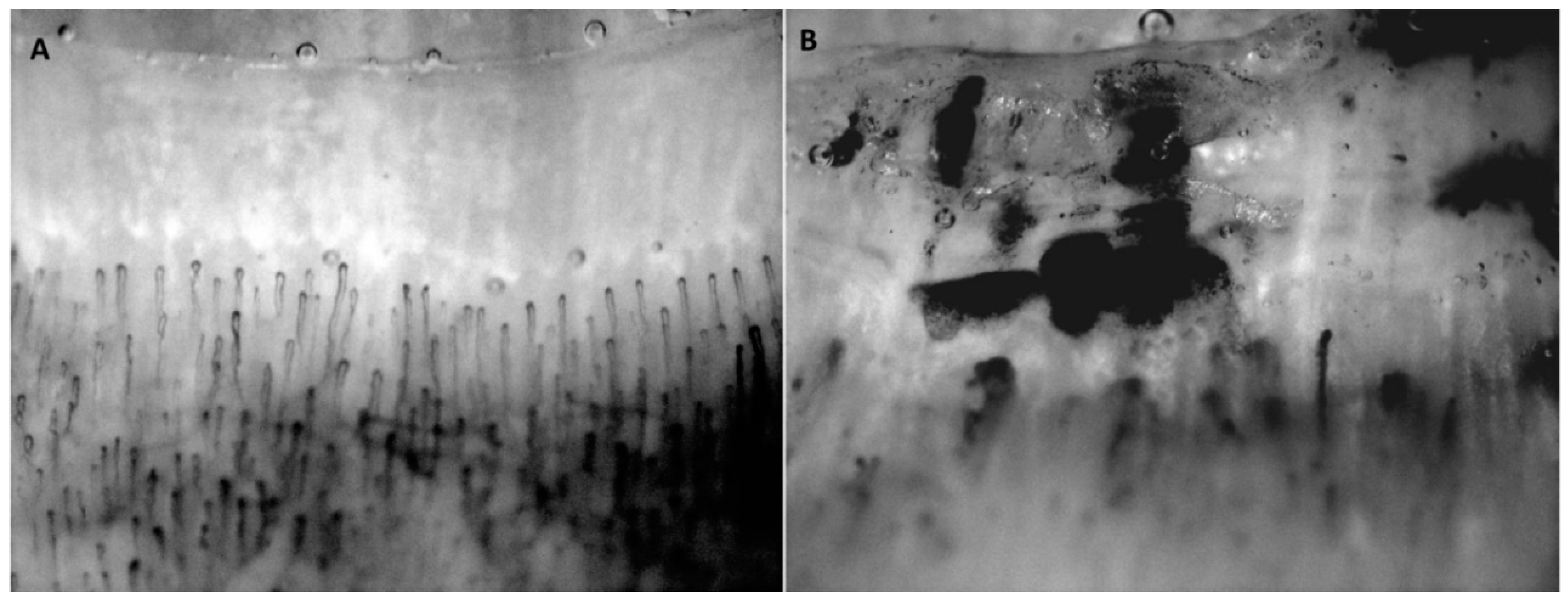
5.1. Hydrogen Sulfide in Experimental Animal Studies
5.2. Hydrogen Sulfide in Human Studies
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Leroy, E.C. Scleroderma (Systemic Sclerosis): Classificiation, Subsets and Pathogenesis. J. Rheumatol. 1988, 15, 202–205. [Google Scholar]
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Almeida, C.; Almeida, I.; Vasconcelos, C. Quality of life in systemic sclerosis. Autoimmun. Rev. 2015, 14, 1087–1096. [Google Scholar] [CrossRef]
- Nikpour, M.; Stevens, W.M.; Herrick, A.L.; Proudman, S.M. Epidemiology of systemic sclerosis. Best Pract. Res. Clin. Rheumatol. 2010, 24, 857–869. [Google Scholar] [CrossRef]
- Ranque, B.; Mouthon, L. Geoepidemiology of systemic sclerosis. Autoimmun. Rev. 2010, 9, A311–A318. [Google Scholar] [CrossRef]
- Chifflot, H.; Fautrel, B.; Sordet, C.; Chatelus, E.; Sibilia, J. Incidence and Prevalence of Systemic Sclerosis: A Systematic Literature Review. Semin. Arthritis Rheum. 2008, 37, 223–235. [Google Scholar] [CrossRef]
- Mostmans, Y.; Cutolo, M.; Giddelo, C.; Decuman, S.; Melsens, K.; Declercq, H.; Vandecasteele, E.; De Keyser, F.; Distler, O.; Gutermuth, J.; et al. The role of endothelial cells in the vasculopathy of systemic sclerosis: A systematic review. Autoimmun. Rev. 2017, 16, 774–786. [Google Scholar] [CrossRef]
- Abdulle, A.E.; Diercks, G.F.H.; Feelisch, M.; Mulder, D.J.; van Goor, H. The Role of Oxidative Stress in the Development of Systemic Sclerosis Related Vasculopathy. Front. Physiol. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Herrick, A.L. The pathogenesis, diagnosis and treatment of Raynaud phenomenon. Nat. Rev. Rheumatol. 2012, 8, 469–479. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Lefer, D.J. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 2014, 114, 730–737. [Google Scholar] [CrossRef]
- Mann, B.E.; Motterlini, R. CO and NO in medicine. Chem. Commun. 2007, 4197–4208. [Google Scholar] [CrossRef]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef]
- Matucci Cerinic, M.; Kahaleh, M.B. Beauty and the beast. The nitric oxide paradox in systemic sclerosis. Rheumatology 2002, 41, 843–847. [Google Scholar] [CrossRef]
- Cotton, S.A.; Herrick, A.L.; Jayson, M.I.V.; Freemont, A.J. Endothelial expression of nitric oxide synthases and nitrotyrosine in systemic sclerosis skin. J. Pathol. 1999, 189, 273–278. [Google Scholar] [CrossRef]
- Allanore, Y.; Borderie, D.; Hilliquin, P.; Hernvann, A.; Levacher, M.; Lemaréchal, H.; Ekindjian, O.G.; Kahan, A. Low levels of nitric oxide (NO) in systemic sclerosis: Inducible NO synthase production is decreased in cultured peripheral blood monocyte/macrophage cells. Rheumatology 2001, 40, 1089–1096. [Google Scholar] [CrossRef]
- Dooley, A.; Gao, B.; Bradley, N.; Abraham, D.J.; Black, C.M.; Jacobs, M.; Bruckdorfer, K.R. Abnormal nitric oxide metabolism in systemic sclerosis: Increased levels of nitrated proteins and asymmetric dimethylarginine. Rheumatology 2006, 45, 676–684. [Google Scholar] [CrossRef]
- Malerba, M.; Radaeli, A.; Ragnoli, B.; Airo’, P.; Corradi, M.; Ponticiello, A.; Zambruni, A.; Grassi, V. Exhaled nitric oxide levels in systemic sclerosis with and without pulmonary involvement. Chest 2007, 132, 575–580. [Google Scholar] [CrossRef]
- Takagi, K.; Kawaguchi, Y.; Hara, M.; Sugiura, T.; Harigai, M.; Kamatani, N. Serum nitric oxide (NO) levels in systemic sclerosis patients: Correlation between NO levels and clinical features. Clin. Exp. Immunol. 2003, 134, 538–544. [Google Scholar] [CrossRef]
- Loscalzo, J.; Welch, G. Nitric oxide and its role in the cardiovascular system. Prog. Cardiovasc. Dis. 1995, 38, 87–104. [Google Scholar] [CrossRef]
- Palmer, R.M.J.; Ashton, D.S.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988, 333, 664–666. [Google Scholar] [CrossRef]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W.S. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef]
- Kröncke, K.D.; Fehsel, K.; Kolb-Bachofen, V. Inducible nitric oxide synthase in human diseases. Clin. Exp. Immunol. 1998, 113, 147–156. [Google Scholar] [CrossRef]
- Marietta, M.A.; Yoon, P.S.; Iyengar, R.; Leaf, C.D.; Wishnok, J.S. Macrophage Oxidation of L-Arginine to Nitrite and Nitrate: Nitric Oxide Is an Intermediate. Biochemistry 1988, 27, 8706–8711. [Google Scholar] [CrossRef]
- Yui, Y.; Hattori, R.; Kosuga, K.; Eizawa, H.; Hiki, K.; Ohkawa, S.; Ohnishi, K.; Terao, S.; Kawai, C. Calmodulin-independent nitric oxide synthase from rat polymorphonuclear neutrophils. J. Biol. Chem. 1991, 266, 3369–3371. [Google Scholar]
- Arnold, W.P.; Mittal, C.K.; Katsuki, S.; Murad, F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. USA 1977, 74, 3203–3207. [Google Scholar] [CrossRef]
- Carvajal, J.A.; Germain, A.M.; Huidobro-Toro, J.P.; Weiner, C.P. Molecular mechanism of cGMP-mediated smooth muscle relaxation. J. Cell. Physiol. 2000, 184, 409–420. [Google Scholar] [CrossRef]
- Sammut, I.A.; Foresti, R.; Clark, J.E.; Exon, D.J.; Vesely, M.J.J.; Sarathchandra, P.; Green, C.J.; Motterlini, R. Carbon monoxide is a major contributor to the regulation of vascular tone in aortas expressing high levels of haeme oxygenase-1. Br. J. Pharmacol. 1998, 125, 1437–1444. [Google Scholar] [CrossRef]
- Thorup, C.; Jones, C.L.; Gross, S.S.; Moore, L.C.; Goligorsky, M.S. Carbon monoxide induces vasodilation and nitric oxide release but suppresses endothelial NOS. Am. J. Physiol. 1999, 277, F882–F889. [Google Scholar] [CrossRef]
- Freedman, R.R.; Girgis, R.; Mayes, M.D. Endothelial and adrenergic dysfunction in Raynaud’s phenomenon and scleroderma. J. Rheumatol. 1999, 26, 2386–2388. [Google Scholar]
- Romero, L.I.; Zhang, D.N.; Cooke, J.P.; Ho, H.K.; Avalos, E.; Herrera, R.; Herron, G.S. Differential expression of nitric oxide by dermal microvascular endothelial cells from patients with scleroderma. Vasc. Med. 2000, 5, 147–158. [Google Scholar] [CrossRef]
- Nara, K.; Konno, D.; Uchida, J.; Kiuchi, Y.; Oguchi, K. Protective effect of nitric oxide against iron-induced neuronal damage. J. Neural Transm. 1999, 106, 835–848. [Google Scholar] [CrossRef]
- O’Donnell, V.B.; Freeman, B.A. Interactions between nitric oxide and lipid oxidation pathways: Implications for vascular disease. Circ. Res. 2001, 88, 12–21. [Google Scholar] [CrossRef]
- Lind, M.; Hayes, A.; Caprnda, M.; Petrovic, D.; Rodrigo, L.; Kruzliak, P.; Zulli, A. Inducible nitric oxide synthase: Good or bad? Biomed. Pharmacother. 2017, 93, 370–375. [Google Scholar] [CrossRef]
- Yamamoto, T.; Katayama, I.; Nishioka, K. Nitric oxide production and inducible nitric oxide synthase expression in systemic sclerosis. J. Rheumatol. 1998, 25, 314–317. [Google Scholar]
- Sud, A.; Khullar, M.; Wanchu, A.; Bambery, P. Increased nitric oxide production in patients with systemic sclerosis. Nitric Oxide Biol. Chem. 2000, 4, 615–619. [Google Scholar] [CrossRef]
- Ewing, J.F.; Maines, M.D. Rapid induction of heme oxygenase 1 mRNA and protein by hyperthermia in rat brain: Heme oxygenase 2 is not a heat shock protein. Proc. Natl. Acad. Sci. USA 1991, 88, 5364–5368. [Google Scholar] [CrossRef]
- Schipper, H.M. Heme oxygenase-1: Transducer of pathological brain iron sequestration under oxidative stress. Ann. N. Y. Acad. Sci. 2004, 1012, 84–93. [Google Scholar] [CrossRef]
- Wang, R.; Wu, L. The chemical modification of K(Ca) channels by carbon monoxide in vascular smooth muscle cells. J. Biol. Chem. 1997, 272, 32804–32809. [Google Scholar] [CrossRef]
- Ndisang, J.; Tabien, H.; Wang, R. Carbon monoxide and hypertension. J. Hypertens. 2004, 22, 1057–1074. [Google Scholar] [CrossRef]
- Andresen, J.; Shafi, N.; Durante, W.; Bryan, R.J. The effect of carbon monoxide and heme oxygenase inhibitors in cerebral vessels of rats and mice. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H223–H230. [Google Scholar] [CrossRef]
- Johnson, F.K.; Johnson, R.A. Carbon monoxide promotes endothelium-dependent constriction of isolated gracilis muscle arterioles. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R536–R541. [Google Scholar] [CrossRef]
- Foresti, R.; Hammad, J.; Clark, J.E.; Johnson, T.R.; Mann, B.E.; Friebe, A.; Green, C.J.; Motterlini, R. Vasoactive properties of CORM-3, a novel water-soluble carbon monoxide-releasing molecule. Br. J. Pharmacol. 2004, 142, 453–460. [Google Scholar] [CrossRef]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef]
- Kim, H.S.; Loughran, P.A.; Rao, J.; Billiar, T.R.; Zuckerbraun, B.S. Carbon monoxide activates NF-κB via ROS generation and Akt pathways to protect against cell death of hepatocytes. AJP Gastrointest. Liver Physiol. 2008, 295, G146–G152. [Google Scholar] [CrossRef]
- Kim, K.M.; Pae, H.O.; Zheng, M.; Park, R.; Kim, Y.M.; Chung, H.T. Carbon monoxide induces heme oxygenase-1 via activation of protein kinase R-like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ. Res. 2007, 101, 919–927. [Google Scholar] [CrossRef]
- Zhou, Z.; Song, R.; Fattman, C.L.; Greenhill, S.; Alber, S.; Oury, T.D.; Choi, A.M.K.; Morse, D. Carbon monoxide suppresses bleomycin-induced lung fibrosis. Am. J. Pathol. 2005, 166, 27–37. [Google Scholar] [CrossRef]
- Peyton, K.J.; Reyna, S.V.; Chapman, G.B.; Ensenat, D.; Liu, X.M.; Wang, H.; Schafer, A.I.; Durante, W. Heme oxygenase-1-derived carbon monoxide is an autocrine inhibitor of vascular smooth muscle cell growth. Blood 2002, 99, 4443–4448. [Google Scholar] [CrossRef]
- Clark, J.E.; Foresti, R.; Sarathchandra, P.; Kaur, H.; Green, C.J.; Motterlini, R. Heme oxygenase-1-derived bilirubin ameliorates postischemic myocardial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H643–H651. [Google Scholar] [CrossRef]
- Choi, Y.K.; Kim, C.K.; Lee, H.; Jeoung, D.; Ha, K.S.; Kwon, Y.G.; Kim, K.W.; Kim, Y.M. Carbon monoxide promotes VEGF expression by increasing HIF-1α protein level via two distinct mechanisms, translational activation and stabilization of HIF-1α protein. J. Biol. Chem. 2010, 285, 32116–32125. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.-I.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Li, L.; Rose, P.; Moore, P.K. Hydrogen Sulfide and Cell Signaling. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Wang, R. Hydrogen sulfide-based therapeutics: Exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 2015, 14, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.T.; Whiteman, M. Hydrogen sulfide and cell signaling: Team player or referee? Plant Physiol. Biochem. 2014, 78, 37–42. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Lu, M.; Hu, L.-F.; Wong, P.T.-H.; Webb, G.D.; Bian, J.-S. Hydrogen Sulfide in the Mammalian Cardiovascular System. Antioxid. Redox Signal. 2012, 17, 141–185. [Google Scholar] [CrossRef]
- Kimura, H. Production and Physiological Effects of Hydrogen Sulfide. Antioxid. Redox Signal. 2014, 20, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Shibuya, N.; Ogasawara, Y.; Kimura, H. Hydrogen sulfide is produced by cystathionine γ-lyase at the steady-state low intracellular Ca2+ concentrations. Biochem. Biophys. Res. Commun. 2013, 431, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Kabil, O.; Banerjee, R. Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef]
- Wang, R. The Gasotransmitter Role of Hydrogen Sulfide. Antioxid. Redox Signal. 2003, 5, 493–501. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Shen, X.; Kevil, C.G. A tale of two gases: NO and H2S, foes or friends for life? Redox Biol. 2013, 1, 313–318. [Google Scholar] [CrossRef]
- Richardson, C.J.; Magee, E.A.M.; Cummings, J.H. A new method for the determination of sulphide in gastrointestinal contents and whole blood by microdistillation and ion chromatography. Clin. Chim. Acta 2000, 293, 115–125. [Google Scholar] [CrossRef]
- Bełtowski, J. Hydrogen sulfide in pharmacology and medicine—An update. Pharmacol. Rep. 2015, 67, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Peter, E.A.; Bir, S.; Wang, R.; Kevil, C.G. Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radic. Biol. Med. 2012, 52, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Furne, J.; Saeed, A.; Levitt, M.D. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. AJP Regul. Integr. Comp. Physiol. 2008, 295, R1479–R1485. [Google Scholar] [CrossRef]
- Whiteman, M.; Moore, P.K. Hydrogen sulfide and the vasculature: A novel vasculoprotective entity and regulator of nitric oxide bioavailability? J. Cell. Mol. Med. 2009, 13, 488–507. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, J.; Li, H.; Xue, M.; Ji, A.; Li, Y. Role of hydrogen sulfide in ischemia-reperfusion injury. Oxid. Med. Cell. Longev. 2015, 2015, 186908. [Google Scholar] [CrossRef]
- Sabry, Z.I.; Shadarevian, S.B.; Cowan, J.W.; Campbell, J.A. Relationship of dietary intake of sulphur amino-acids to urinary excretion of inorganic sulphate in man. Nature 1965, 206, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Houterman, S.; Van Faassen, A.; Ocke, M.C.; Habets, L.H.M.; Van Dieijen-Visser, M.P.; Bueno-de-Mesquita, B.H.; Janknegt, R.A. Is urinary sulfate a biomarker for the intake of animal protein and meat? Cancer Lett. 1997, 114, 295–296. [Google Scholar] [CrossRef]
- van den Berg, E.; Pasch, A.; Westendorp, W.H.; Navis, G.; Brink, E.J.; Gans, R.O.B.; van Goor, H.; Bakker, S.J.L. Urinary Sulfur Metabolites Associate with a Favorable Cardiovascular Risk Profile and Survival Benefit in Renal Transplant Recipients. J. Am. Soc. Nephrol. 2014, 25, 1303–1312. [Google Scholar] [CrossRef]
- van den Born, J.C.; Frenay, A.-R.S.; Koning, A.M.; Bachtler, M.; Riphagen, I.J.; Minovíc, I.; Feelisch, M.; Dekker, M.M.; Bulthuis, M.L.C.; Gansevoort, R.T.; et al. Urinary Excretion of Sulfur Metabolites and Risk of Cardiovascular Events and All-Cause Mortality in the General Population. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef]
- Ali, M.Y.; Ping, C.Y.; Mok, Y.-Y.; Ling, L.; Whiteman, M.; Bhatia, M.; Moore, P.K. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br. J. Pharmacol. 2006, 149, 625–634. [Google Scholar] [CrossRef]
- Lim, J.J.; Liu, Y.-H.; Khin, E.S.W.; Bian, J.-S. Vasoconstrictive effect of hydrogen sulfide involves downregulation of cAMP in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2008, 295, C1261–C1270. [Google Scholar] [CrossRef]
- Kubo, S.; Kurokawa, Y.; Doe, I.; Masuko, T.; Sekiguchi, F.; Kawabata, A. Hydrogen sulfide inhibits activity of three isoforms of recombinant nitric oxide synthase. Toxicology 2007, 241, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Deitch, E.A.; Szabó, C. Hydrogen sulfide decreases adenosine triphosphate levels in aortic rings and leads to vasorelaxation via metabolic inhibition. Life Sci. 2008, 83, 589–594. [Google Scholar] [CrossRef]
- Holwerda, K.M.; Karumanchi, S.A.; Lely, A.T. Hydrogen sulfide: Role in vascular physiology and pathology. Curr. Opin. Nephrol. Hypertens. 2015, 24, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Testai, L.; Breschi, M.C.; Lawson, K.; McKay, N.G.; Miceli, F.; Taglialatela, M.; Calderone, V. Vasorelaxation by hydrogen sulphide involves activation of Kv7 potassium channels. Pharmacol. Res. 2013, 70, 27–34. [Google Scholar] [CrossRef]
- Jackson-Weaver, O.; Osmond, J.M.; Riddle, M.A.; Naik, J.S.; Bosc, L.V.G.; Walker, B.R.; Kanagy, N.L. Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca2+-activated K+ channels and smooth muscle Ca2+ sparks. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1446–H1454. [Google Scholar] [CrossRef]
- Nicholls, P.; Kim, J. Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Can. J. Biochem. 1982, 60, 613–623. [Google Scholar] [CrossRef]
- Cooper, C.E.; Brown, G.C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: Chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 2008, 40, 533–539. [Google Scholar] [CrossRef]
- Dongó, E.; Beliczai-Marosi, G.; Dybvig, A.S.; Kiss, L. The mechanism of action and role of hydrogen sulfide in the control of vascular tone. Nitric Oxide Biol. Chem. 2018, 81, 75–87. [Google Scholar] [CrossRef]
- Ahmad, F.U.D.; Sattar, M.A.; Rathore, H.A.; Tan, Y.C.; Akhtar, S.; Jin, O.H.; Pei, Y.P.; Abdullah, N.A.; Johns, E.J. Hydrogen sulphide and tempol treatments improve the blood pressure and renal excretory responses in spontaneously hypertensive rats. Ren. Fail. 2014, 598–605. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, L.-K.; Zhang, C.-Y.; Zeng, X.-J.; Yan, H.; Jin, H.-F.; Tang, C.-S.; Du, J.-B. Regulatory effect of hydrogen sulfide on vascular collagen content in spontaneously hypertensive rats. Hypertens. Res. 2008, 31, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, H.; Van Den Born, J.C.; Hillebrands, J.L.; Joles, J.A. Hydrogen sulfide in hypertension. Curr. Opin. Nephrol. Hypertens. 2016, 25, 107–113. [Google Scholar] [CrossRef]
- Nagpure, B.V.; Bian, J.-S. Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System. Oxid. Med. Cell. Longev. 2016, 2016, 1–16. [Google Scholar] [CrossRef]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.L.; Lumsden, N.G.; Farry, J.; Jefferis, A.-M.; Kemp-Harper, B.K.; Chin-Dusting, J.P.F. Nitroxyl: A vasodilator of human vessels that is not susceptible to tolerance. Clin. Sci. 2015, 129, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Cortese-Krott, M.M.; Koning, A.; Kuhnle, G.G.C.; Nagy, P.; Bianco, C.L.; Pasch, A.; Wink, D.A.; Fukuto, J.M.; Jackson, A.A.; van Goor, H.; et al. The Reactive Species Interactome: Evolutionary Emergence, Biological Significance, and Opportunities for Redox Metabolomics and Personalized Medicine. Antioxid. Redox Signal. 2017, 684–712. [Google Scholar] [CrossRef]
- Cai, W.J.; Wang, M.J.; Moore, P.K.; Jin, H.M.; Yao, T.; Zhu, Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 2007, 29–40. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 21972–21977. [Google Scholar] [CrossRef]
- Szabó, C.; Papapetropoulos, A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br. J. Pharmacol. 2011, 164, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, K.M.; Burke, S.D.; Faas, M.M.; Zsengeller, Z.; Stillman, I.E.; Kang, P.M.; van Goor, H.; McCurley, A.; Jaffe, I.Z.; Karumanchi, S.A.; et al. Hydrogen Sulfide Attenuates sFlt1-Induced Hypertension and Renal Damage by Upregulating Vascular Endothelial Growth Factor. J. Am. Soc. Nephrol. 2014, 25, 717–725. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007. [Google Scholar] [CrossRef] [PubMed]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Modis, K.; Panopoulos, P.; Asimakopoulou, A.; Gero, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-J.; Cai, W.-J.; Zhu, Y.-C. Mechanisms of angiogenesis: Role of hydrogen sulphide. Clin. Exp. Pharmacol. Physiol. 2010, 37, 764–771. [Google Scholar] [CrossRef]
- Geng, B.; Cui, Y.; Zhao, J.; Yu, F.; Zhu, Y.; Xu, G.; Zhang, Z.; Tang, C.; Du, J. Hydrogen sulfide downregulates the aortic L-arginine/nitric oxide pathway in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1608–R1618. [Google Scholar] [CrossRef]
- Ono, K.; Akaike, T.; Sawa, T.; Kumagai, Y.; Wink, D.A.; Tantillo, D.J.; Hobbs, A.J.; Nagy, P.; Xian, M.; Lin, J.; et al. Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: Implications of their possible biological activity and utility. Free Radic. Biol. Med. 2014, 77, 82–94. [Google Scholar] [CrossRef]
- Predmore, B.L.; Lefer, D.J.; Gojon, G. Hydrogen Sulfide in Biochemistry and Medicine. Antioxid. Redox Signal. 2012, 17, 119–140. [Google Scholar] [CrossRef]
- Xie, Z.-Z.; Liu, Y.; Bian, J.-S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxid. Med. Cell. Longev. 2016, 2016, 6043038. [Google Scholar] [CrossRef] [PubMed]
- Chutkow, W.A.; Lee, R.T. Thioredoxin regulates adipogenesis through thioredoxin-interacting protein (Txnip) protein stability. J. Biol. Chem. 2011, 286, 29139–29145. [Google Scholar] [CrossRef]
- Moreno, M.-L.; Escobar, J.; Izquierdo-Álvarez, A.; Gil, A.; Pérez, S.; Pereda, J.; Zapico, I.; Vento, M.; Sabater, L.; Marina, A.; et al. Disulfide stress: A novel type of oxidative stress in acute pancreatitis. Free Radic. Biol. Med. 2014, 70, 265–277. [Google Scholar] [CrossRef]
- Luo, Y.; He, F.; Hu, L.; Hai, L.; Huang, M.; Xu, Z.; Zhang, J.; Zhou, Z.; Liu, F.; Dai, Y.S. Transcription factor Ets1 regulates expression of thioredoxin-interacting protein and inhibits insulin secretion in pancreatic β-cells. PLoS ONE 2014, 9, e99049. [Google Scholar] [CrossRef]
- Gan, L.; Johnson, J.A. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef]
- Sgonc, R.; Gruschwitz, M.S.; Dietrich, H.; Recheis, H.; Gershwin, M.E.; Wick, G. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J. Clin. Investig. 1996, 98, 785–792. [Google Scholar] [CrossRef]
- Herrick, A.L. Pathogenesis of Raynaud’s phenomenon. Revmatologiia 2005, 13, 62–65. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Vascular pathology of homocysteinemia: Implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969, 56, 111–128. [Google Scholar]
- Caramaschi, P.; Volpe, A.; Canestrini, S.; Bambara, L.M.; Faccini, G.; Carletto, A.; Biasi, D. Correlation between homocysteine plasma levels and nailfold videocapillaroscopic patterns in systemic sclerosis. Clin. Rheumatol. 2007, 26, 902–907. [Google Scholar] [CrossRef]
- Caramaschi, P.; Martinelli, N.; Biasi, D.; Carletto, A.; Faccini, G.; Volpe, A.; Ferrari, M.; Scambi, C.; Bambara, L.M. Homocysteine plasma concentration is related to severity of lung impairment in scleroderma. J. Rheumatol. 2003, 30, 298–304. [Google Scholar]
- Marasini, B.; Casari, S.; Bestetti, A.; Maioli, C.; Cugno, M.; Zeni, S.; Turri, O.; Guagnellini, E.; Biondi, M.L. Homocysteine concentration in primary and systemic sclerosis associated Raynaud’s phenomenon. J. Rheumatol. 2000, 27, 2621–2623. [Google Scholar]
- Hankey, G.J.; Eikelboom, J.W. Homocysteine and vascular disease. Lancet 1999, 354, 407–413. [Google Scholar] [CrossRef]
- Szamosi, S.; Csiki, Z.; Szomják, E.; Szolnoki, E.; Szoke, G.; Szekanecz, Z.; Szegedi, G.; Shoenfeld, Y.; Szucs, G. Plasma homocysteine levels, the prevalence of methylenetetrahydrofolate reductase gene C677T polymorphism and macrovascular disorders in systemic sclerosis: Risk factors for accelerated macrovascular damage? Clin. Rev. Allergy Immunol. 2009, 36, 145–149. [Google Scholar] [CrossRef]
- Mahalle, N.; Kulkarni, M.V.; Garg, M.K.; Naik, S.S. Vitamin B12 deficiency and hyperhomocysteinemia as correlates of cardiovascular risk factors in Indian subjects with coronary artery disease. J. Cardiol. 2013, 61, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Osada, J.; Aratani, Y.; Kluckman, K.; Reddick, R.; Malinow, M.R.; Maeda, N. Mice deficient in cystathionine beta-synthase: Animal models for mild and severe homocyst(e)inemia. Proc. Natl. Acad. Sci. USA 1995, 92, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, R.T.; Forgione, M.A.; Cap, A.; Leopold, J.A.; Rudd, M.A.; Trolliet, M.; Heydrick, S.; Stark, R.; Klings, E.S.; Moldovan, N.I.; et al. Endothelial dysfunction in a murine model of mild hyperhomocyst(e)inemia. J. Clin. Investig. 2000, 106, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Yang, F.; Tan, H.; Liao, D.; Bryan, R.M.; Randhawa, J.K.; Rumbaut, R.E.; Durante, W.; Schafer, A.I.; Yang, X.; et al. Hyperhomocystinemia impairs endothelial function and eNOS activity via PKC activation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2515–2521. [Google Scholar] [CrossRef]
- Zhang, F.; Slungaard, A.; Vercellotti, G.M.; Iadecola, C. Superoxide-dependent cerebrovascular effects of homocysteine. Am. J. Physiol. 1998, 274 Pt 2, R1704–R1711. [Google Scholar] [CrossRef]
- Upchurch, G.R.; Welche, G.N.; Fabian, A.J.; Freedman, J.E.; Johnson, J.L.; Keaney, J.F.; Loscalzo, J. Homocyst(e)ine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. J. Biol. Chem. 1997, 272, 17012–17017. [Google Scholar] [CrossRef]
- Weiss, N.; Zhang, Y.Y.; Heydrick, S.; Bierl, C.; Loscalzo, J. Overexpression of cellular glutathione peroxidase rescues homocyst(e)ine-induced endothelial dysfunction. Proc. Natl. Acad. Sci. USA 2001, 98, 12503–12508. [Google Scholar] [CrossRef] [PubMed]
- Sawle, P.; Foresti, R.; Green, C.J.; Motterlini, R. Homocysteine attenuates endothelial haem oxygenase-1 induction by nitric oxide (NO) and hypoxia. FEBS Lett. 2001, 508, 403–406. [Google Scholar] [CrossRef]
- Loscalzo, J. The oxidant stress of hyperhomocyst(e)inemia. J. Clin. Investig. 1996, 98, 5–7. [Google Scholar] [CrossRef]
- Yao, K. Effects of several unusual sulfur-containing amino acids on rat liver cystathionine-gamma-lyase. Physiol. Chem. Phys. 1975, 7, 401–408. [Google Scholar]
- Chang, L.; Geng, B.; Yu, F.; Zhao, J.; Jiang, H.; Du, J.; Tang, C. Hydrogen sulfide inhibits myocardial injury induced by homocysteine in rats. Amino Acids 2008, 34, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Pizzorni, C.; Tuccio, M.; Burroni, A.; Craviotto, C.; Basso, M.; Seriolo, B.; Sulli, A. Nailfold videocapillaroscopic patterns and serum autoantibodies in systemic sclerosis. Rheumatology 2004, 43, 719–726. [Google Scholar] [CrossRef]
- Motegi, S.; Toki, S.; Yamada, K.; Uchiyama, A.; Ishikawa, O. Elevated plasma homocysteine level is possibly associated with skin sclerosis in a series of Japanese patients with systemic sclerosis. J. Dermatol. 2014, 41, 986–991. [Google Scholar] [CrossRef]
- Chiku, T.; Padovani, D.; Zhu, W.; Singh, S.; Vitvitsky, V.; Banerjee, R. H2S biogenesis by human cystathionine γ-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 2009, 284, 11601–11612. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Schett, G.; Gay, S.; Distler, O.; Distler, J.H. Hypoxia. Hypoxia in the pathogenesis of systemic sclerosis. Arthritis Res. Ther. 2009, 11, 220. [Google Scholar] [CrossRef]
- Wang, M.; Guo, Z.; Wang, S. Regulation of cystathionine γ-lyase in mammalian cells by hypoxia. Biochem. Genet. 2014, 52, 29–37. [Google Scholar] [CrossRef]
- Cindrova-Davies, T.; Herrera, E.A.; Niu, Y.; Kingdom, J.; Giussani, D.A.; Burton, G.J. Reduced cystathionine γ-lyase and increased miR-21 expression are associated with increased vascular resistance in growth-restricted pregnancies: Hydrogen sulfide as a placental vasodilator. Am. J. Pathol. 2013, 182, 1448–1458. [Google Scholar] [CrossRef]
- Wu, B.; Teng, H.; Yang, G.; Wu, L.; Wang, R. Hydrogen sulfide inhibits the translational expression of hypoxia-inducible factor-1 alpha. Br. J. Pharmacol. 2012, 167, 1492–1505. [Google Scholar] [CrossRef]
- Manetti, M.; Guiducci, S.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Impaired Angiogenesis in Systemic Sclerosis: The Emerging Role of the Antiangiogenic VEGF165b Splice Variant. Trends Cardiovasc. Med. 2011, 21, 204–2010. [Google Scholar] [CrossRef]
- D’Alessio, S.; Fibbi, G.; Cinelli, M.; Guiducci, S.; Del Rosso, A.; Margheri, F.; Serrati, S.; Pucci, M.; Kahaleh, B.; Fan, P.; et al. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004, 50, 3275–3285. [Google Scholar] [CrossRef]
- Bhatia, M. Role of Hydrogen Sulfide in the Pathology of Inflammation. Scientifica 2012, 2012, 159680. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bhatia, M.; Zhu, Y.Z.; Zhu, Y.C.; Ramnath, R.D.; Wang, Z.J.; Anuar, F.B.M.; Whiteman, M.; Salto-Tellez, M.; Moore, P.K. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005, 19, 1196–1198. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, R.C.O.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef]
- Zhang, H.; Moochhala, S.M.; Bhatia, M. Endogenous Hydrogen Sulfide Regulates Inflammatory Response by Activating the ERK Pathway in Polymicrobial Sepsis. J. Immunol. 2008, 181, 4320–4331. [Google Scholar] [CrossRef]
- Rinaldi, L.; Gobbi, G.; Pambianco, M.; Micheloni, C.; Mirandola, P.; Vitale, M. Hydrogen sulfide prevents apoptosis of human PMN via inhibition of p38 and caspase 3. Lab. Investig. 2006, 86, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Armstrong, J.S.; Chu, S.H.; Jia-Ling, S.; Wong, B.S.; Cheung, N.S.; Halliwell, B.; Moore, P.K. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite “scavenger”? J. Neurochem. 2004, 90, 765–768. [Google Scholar] [CrossRef]
- Whiteman, M.; Cheung, N.S.; Zhu, Y.Z.; Chu, S.H.; Siau, J.L.; Wong, B.S.; Armstrong, J.S.; Moore, P.K. Hydrogen sulphide: A novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem. Biophys. Res. Commun. 2005, 326, 794–798. [Google Scholar] [CrossRef]
- Majors, A.; Ehrhart, L.A.; Pezacka, E.H. Homocysteine as a Risk Factor for Vascular Disease: Enhanced Collagen Production and Accumulation by Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2074–2081. [Google Scholar] [CrossRef]
- Mayer, O.; Filipovský, J.; Dolejšová, M.; Cífková, R.; Šimon, J.; Bolek, L. Mild hyperhomocysteinaemia is associated with increased aortic stiffness in general population. J. Hum. Hypertens. 2006, 20, 267–271. [Google Scholar] [CrossRef]
- Song, K.; Li, Q.; Yin, X.Y.; Lu, Y.; Liu, C.F.; Hu, L.F. Hydrogen sulfide: A therapeutic candidate for fibrotic disease? Oxid. Med. Cell. Longev. 2015, 2015, 458720. [Google Scholar] [CrossRef]
- Wang, Z.; Yin, X.; Gao, L.; Feng, S.; Song, K.; Li, L.; Lu, Y.; Shen, H. The protective effect of hydrogen sulfide on systemic sclerosis associated skin and lung fibrosis in mice model. Springerplus 2016, 5, 1084. [Google Scholar] [CrossRef]
- El-Seweidy, M.M.; Sadik, N.A.H.; Shaker, O.G. Role of sulfurous mineral water and sodium hydrosulfide as potent inhibitors of fibrosis in the heart of diabetic rats. Arch. Biochem. Biophys. 2011, 506, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Pan, S.; Li, J.; Dong, X.; Kang, K.; Zhao, M.; Jiang, X.; Kanwar, J.R.; Qiao, H.; Jiang, H.; et al. Hydrogen sulfide attenuates carbon tetrachloride-induced hepatotoxicity, liver cirrhosis and portal hypertension in rats. PLoS ONE 2011, 6, e25943. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Wang, F.; Li, Q.; Shi, Y.B.; Zheng, H.F.; Peng, H.; Shen, H.Y.; Liu, C.F.; Hu, L.F. Hydrogen sulfide inhibits the renal fibrosis of obstructive nephropathy. Kidney Int. 2014, 85, 1318–1329. [Google Scholar] [CrossRef]
- Jung, K.J.; Jang, H.S.; Kim, J.I.; Han, S.J.; Park, J.W.; Park, K.M. Involvement of hydrogen sulfide and homocysteine transsulfuration pathway in the progression of kidney fibrosis after ureteral obstruction. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1989–1997. [Google Scholar] [CrossRef]
- Bos, E.M.; Van Goor, H.; Joles, J.A.; Whiteman, M.; Leuvenink, H.G.D. Hydrogen sulfide: Physiological properties and therapeutic potential in ischaemia. Br. J. Pharmacol. 2015, 172, 1479–1493. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A. Heme Oxygenase-1/Carbon Monoxide: From Basic Science to Therapeutic Applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta Proteins Proteom. 2005, 1703, 93–109. [Google Scholar] [CrossRef]
- Xiao, L.; Dong, J.H.; Jin, S.; Xue, H.M.; Guo, Q.; Teng, X.; Wu, Y.M. Hydrogen Sulfide Improves Endothelial Dysfunction via Downregulating BMP4/COX-2 Pathway in Rats with Hypertension. Oxid. Med. Cell. Longev. 2016, 2016, 8128957. [Google Scholar] [CrossRef]
- Jin, S.; Teng, X.; Xiao, L.; Xue, H.; Guo, Q.; Duan, X.; Chen, Y.; Wu, Y. Hydrogen sulfide ameliorated L-NAME-induced hypertensive heart disease by the Akt/eNOS/NO pathway. Exp. Biol. Med. 2017, 242, 1831–1841. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Radpour, R.; Bachtler, M.; van Goor, H.; Smith, E.R.; Lister, A.; Odermatt, A.; Feelisch, M.; Pasch, A. Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis 2017, 265, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, C.K.; Calvert, J.W. Hydrogen sulfide and ischemia-reperfusion injury. Pharmacol. Res. 2010, 62, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fu, H.; Zhang, H.; Xu, F.; Zou, Z.; Liu, M.; Wang, Q.; Miao, M.; Shi, X. Hydrogen Sulfide Preconditioning Protects Rat Liver against Ischemia/Reperfusion Injury by Activating Akt-GSK-3β Signaling and Inhibiting Mitochondrial Permeability Transition. PLoS ONE 2013, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Johansen, D.; Ytrehus, K.; Baxter, G.F. Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury. Evidence for a role of KATP channels. Basic Res. Cardiol. 2006, 101, 53–60. [Google Scholar] [CrossRef]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.-L.; Huang, X.-W.; Wang, Y.-G.; Cao, Y.-X.; Zhang, C.-C.; Zhu, Y.-C. Hydrogen sulfide protects cardiomyocytes from hypoxia/reoxygenation-induced apoptosis by preventing GSK-3beta-dependent opening of mPTP. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, 1310–1319. [Google Scholar] [CrossRef]
- Tripatara, P.; Patel, N.S.A.; Collino, M.; Gallicchio, M.; Kieswich, J.; Castiglia, S.; Benetti, E.; Stewart, K.N.; Brown, P.A.; Yaqoob, M.M.; et al. Generation of endogenous hydrogen sulfide by cystathionine γ-lyase limits renal ischemia/reperfusion injury and dysfunction. Lab. Investig. 2008, 88, 1038–1048. [Google Scholar] [CrossRef]
- Xu, Z.; Prathapasinghe, G.; Wu, N.; Hwang, S.-Y.; Siow, Y.L.; O, K. Ischemia-reperfusion reduces cystathionine-β-synthase-mediated hydrogen sulfide generation in the kidney. Am. J. Physiol. 2009, 297, F27–F35. [Google Scholar] [CrossRef]
- Blackstone, E.; Morrison, M.; Roth, M.B. H2S induces a suspended animation-like state in mice. Science 2005, 308, 518. [Google Scholar] [CrossRef]
- Blackstone, E.; Roth, M.B. Suspended animation-like state protects mice from lethal hypoxia. Shock 2007, 27, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Gooding, K.M.; Whatmore, J.L.; Ball, C.I.; Mawson, D.; Skinner, K.; Tooke, J.E.; Shore, A.C. Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia 2010, 53, 1722–1726. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Jin, H.; Sun, L.; Chen, S.; Huang, Y.; Liu, J.; Li, Z.; Zhao, M.; Sun, Y.; Tang, C.; et al. Hydrogen sulfide alleviates myocardial collagen remodeling in association with inhibition of TGF-beta/Smad signaling pathway in spontaneously hypertensive rats. Mol. Med. 2015, 20, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Drapala, A.; Ufnal, M. Exogenous hydrogen sulfide causes different hemodynamic effects in normotensive and hypertensive rats via neurogenic mechanisms. Pharmacol. Rep. 2014, 66, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chen, S.; Wang, Y.; Liu, J.; Yao, Q.; Huang, Y.; Li, H.; Zhu, M.; Wang, S.; Li, L.; et al. Down-regulated CBS/H2S pathway is involved in high-salt-induced hypertension in Dahl rats. Nitric Oxide Biol. Chem. 2015, 46, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Tomasova, L.; Konopelski, P.; Ufnal, M. Gut bacteria and hydrogen sulfide: The new old players in circulatory system homeostasis. Molecules 2016, 21, 1558. [Google Scholar] [CrossRef]
- Tomasova, L.; Dobrowolski, L.; Jurkowska, H.; Wróbel, M.; Huc, T.; Ondrias, K.; Ostaszewski, R.; Ufnal, M. Intracolonic hydrogen sulfide lowers blood pressure in rats. Nitric Oxide Biol. Chem. 2016, 60, 50–58. [Google Scholar] [CrossRef]
- Freyer, D.R.; Chen, L.; Krailo, M.D.; Knight, K.; Villaluna, D.; Bliss, B.; Pollock, B.H.; Ramdas, J.; Lange, B.; Van Hoff, D.; et al. Effects of sodium thiosulfate versus observation on development of cisplatin-induced hearing loss in children with cancer (ACCL0431): A multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 63–74. [Google Scholar] [CrossRef]
- Whiteman, M.; Le Trionnaire, S.; Chopra, M.; Fox, B.; Whatmore, J. Emerging role of hydrogen sulfide in health and disease: Critical appraisal of biomarkers and pharmacological tools. Clin. Sci. 2011, 121, 459–488. [Google Scholar] [CrossRef]
- Cicone, J.S.; Petronis, J.B.; Embert, C.D.; Spector, D.A. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am. J. Kidney Dis. 2004, 43, 1104–1108. [Google Scholar] [CrossRef]
- Bourgeois, P.; De Haes, P. Sodium thiosulfate as a treatment for calciphylaxis: A case series. J. Dermatolog. Treat. 2016, 27, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Zitt, E.; König, M.; Vychytil, A.; Auinger, M.; Wallner, M.; Lingenhel, G.; Schilcher, G.; Rudnicki, M.; Salmhofer, H.; Lhotta, K. Use of sodium thiosulphate in a multi-interventional setting for the treatment of calciphylaxis in dialysis patients. Nephrol. Dial. Transplant. 2013, 28, 1232–1240. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdulle, A.E.; Van Goor, H.; Mulder, D.J. Hydrogen Sulfide: A Therapeutic Option in Systemic Sclerosis. Int. J. Mol. Sci. 2018, 19, 4121. https://doi.org/10.3390/ijms19124121
Abdulle AE, Van Goor H, Mulder DJ. Hydrogen Sulfide: A Therapeutic Option in Systemic Sclerosis. International Journal of Molecular Sciences. 2018; 19(12):4121. https://doi.org/10.3390/ijms19124121
Chicago/Turabian StyleAbdulle, Amaal Eman, Harry Van Goor, and Douwe J. Mulder. 2018. "Hydrogen Sulfide: A Therapeutic Option in Systemic Sclerosis" International Journal of Molecular Sciences 19, no. 12: 4121. https://doi.org/10.3390/ijms19124121
APA StyleAbdulle, A. E., Van Goor, H., & Mulder, D. J. (2018). Hydrogen Sulfide: A Therapeutic Option in Systemic Sclerosis. International Journal of Molecular Sciences, 19(12), 4121. https://doi.org/10.3390/ijms19124121