Potential Antitumor Activity of 2-O-α-d-Glucopyranosyl-6-O-(2-Pentylheptanoyl)-l-Ascorbic Acid

Abstract
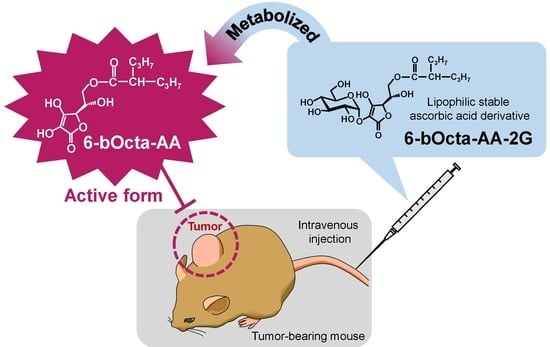
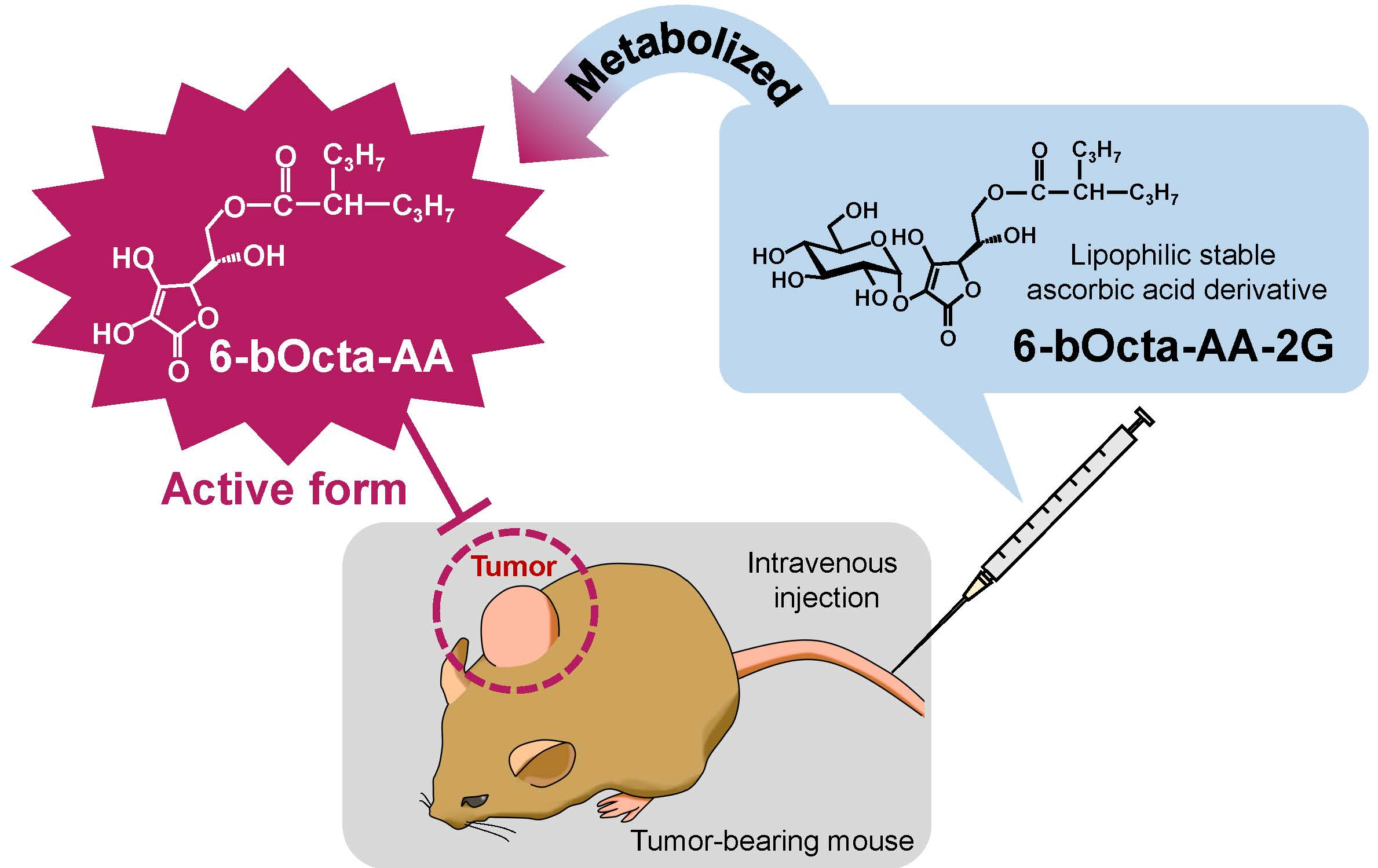
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
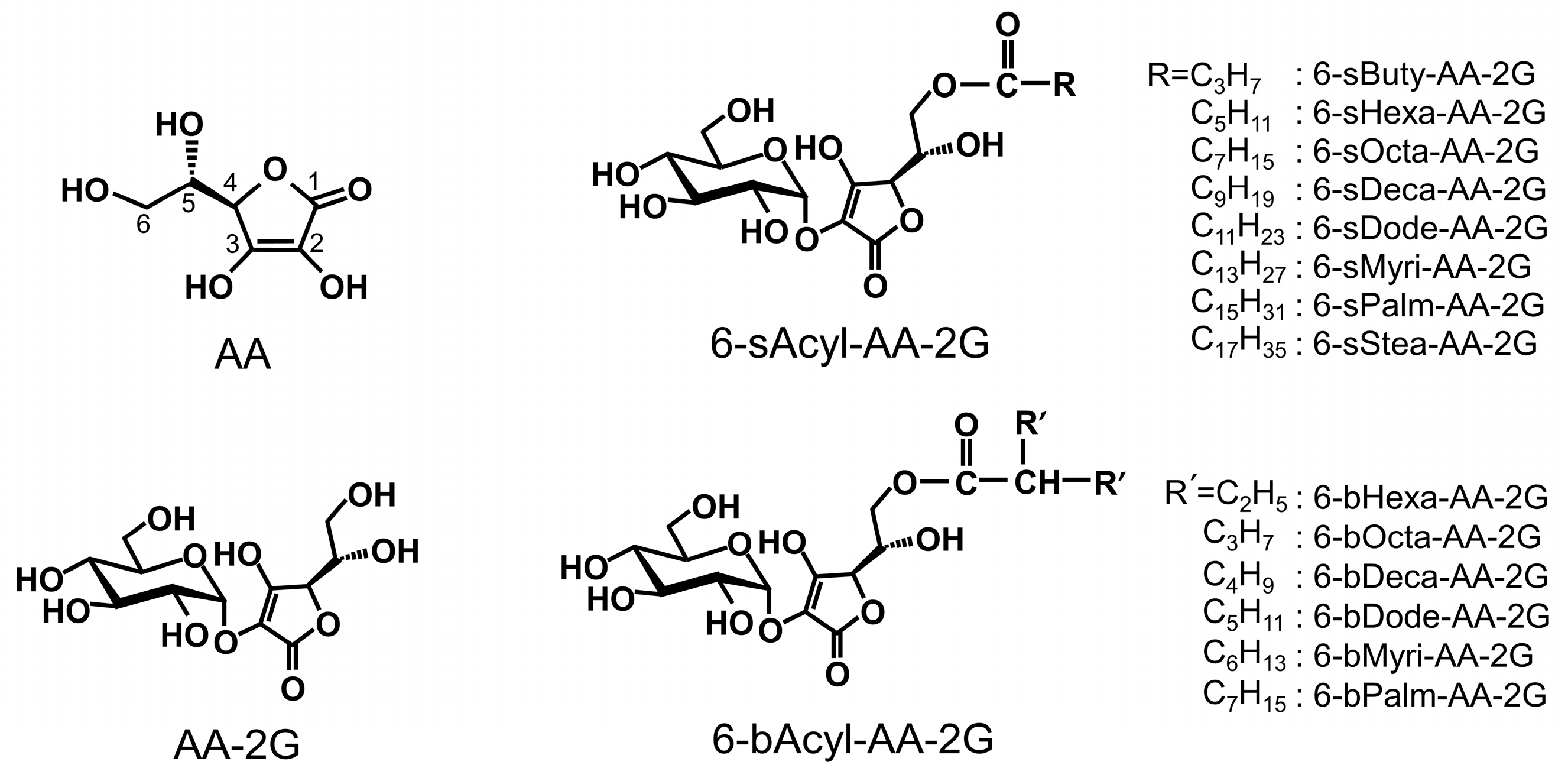
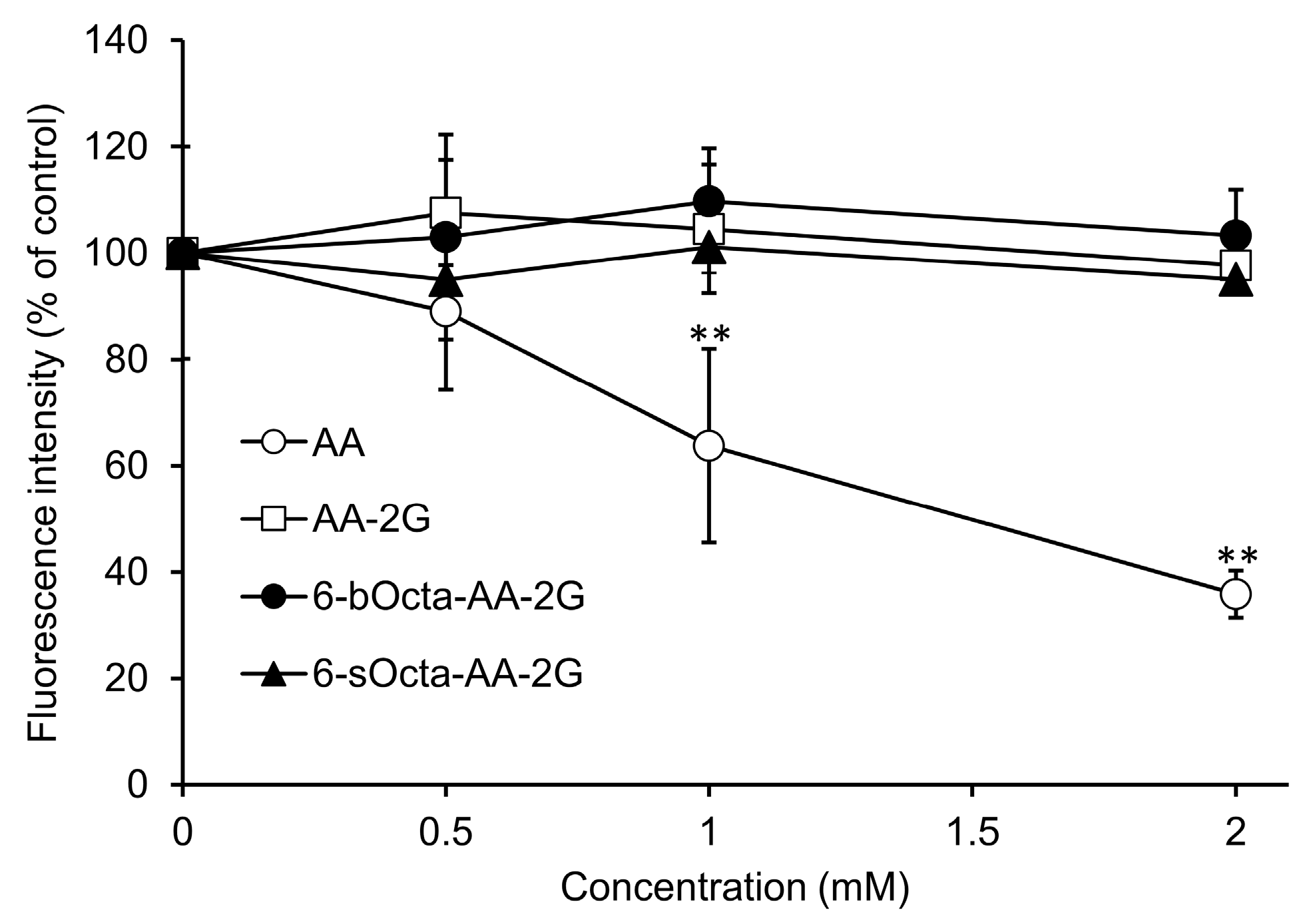
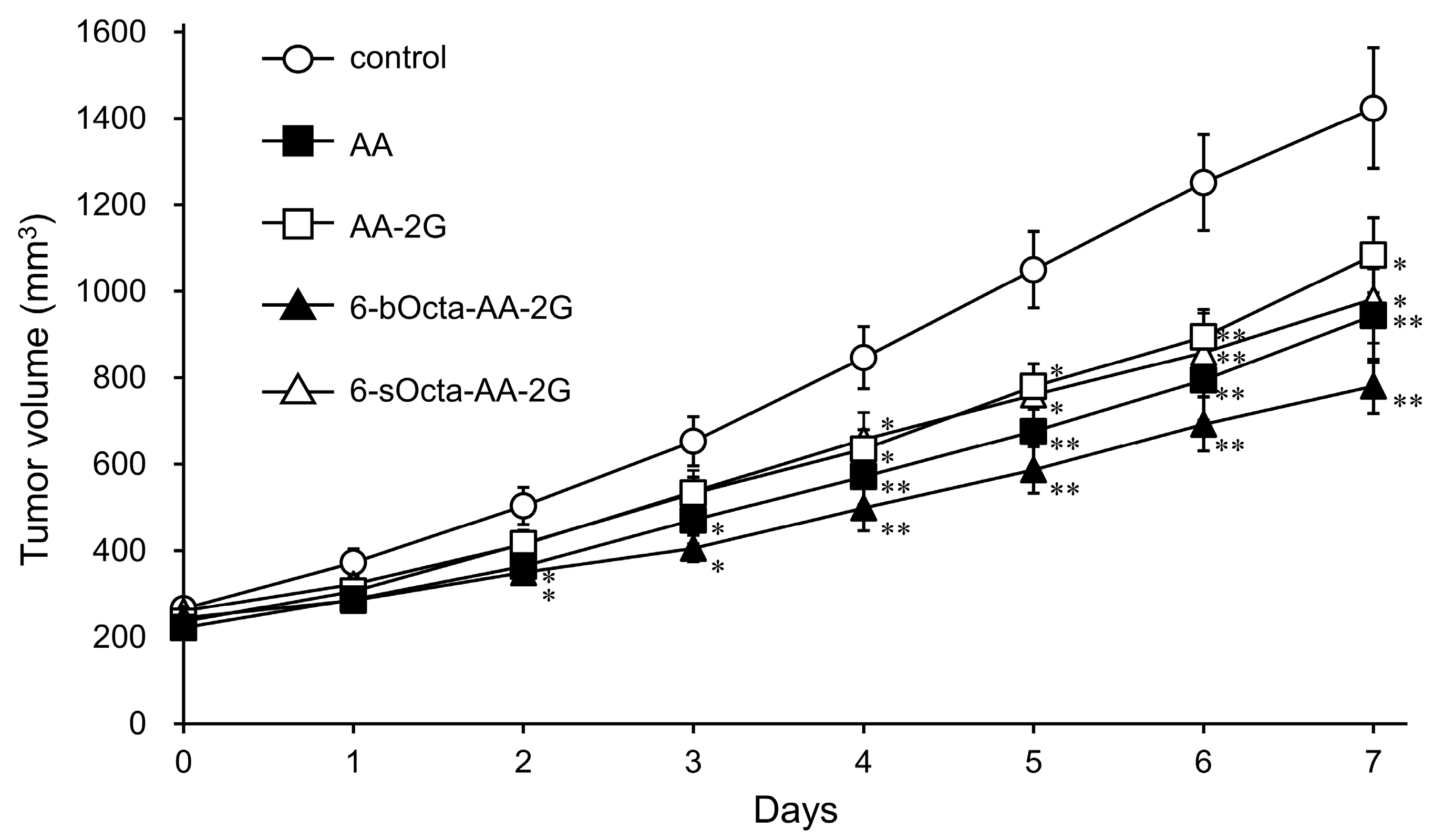
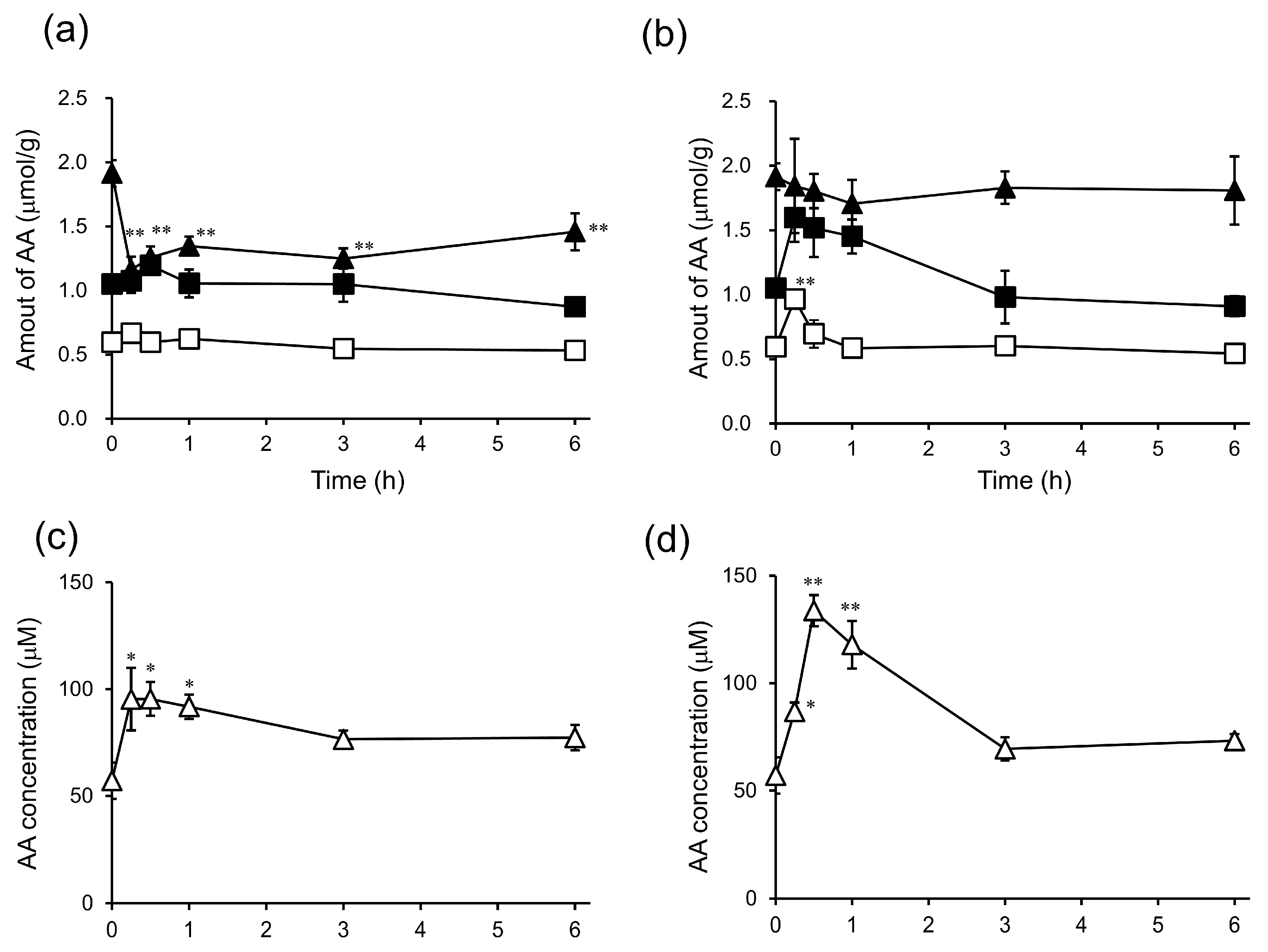
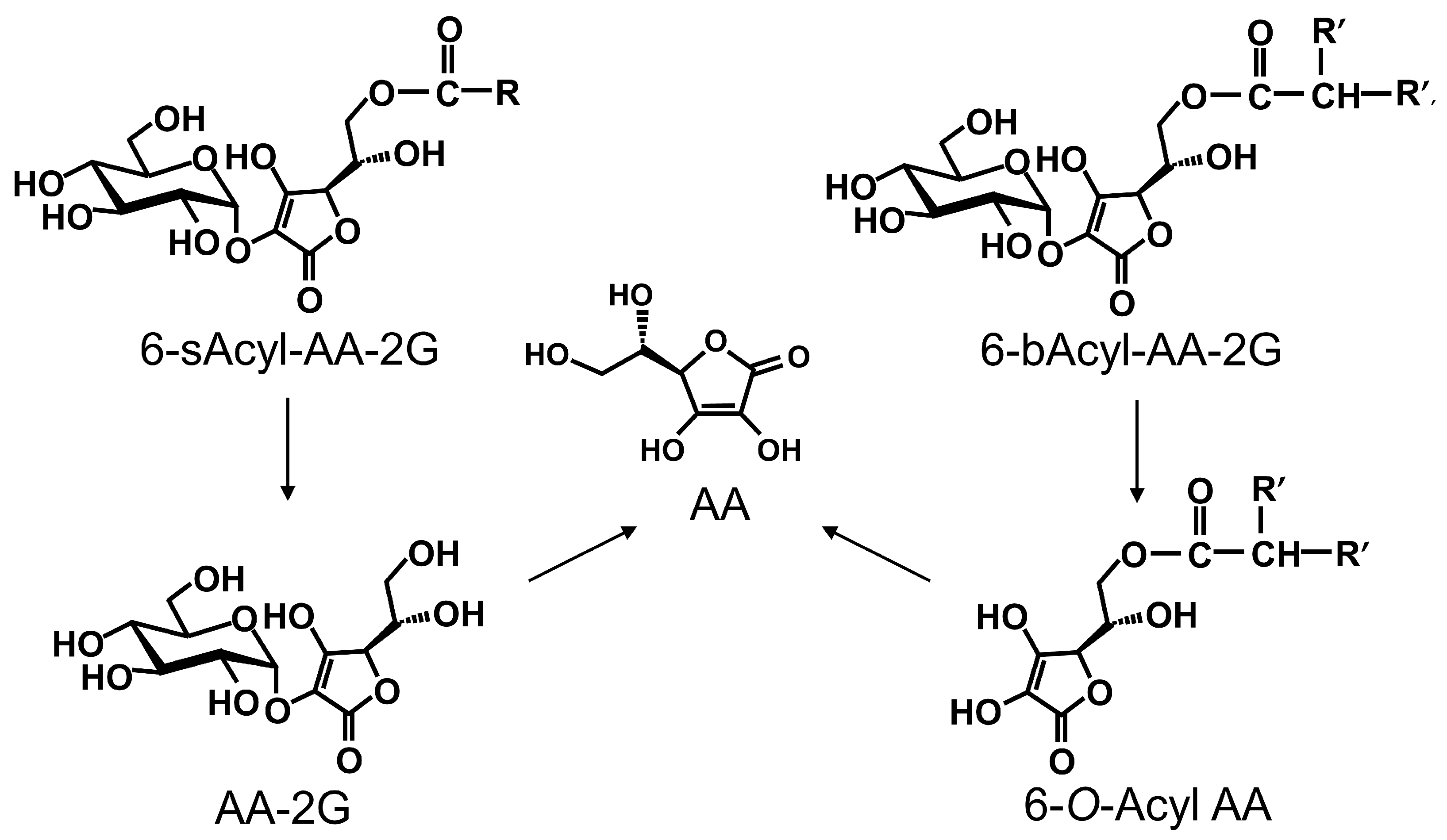
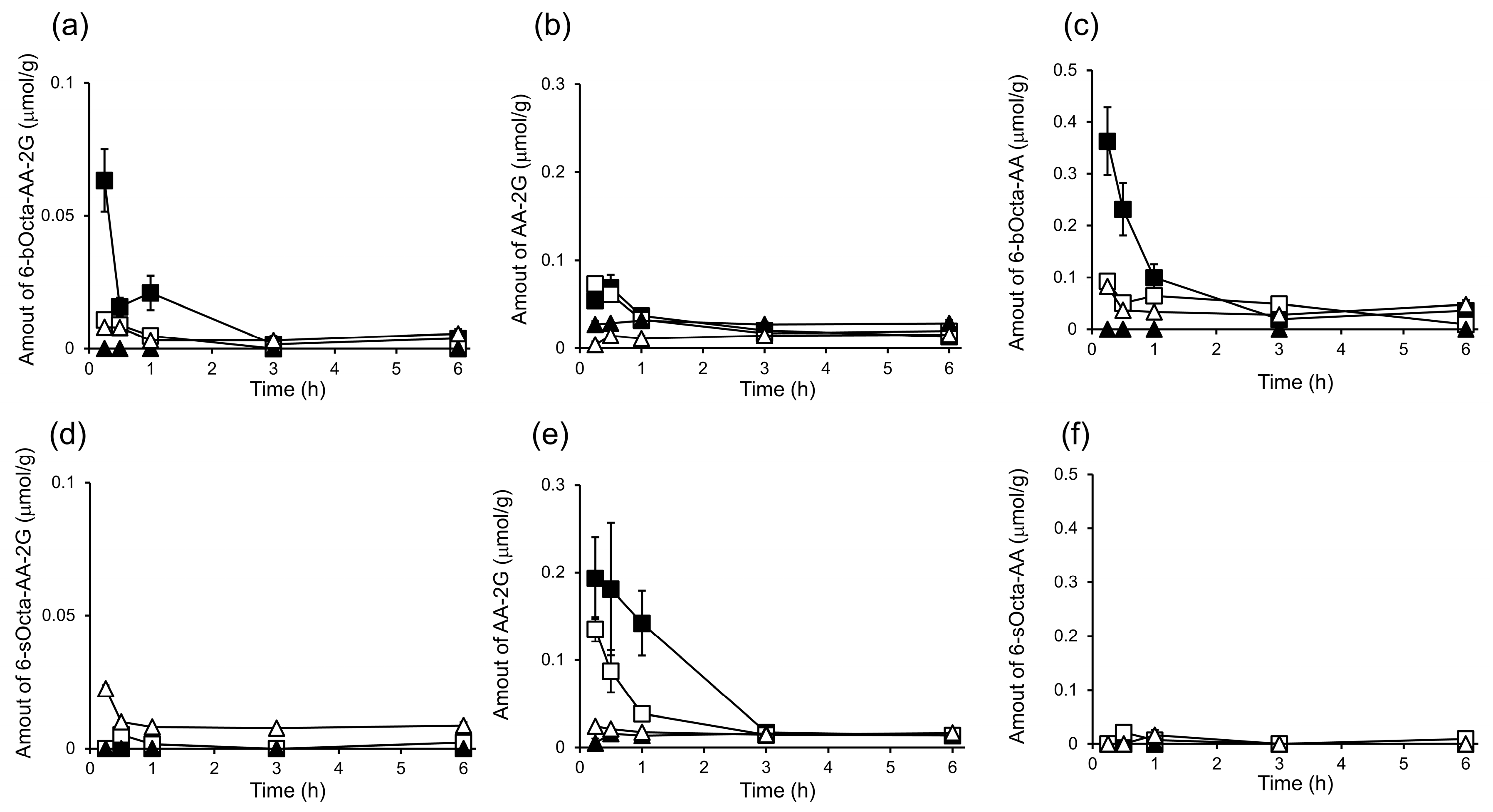
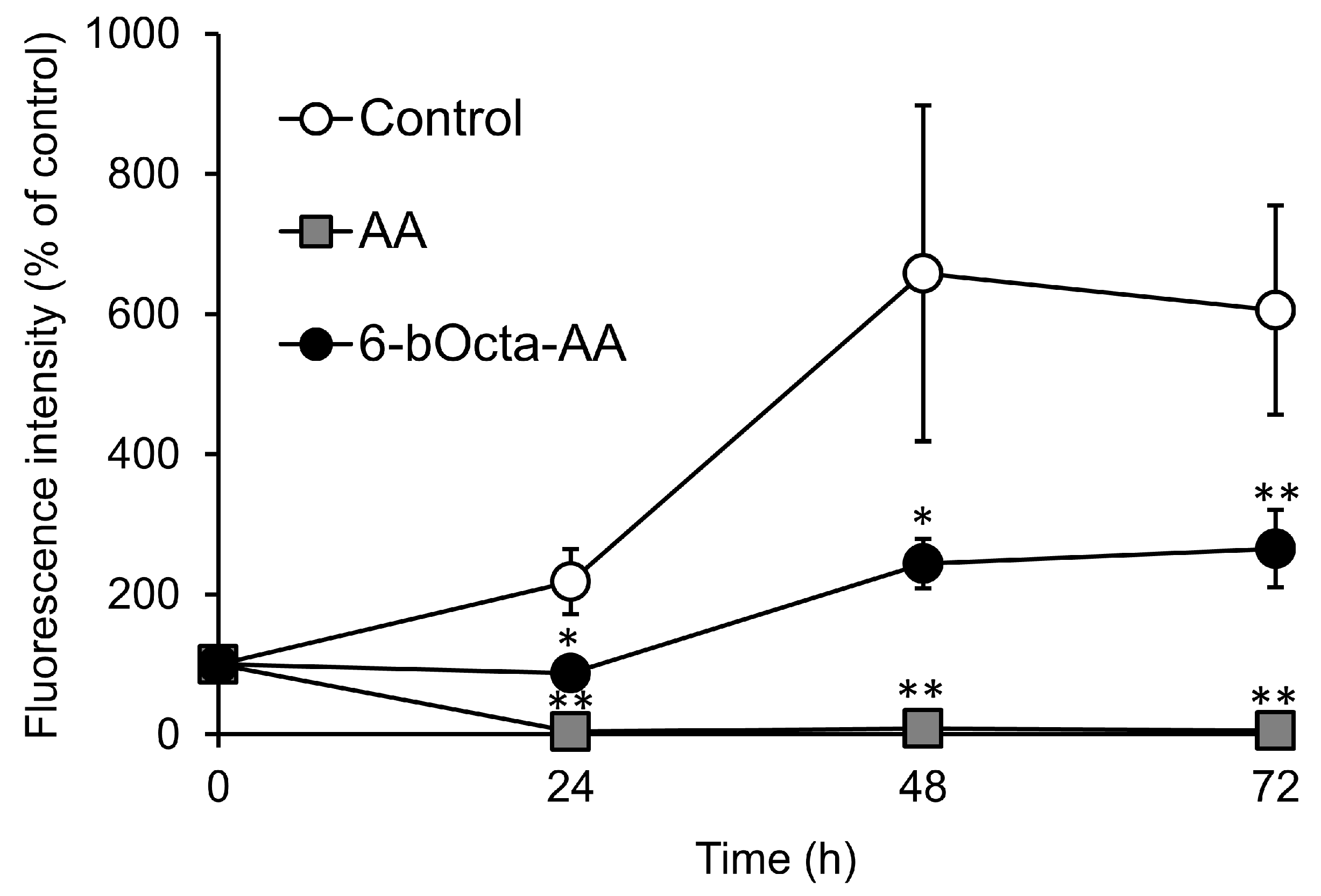
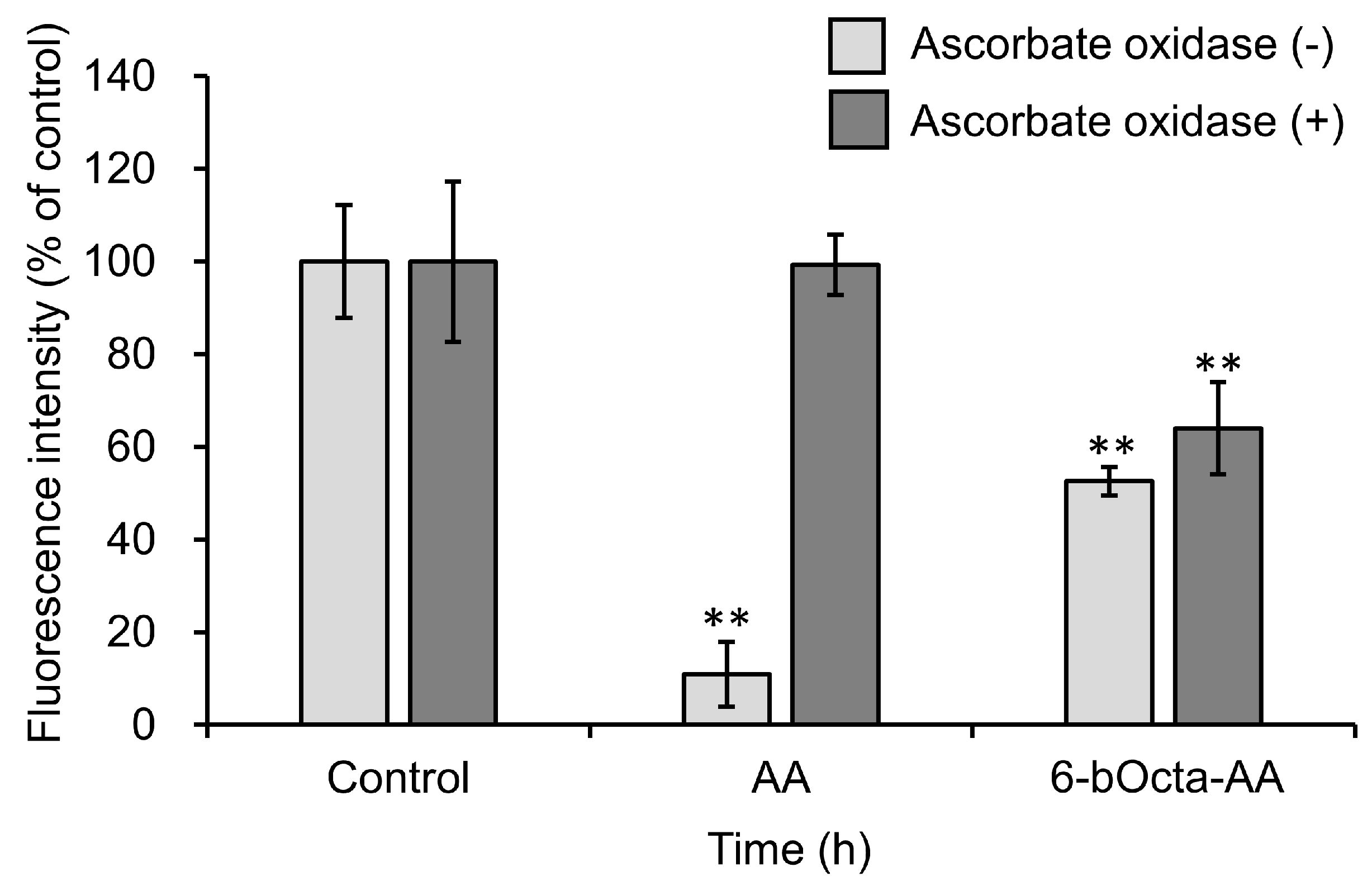
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of 6-O-(2-Propylpentanoyl)-l-Ascorbic Acid
3.3. Cell Lines
3.4. Evaluation of Cytotoxic Activity
3.5. Evaluation of Cytotoxic Activity in Short-Term Exposure of 6-bOcta-AA and AA
3.6. Evaluation of Antitumor Activity in Tumor-Bearing Mice
3.7. In Vivo Biodistribution and Clearance of 6-bOcta-AA-2G, 6-sOcta-AA-2G and Their Metabolites
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Reevaluation of prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1978, 75, 4535–4542. [Google Scholar] [CrossRef]
- Hoffer, L.J.; Levine, M.; Assouline, S.; Melnychuk, D.; Padayatty, S.J.; Rosadiuk, K.; Rousseau, C.; Robitaille, L.; Miller, W.H., Jr. Phase I clinical trial of IV ascorbic acid in advanced malignancy. Ann. Oncol. 2008, 19, 1969–1974. [Google Scholar] [CrossRef] [PubMed]
- Parrow, N.L.; Leshin, J.A.; Levine, M. Parenteral ascorbate as a cancer therapeutic: A reassessment based on pharmacokinetics. Antioxid. Redox Signal. 2013, 19, 2141–2156. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed]
- Oberley, T.D.; Oberley, L.W. Antioxidant enzyme levels in cancer. Histol. Histopathol. 1997, 12, 525–535. [Google Scholar] [PubMed]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 function blockes aberrant self-renewal and leukemia progression. Cell 2017, 170, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra18. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, I.; Tanaka, M.; Muto, N. Enhancement of in vitro antibody production of murine splenocytes by ascorbic acid 2-O-α-glucoside. Int. J. Immunopharmacol. 1993, 15, 319–325. [Google Scholar] [CrossRef]
- Ichiyama, K.; Mitsuzumi, H.; Zhong, M.; Tai, A.; Tsuchioka, A.; Kawai, S.; Yamamoto, I.; Gohda, E. Promotion of IL-4-and IL-5-dependent differentiation of anti-μ-primed B cells by ascorbic acid 2-glucoside. Immunol. Lett. 2009, 122, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, I.; Suga, S.; Mitoh, Y.; Tanaka, M.; Muto, N. Antiscorbutic activity of l-ascorbic acid 2-glucoside and its availability as a vitamin C supplement in normal rats and guinea pigs. J. Pharmacobiodyn. 1990, 13, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, I.; Muto, N.; Murakami, K.; Akiyama, J. Collagen synthesis in human skin fibroblasts is stimulated by a stable form of ascorbate, 2-O-α-d-glucopyranosyl-l-ascorbic acid. J. Nutr. 1992, 122, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Tai, A. 2-O-α-d-Glucopyranosyl-l-ascorbic acid as an antitumor agent for infusion therapy. Biochem. Biophys. Rep. 2017, 22, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Sun, A.Y.; Chen, Q.; Espey, M.G.; Drisko, J.; Levine, M. Vitamin C: Intravenous use by complementary and alternative medicine practitioners and adverse effects. PLoS ONE 2010, 5, e11414. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, C.M.; Levin, R.D.; Spector, T.; Lis, C.G. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother. Pharmacol. 2013, 72, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Rumsey, S.C.; Daruwala, R.; Park, J.B.; Wang, Y. Criteria and recommendations for vitamin C intake. JAMA 1999, 281, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- McAllister, C.J.; Scowden, E.B.; Dewberry, F.L.; Richman, A. Renal failure secondary to massive infusion of vitamin C. JAMA 1984, 252, 1684. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, I.; Tai, A.; Fujinami, Y.; Sasaki, K.; Okazaki, S. Synthesis and characterization of a series of novel monoacylated ascorbic acid derivatives, 6-O-acyl-2-O-α-d-glucopyranosyl-l-ascorbic acids, as skin antioxidants. J. Med. Chem. 2002, 45, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.; Kawasaki, D.; Sasaki, K.; Gohda, E.; Yamamoto, I. Synthesis and characterization of 6-O-acyl-2-O-α-d-glucopyranosyl-l-ascorbic acids with a branched-acyl chain. Chem. Pharm. Bull. 2003, 51, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.; Goto, A.; Ishiguro, Y.; Suzuki, K.; Nitoda, T.; Yamamoto, I. Permeation and metabolism of a series of novel lipophilic ascorbic acid derivatives, 6-O-acyl-2-O-α-d-glucopyranosyl-l-ascorbic acids with a branched-acyl chain, in a human living skin equivalent model. Bioorg. Med. Chem. Lett. 2004, 14, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.; Fujinami, Y.; Matsumoto, K.; Kawasaki, D.; Yamamoto, I. Bioavailability of a series of novel acylated ascorbic acid derivatives, 6-O-acyl-2-O-α-d-glucopyranosyl-l-ascorbic acids, as an ascorbic acid supplement in rats and guinea pigs. Biosci. Biotechnol. Biochem. 2002, 66, 1628–1634. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Morishita, Y.; Matsuno, H.; Aota, Y.; Ito, H.; Tai, A. Anti-allergic activity of monoacylated ascorbic acid 2-glucosides. Molecules 2017, 22, 2202. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yazama, F.; Tai, A. Oxidative stress-mediated antitumor activity of erythorbic acid in high doses. Biochem. Biophys. Rep. 2015, 3, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.; Kawasaki, D.; Goto, S.; Gohda, E.; Yamamoto, I. Vitamin C activity in guinea pigs of 6-O-acyl-2-O-α-d-glucopyranosyl-l-ascorbic acids with a branched-acyl chain. Biosci. Biotechnol. Biochem. 2003, 67, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Yamamoto, R. Synthesis of esters of ascorbic acid and their physicochemical properties. Yakugaku Zasshi 1966, 86, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Haranaka, K. Antitumor activity of murine tumor necrosis factor (TNF) against transplanted murine tumors and heterotransplanted human tumors in nude mice. Int. J. Cancer 1984, 34, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.; Gohda, E. Determination of ascorbic acid and its related compounds in foods and beverages by hydrophilic interaction liquid chromatography. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 853, 214–220. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miura, K.; Haraguchi, M.; Ito, H.; Tai, A. Potential Antitumor Activity of 2-O-α-d-Glucopyranosyl-6-O-(2-Pentylheptanoyl)-l-Ascorbic Acid. Int. J. Mol. Sci. 2018, 19, 535. https://doi.org/10.3390/ijms19020535
Miura K, Haraguchi M, Ito H, Tai A. Potential Antitumor Activity of 2-O-α-d-Glucopyranosyl-6-O-(2-Pentylheptanoyl)-l-Ascorbic Acid. International Journal of Molecular Sciences. 2018; 19(2):535. https://doi.org/10.3390/ijms19020535
Chicago/Turabian StyleMiura, Kaori, Misaki Haraguchi, Hideyuki Ito, and Akihiro Tai. 2018. "Potential Antitumor Activity of 2-O-α-d-Glucopyranosyl-6-O-(2-Pentylheptanoyl)-l-Ascorbic Acid" International Journal of Molecular Sciences 19, no. 2: 535. https://doi.org/10.3390/ijms19020535
APA StyleMiura, K., Haraguchi, M., Ito, H., & Tai, A. (2018). Potential Antitumor Activity of 2-O-α-d-Glucopyranosyl-6-O-(2-Pentylheptanoyl)-l-Ascorbic Acid. International Journal of Molecular Sciences, 19(2), 535. https://doi.org/10.3390/ijms19020535