1. Introduction
Influenza virus hemagglutinin is a trimeric, single-span transmembrane protein and is solely involved in membrane fusion between the viral envelope and the endosomal membrane of the infected host cell. It is composed of two subunits, HA1 and HA2, which are created by the proteolytic cleavage of hemagglutinin precursor HA0. The kind of the HA-processing subtilisin-like proteases has been firmly confirmed in the case of avian influenza viruses. In the case of human strains, the exact nature of cleaving enzymes remains unknown, but other proteases, such as type II transmembrane serine proteases and human airway trypsin-like protease (HAT), were pointed out as being involved [
1]. However, regardless of the protease family, as a result of cleavage reaction, the N-terminal part of HA2 subunit is ended with a free amine group.
Since the early observations by Lear and de Grado [
2] showing that membrane fusion of lipid vesicles can be accomplished by a short N-terminal synthetic fragment of HA2 subunit, such fusion peptides (HAfp) have been extensively used as full-length hemagglutinin mimicking species in the studies on membrane fusion. In the same seminal paper, the authors showed that HAfp1-20 was able to induce membrane fusion, in contrast to HAfp1-16 which appeared to be a poor fusogenic agent, even at concentrations saturating binding to POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) vesicles. This issue indicated that the length of a fusion peptide, possibly warranting its ability to reach the active conformation, may be an important factor to consider in the studies devoted to the mechanism of membrane fusion. Indeed, the most commonly studied HAfp1-20 was shown to have an open, “boomerang” conformation [
3,
4], in contrast to HAfp1-23, comprising the three conservative C-terminal residues (W21-Y22-G23), and adopting a distinct structure of a tight helical hairpin ([
5,
6,
7,
8,
9], and reviewed in [
10]). In agreement with the above observations, in our recent paper, we showed that the presence of the three C-terminal W21-Y22-G23 residues promotes the formation of a helical hairpin, which orients itself perpendicularly to the membrane plane and induces more disorder in the surrounding lipids than the less structured HAfp1-20 [
11].
Irrespective to peptide length, NMR experiments showed an elevated pK for G1 N-terminal amino group: 8.69 for HAfp1-20, measured in the presence of dioleoyl phosphocholine (PC) lipids [
12], and 8.8 for HAfp1-23, measured in the presence of dodecylphosphatidyl (DPC) detergents [
6]. These values are considerably higher than expected in a hydrophobic environment (8.00 ± 0.03) [
13]. In the helical hairpin formed by HAfp1-23, the N-terminal amine group of G1 was shown to form hydrogen bonds with carbonyl oxygen atoms of W21 and G23 and to be involved in charge-dipole interactions [
5,
6]. The assignment of protonation state of G1 was somewhat ambiguous in the molecular dynamics simulations performed hitherto. For instance, the N-terminus of the peptide was shown to be localized close to the amphiphatic membrane interface regardless of the protonation state of HAfp1-20 [
14]. The N-terminal charge was also shown to have an influence on the peptide orientation [
15,
16]. Apart from the mainly interfacial location of HAfp1-20, transmembrane configurations have been also reported for HAfp1-20 peptide with positively charged N-terminus [
16]. On the experimental side, broad comparative studies are missing, because either the neutral (acylated) form of the peptide was studied [
17], or peptides with both kinds of termini were compared, however they contained mutations for negatively charged glutamic acid residues [
18,
19].
In the light of our recent paper [
11], here we address the influence of the N-terminal positive charge on peptide configuration within the lipid bilayer, its impact on local membrane structure, and overall fusogenic activity. We compare HAfp1-20 and HAfp1-23 peptides either with N-terminal charged amine groups, or with neutral, acetylated N-termini, by means of experimental fluorescence techniques as well as full-atom, explicit solvent molecular dynamics simulations. For the sake of simplicity, we abbreviate the peptide names to indicate their length and the N-terminal group: 23N+ (HAfp1-23, unmodified amine N-terminus), 23Ac (HAfp1-23, acetylated N-terminus), 20N+ (HAfp1-20, unmodified amine N-terminus), 20Ac (HAfp1-20, acetylated N-terminus). Here, we demonstrate that the N-terminal charge is an important factor contributing to the activity of influenza fusion peptides owing to its role in the stabilization of helical hairpin conformation and peptide positioning in transmembrane configuration.
3. Discussion
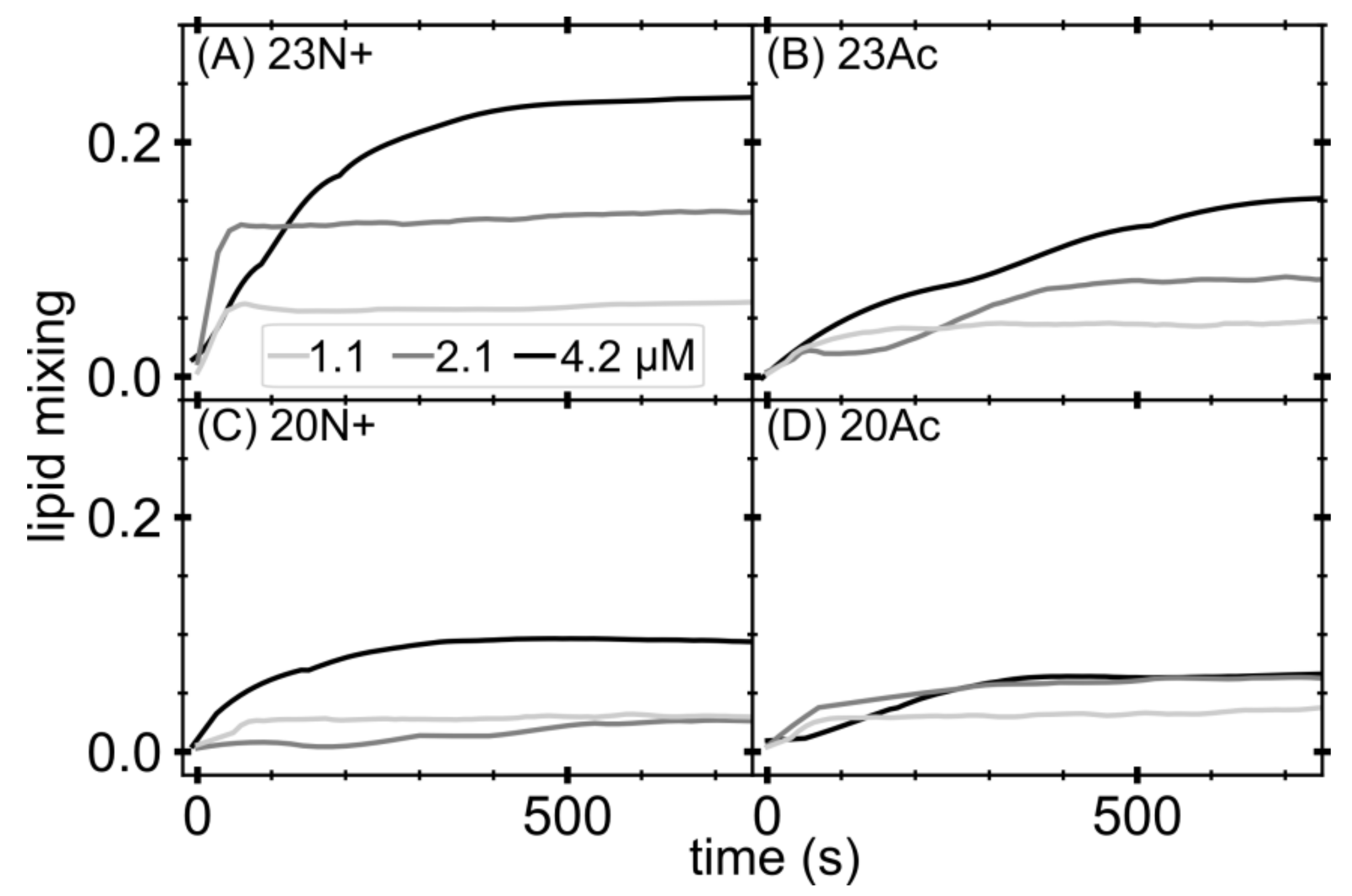
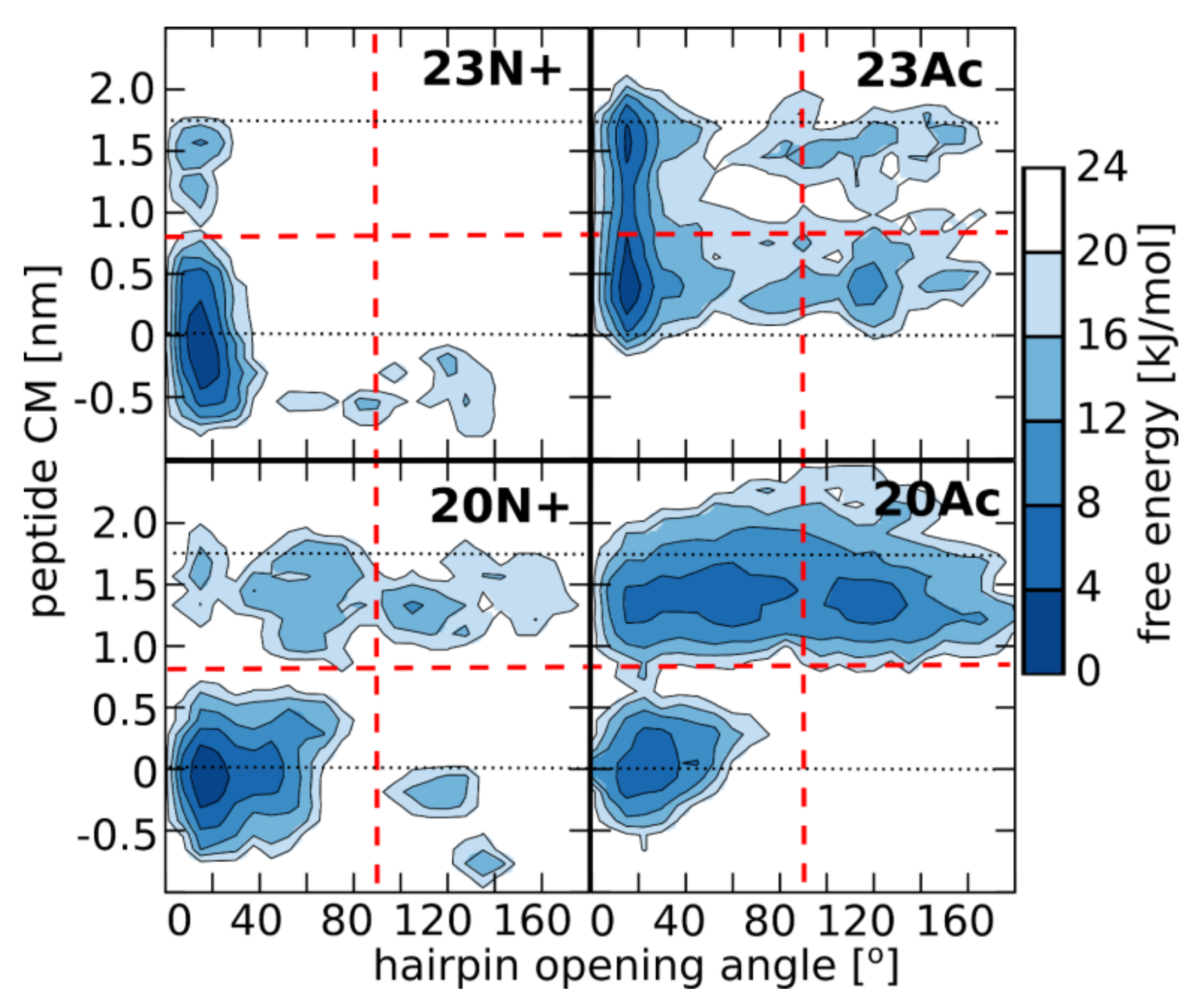
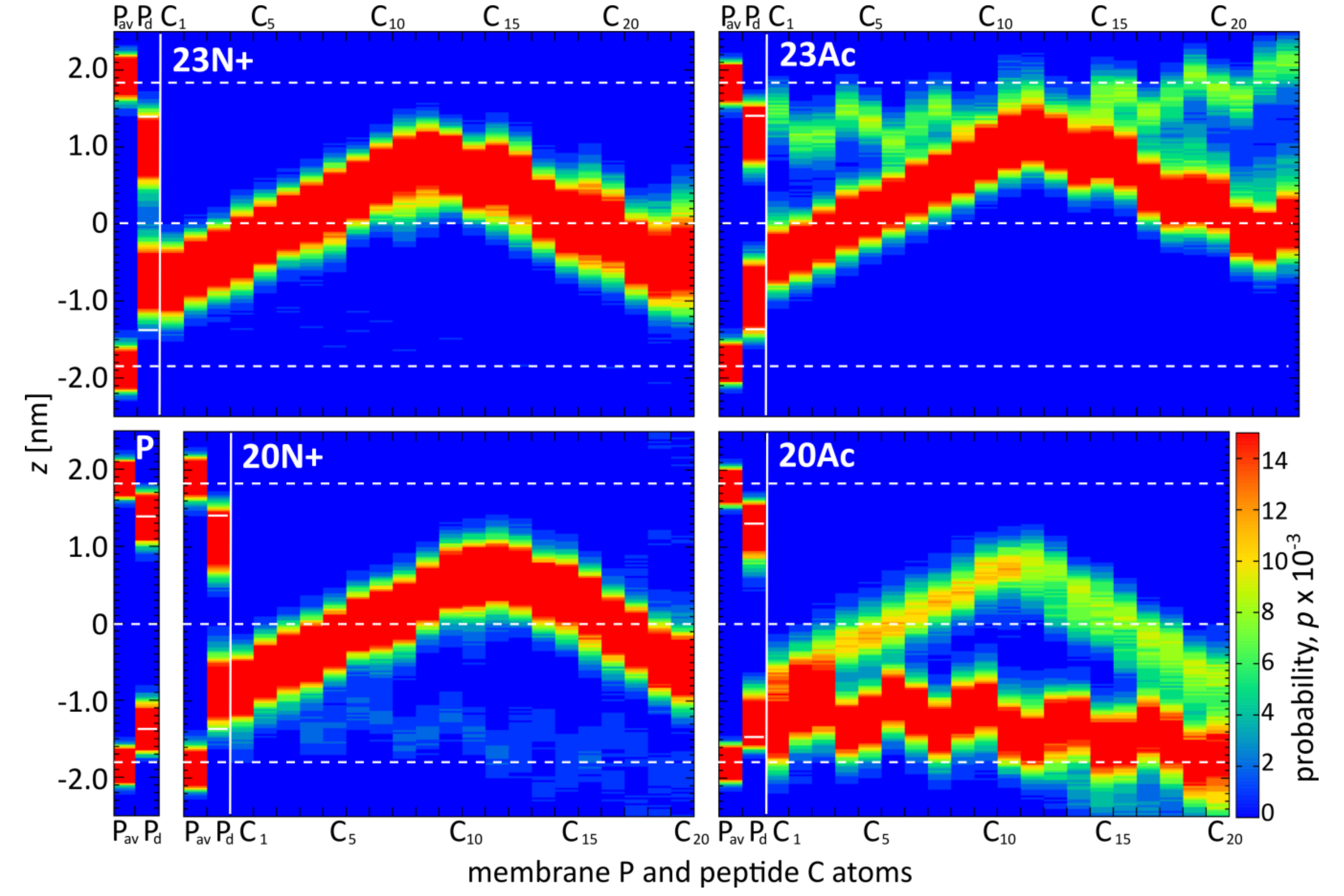
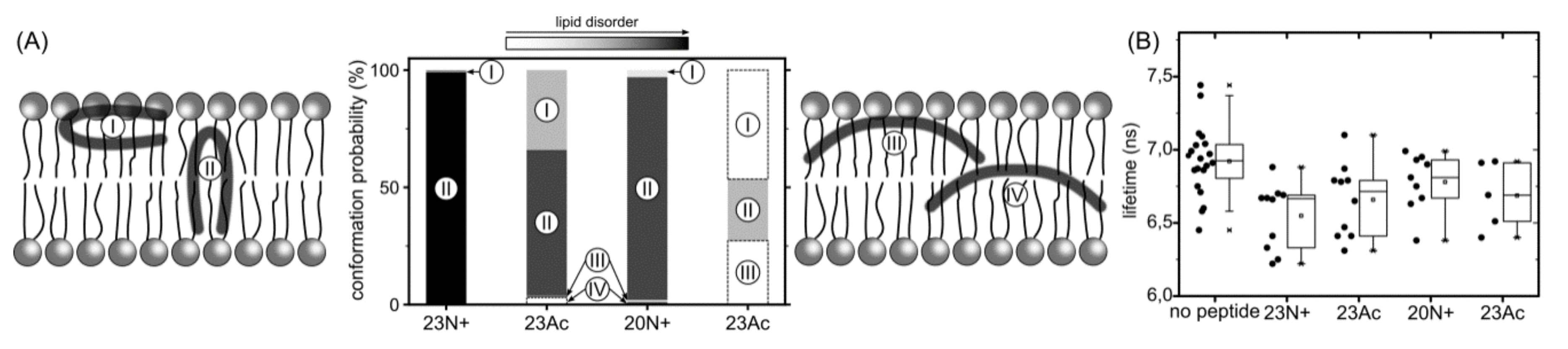
In the present study, we investigate the role of influenza fusion peptide N-terminus for its interaction with lipid bilayer and fusogenic activity, using experiments on phospholipid liposomes and atomistic molecular dynamics simulations. We find that the presence of a charged unmodified amino terminus group positively contributes to fusogenic activity in comparison to neutral, acetylated N-terminus. In parallel, the charged N terminus is observed to promote the formation of a tight helical hairpin peptide conformation, which is deeply buried within the membrane, in membrane-spanning orientation. According to our results, this configuration has the greatest potential to perturb lipid structure, likely explaining greater fusogenic activity peptides with unmodified amino terminus.
Indeed, the positive correlation between the depth of fusion peptide membrane insertion and fusogenity which was also observed for HIV fusion peptide [
22]. Furthermore, deeper peptide insertion into membrane was found to perturb lateral bilayer organization leading to increased lipid mixing with a proximal membrane, which is thought to be a prerequisite for the formation of a fusion pore [
23,
24,
25]. In agreement with our findings, peptide orientation closer to membrane normal was observed for wild-type HAfp1-25 peptide in DMPC:DMPG 1:1 (mol:mol) by attenuated total reflectance (ATR)-FTIR measurements [
26]. Similar membrane-spanning perpendicular configuration of HAfp1-23 was reported also for molecular dynamics simulations performed as self-assembly of DMPC lipids [
27]. We speculate that a closed hairpin deeply embedded in the membrane is the most fusogenic form of the fusion peptide also in vivo, in the hemagglutinin trimer, however the insertion depth may depend also on the lipid composition of membrane. Factors such as cholesterol content, percentage of saturated lipids and presence of negatively charged lipids may lead to more shallow, yet still effectively lipid-perturbing, peptide location.
Since the early works on fusion peptide-induced membrane fusion, it has been noticed that the binding energy is not necessarily related to the peptide activity [
2]. Although peptide length-dependent correlation between binding energy and fusogenity has been found for HAfp1-n (
n = 8, 13, 16, and 20) peptides [
28], the effects of N-terminal mutations do not indicate a simple energy-function relationship. For instance, binding energy for fusion-blocking HAfp1-20 G1V mutant has been shown to be ~2.75 kJ/mol more favorable than for HAfp1-16 peptide showing some fusogenic activity [
29]. At the same time, fusion-defective peptides were more self-associated at the membrane surface compared to wild-type peptides [
29]. Here we observe a similar effect for acetylated peptides: while more favorably surface-bound, they have smaller impact on the disorder of lipid acyl chains, that is presumably needed for membrane fusion. Such a relationship seems to be true not only in the case of influenza fusion peptides, but also for cell-penetrating peptides among which examples of surface-bound and less active peptides have been described (reviewed in [
30]).
The lack of clear correlation between peptide-membrane affinity and fusiogenic potential may, in part, be explained by the fact that experimentally determined partition coefficients served to estimate binding free energies, reflect free energy change between the states that correspond to: (a) peptide in solution plus unperturbed membrane; and (b) peptide bound into perturbed membrane. Accordingly, the overall binding free energy of membrane-perturbing peptides accounts also for unfavorable contributions due to disruption of equilibrium lipid structure. In other words, for a given concentration of fusion peptides in solution, fewer bound, but more membrane-perturbing units may induce fusion more effectively than multiple membrane associated, but less active structures. This notion seems to be well illustrated by the comparison of 23N+ and 23Ac peptides. The former one has less favorable binding free energy, but remains centrally located within the membrane, and its charged N terminus leads to significant intrusions of phosphate groups into the nonpolar core. The latter one fits well into the membrane with relatively buried neutral termini and solvent-exposed kink region. Its acetylated terminal group, however, is not able to establish hydrogen bonds with phosphate atoms of phospholipids, and hence, less effectively perturbs the membrane structure. This issue may be an important factor in the studies of other viral class I peptides also obtained as a result of proteolytic cleavage and therefore having a protonated free amine group (reviewed in [
31]). To the best of our knowledge, such systematic studies on viral fusion peptides do not exist and probably would contribute to better understanding of protein-mediated membrane fusion mechanisms.
Apart from being involved in the interactions with environment, the N-terminal NH
3+ group apparently contributes to hairpin structure stabilization by maintaining hydrogen bonds with C-terminal residues 21–23 [
5]. As indicated by surprisingly low membrane affinity of the 20Ac peptide, which adopts the most open conformation out of all peptides considered here, such closed hairpin structure may provide more compatible distribution of surface groups for peptide interaction with membrane environment.
4. Materials and Methods
4.1. Materials
The peptides were custom ordered with purity >95% (Genemed Synthesis, Inc., San Antonio, TX, USA and Lipopharm, Gdańsk, Poland). Sequences were as following: HAfp1-23N: GLFGAIAGFIEGGWQGMVDGWYG-amide, HAfp1-20N: GLFGAIAGFIEGGWQGMVDG-amide. Acetylated peptide (Ac) had the N-terminal amine substituted by amide group. Stocks were prepared from weighted amounts dissolved in DMSO as 300–500 µM solutions. Concentrations were checked by UV spectroscopy using the extinction coefficient at 280 nm of 12.490 M−1 cm−1 for all peptides. POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine), DOPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine), and C6-NBD-PC (1-palmitoyl-2-(6-((7-nitro-2-1,3-benzoxadiazol-4-yl)amino)hexanoyl)-sn-glycero-3-phosphocholine)were purchased from Avanti Polar Lipids Ltd. (Alabaster, AL, USA) and used with no further purifications. (N-(7-Nitrobenz-2-Oxa-1,3-Diazol-4-yl)-1,2-Dihexadecanoyl-sn-Glycero-3-Phosphoethanolamine (NBD-C6-PE) and Lissamine™ Rhodamine B 1,2-Dihexadecanoyl-sn-Glycero-3-Phosphoethanolamine (N-Rh-PE) used in fusion assays were from ThermoFisher Scientific (Waltham, MA, USA). All other chemicals were from Sigma Aldrich (Saint Louis, MO, USA). All experiments were performed in buffer pH 5.0 (10 mM citric acid/NaOH, 150 mM NaCl), and additional binding experiments at pH 7.4 in 10 mM Hepes/NaOH, 150 mM NaCl.
4.2. Liposome Preparation
Desired amounts of lipids in chloroform or chloroform/methanol 2/1 (v/v) were dried under a stream of nitrogen and overnight under vacuum, followed by rehydration with appropriate buffer to 2–10 mg/mL concentration. For LUV preparation, the dispersion was frozen and thawed in liquid nitrogen and 40 °C water bath at least 6 times, followed by extrusion (21 times) through polycarbonate filters with 100 nm pores (Whatman) using Avanti Mini Extruder (Avanti Polar Lipids Ltd., Alabaster, AL, USA). For SUV preparation, the dispersion was sonicated with a tip sonicator (VibraCell VCX130, Sonics and Materials, Newtown, CT, USA) in 7–20 pulses lasting 10 s separated by 10 s breaks until the solution was clear. GUVs were prepared by electroformation using Pt wire electrodes in homemade Teflon chambers. We applied 5 µl of 1 mg/mL lipid mixture in chloroform on each wire that was cleaned beforehand in ethanol in an ultrasonic bath, followed by 15 min of drying at 37 °C for solvent evaporation. The chamber was filled with 350 µL of 0.3 M sucrose and an AC-current of 3 V (peak-to-peak) was applied in two steps: 10 Hz for 2 h and 2 Hz for 0.5 h. Forty microliters of GUVs solution was transferred to a chambered 8-well cover glass #1 (Nunc LabTek II Chamber Slide System, ThermoFisher Scientific Waltham, MA, USA) or homemade chambers consisting of cut 1.5 mL Eppendorf tubes glued (Norland Optical Adhesive 63, Norland Products, Cranbury, NJ, USA) to a glass cover slip (22 mm × 22 mm, #1, Carl Roth, Karlsruhe, Germany). Each chamber contained 125 µL (or 255 µL in Nunc chambers) of buffer. The concentration of added peptides in the experiments with GUVs was in the range of 2.6–7.5 µM.
4.3. Lipid Fusion in LUVs
Lipid mixing of membrane fusion was measured by FRET using a Cary Eclipse (Varian, Agilent, Santa Clara, CA, USA) spectrofluorometer. For each lipid composition, unlabeled and labeled LUVs were prepared. To prepare the labeled LUVs, we included 1 mol% NBD-PE and N-Rh-PE in the lipid mixture before drying the lipids in the liposome preparation procedure. Unlabeled and labeled LUVs were mixed at a 9:1 ratio in pH 5.0 buffer. The total lipids concentration was 0.2 mM. After the equilibration of the vesicles, an appropriate amount of peptides from a stock solution was added to give final concentrations of 1.1, 2.1 and 4.2 μM. Then, 10% Triton X-100 was added to achieve a final concentration of 1% after fusion had been completed. Fluorescence intensity of the acceptor (excitation with 463 nm and emission at 590 nm) before the addition of peptides and after the addition of Triton X-100 was defined as 0% and 100% fusion, respectively. Experiments were performed in triplicates and averaged signal is shown.
4.4. FLIM Imaging
For FLIM laser scanning microscopy, an upgrade kit (PicoQuant, Berlin, Germany) installed on Zeiss LSM 710 (Carl Zeiss, Jena, Germany) was applied. The 256 × 256 pixel images of membrane dyes in GUVs were collected by excitation with a pulsed diode laser (pulse frequency: 25 MHz) with a wavelength of 485 nm focused by C-Apochromat 40×, 1.2 NA water immersion objective (Carl Zeiss, Jena, Germany). Emission of C6-NBD-PC was recorded using a 520/35 bandpass filter. Single photons were registered with a single photon avalanche photo diode and registered using a time-correlated single photon counting (TCSPC) approach. The decay curves were analyzed in the range not affected by the instrument response function (“tail-fit”). A non-linear least squares iterative fitting procedure was applied to obtain the fluorescence lifetimes of QDs by fitting a sum of two exponential decays. The average lifetime was calculated as τav = (ΣAiτi2/ΣAiτi).
4.5. Tryptophan Fluorescence
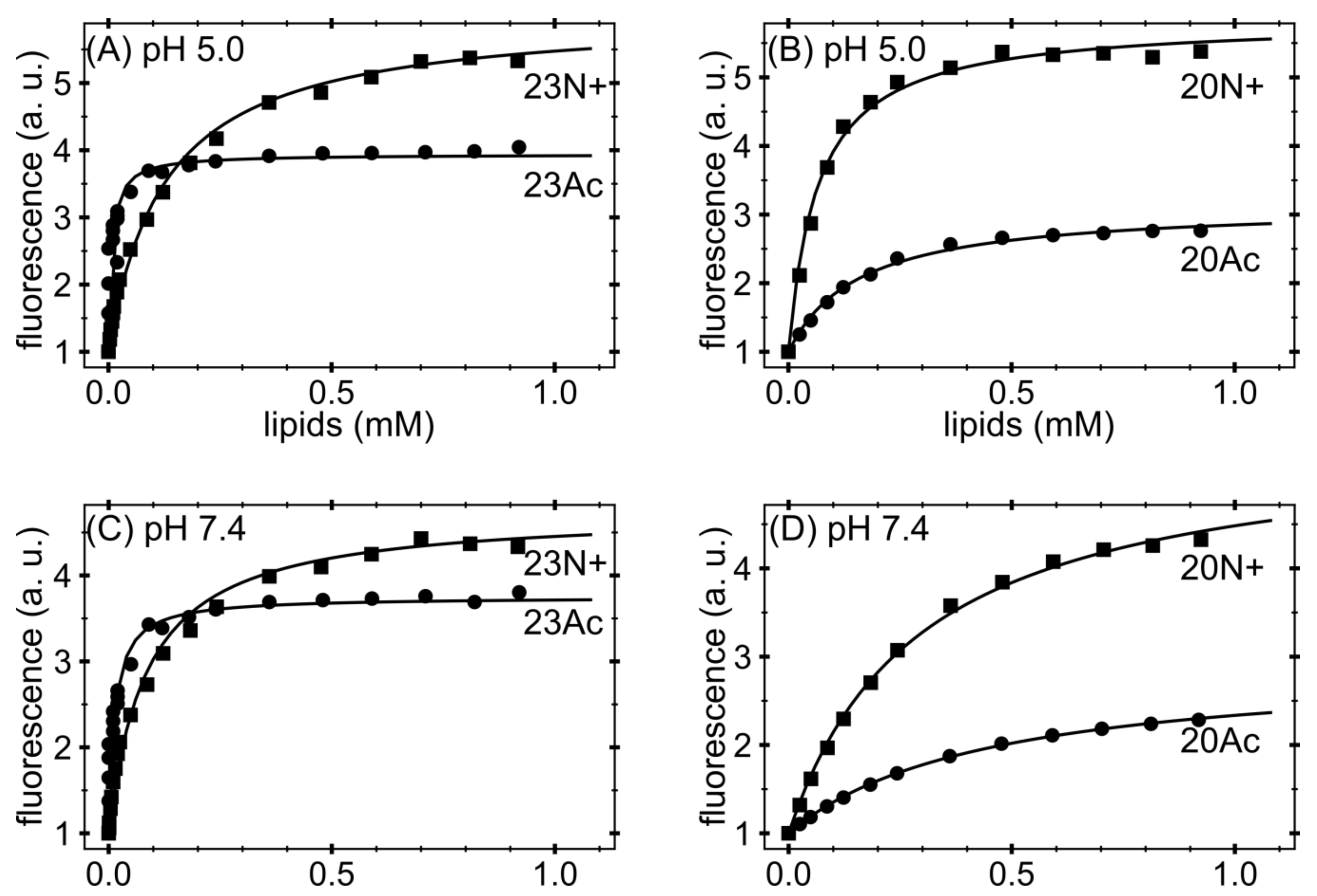
Fluorescence measurements were made with a Carry Eclipse (Varian, Agilent, Santa Clara, CA, USA) spectrofluorometer with an excitation wavelength of 280 nm. Excitation slits were not wider than 2.5 nm; emission slits were 4 nm. Photomultiplier voltage was 800 V. Spectra were measured using a 4 mm × 10 mm cuvettes (Hellma USA Inc., Plainview, NY, USA) in the emission region of 295–500 nm with an increment of 1 nm. Peptide solutions were used in 2–10 μM concentrations in 1000 μL volume, titrated with increasing portions of SUV suspension up to ~1mM in 13–20 steps. Normally, for each lipid concentration 3 spectra were averaged to achieve an adequate signal-to-noise ratio. Titration was performed with mild stirring and the cuvette was in the contact with a thermostat, assuring constant temperature of 22.0 × 0.5 °C. From each spectrum background was subtracted (by measuring blank titration). The titration curves were constructed as normalized intensity values for the wavelength for which the maximum spectral shift was observed between free and liposome-bound peptide (~328 nm). Such procedure was shown to govern a linear response of the signal in respect to the titrated peptide [
20]. The titration curves were further corrected for SUV scattering according to [
20] by using the tryptophan (
N-acetyl-
l-tryptophanamide) fluorescence registered under the same conditions in the presence of SUV solution at concentration [L] according to the equation:
To corrected data point, non-linear hyberbolic curve was fitted according to the equation:
where I denotes asymptotic intensity value, [
W] is the molar water concentration (=55.55 M) and
Kx is the molar partition coefficient. Gibbs free energies were calculated as:
4.6. Molecular Dynamics Simulations
Molecular dynamics simulations conducted in this work were based on the same methodology as in our previous study [
11]. Briefly, we used a temperature replica exchange method implemented in GROMACS 2016 package [
32], with amber99*ILDNP force field for peptides [
33], Lipd14 force field for POPC molecules [
34], and TIP3P model [
35] for water. Peptide sequences of GLFGAIAGFIEGGWQGMVDG(WYG) included protonated E11, charged or acetylated N-terminus, and amidated C-terminus, reflecting the experimental setup, and protonation state expected for pH = 5 [
36]. The systems including one peptide, 128 POPC phospholipids, and ~4000 water molecules were simulated using periodic boundary conditions, constrained bond lengths, 2 fs time step, 1 nm cutoff for van der Waals interactions with long range correction, particle mesh Ewald method with 0.12 nm grid spacing for electrostatic interactions. Pressure of 1 atm was maintained with anisotropic Parinello-Rahman barostat. Following spontaneous assembly procedure [
11], and 100 ns equilibration runs, each system was simulated using 24 replicas with temperatures ranging from 310 to 376 K, with spacing determined to provide equal exchange probabilities [
36]. Replica exchange simulation was conducted for 200 ns for each system, 150 ns of which at T = 310 K was used for analysis.
SCD order parameters were calculated using the algorithm implemented in g_lomepro program [
37], while hydrogen bond analysis was carried out using gmx hbond Gromacs tool.
5. Conclusions
Based on atomistic molecular dynamics simulations of peptide–membrane systems and experiments on phospholipid liposomes, our study provides insights into the influence of influenza fusion peptide N-terminal group on its interaction with lipid membrane and the resulting fusogenic activity. According to our observations, the most fusogenic form of the fusion peptide corresponds to a tight helical hairpin, deeply buried within the membrane, and adopting membrane-spanning orientation. As expected, we find that an unmodified, charged N-terminal amino group contributes positively to the fusogenic activity in comparison with neutral, acetylated N-terminus. We ascribe the following roles to the charged N-terminus: (a) stabilization of the closed hairpin peptide form by hydrogen bonds with residues 21–23; (b) stabilization of perpendicular, centrally located membrane spanning hairpin configuration by maintaining polar contacts with aqueous environment and membrane phosphate groups, which, together with analogous interactions on the kink side, contribute to a pair of forces that fix hairpin ends to opposite sides of the bilayer; and (c) influence on lipid disorder by promoting phosphate groups intrusions into the nonpolar membrane core.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}