Key Anti-Fibrosis Associated Long Noncoding RNAs Identified in Human Hepatic Stellate Cell via Transcriptome Sequencing Analysis

Abstract
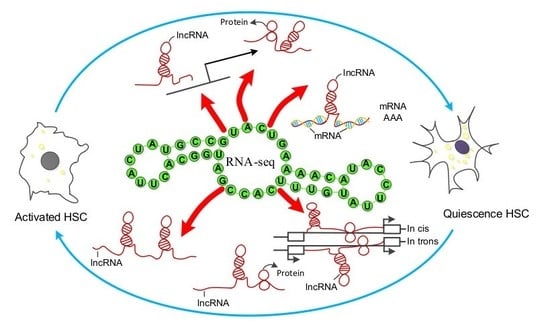
:
1. Introduction
2. Results
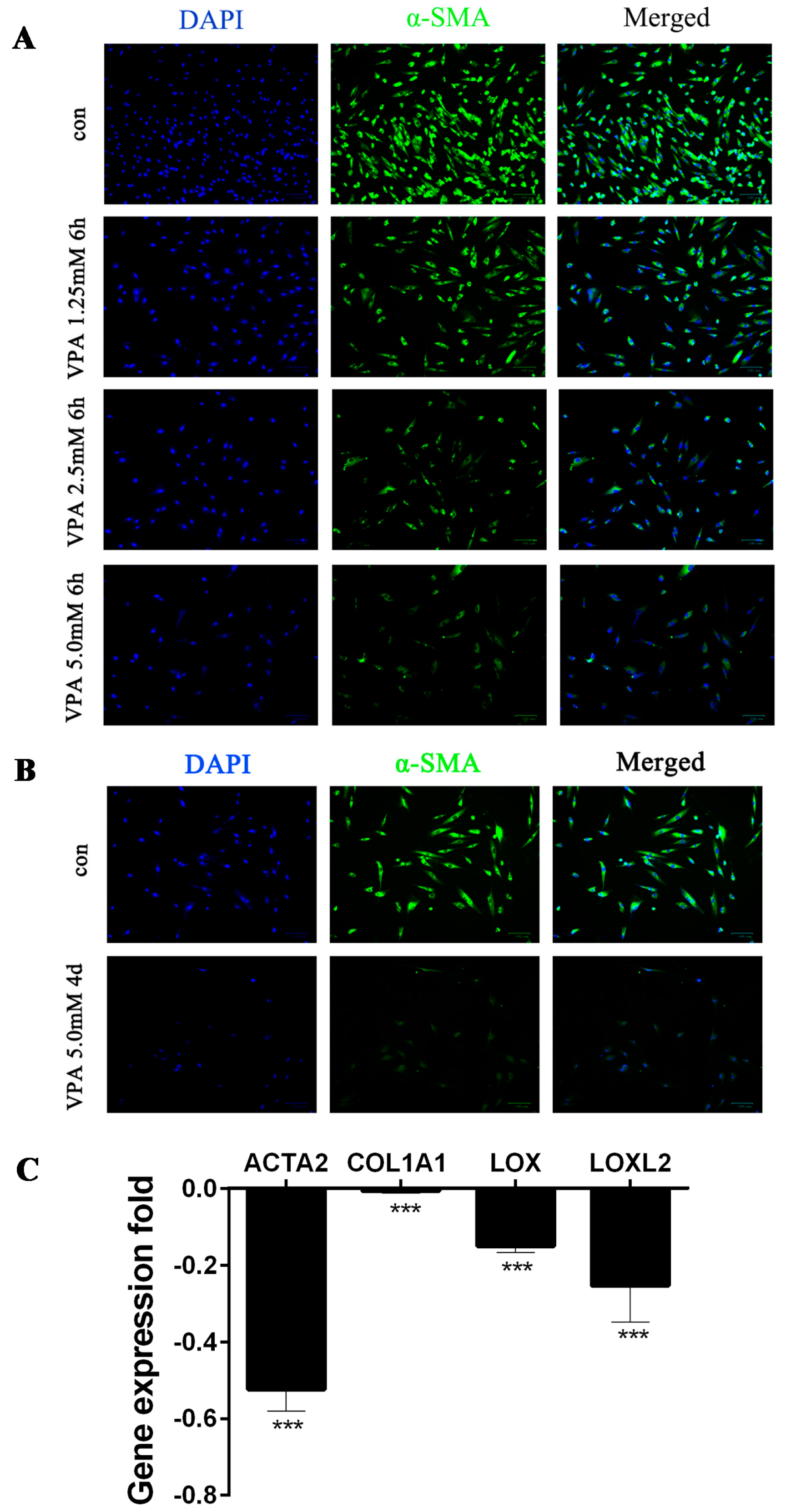
2.1. VPA-Mediated Induction of hHSC Myofibroblasts Conversion into an Inactive Phenotype Subsection
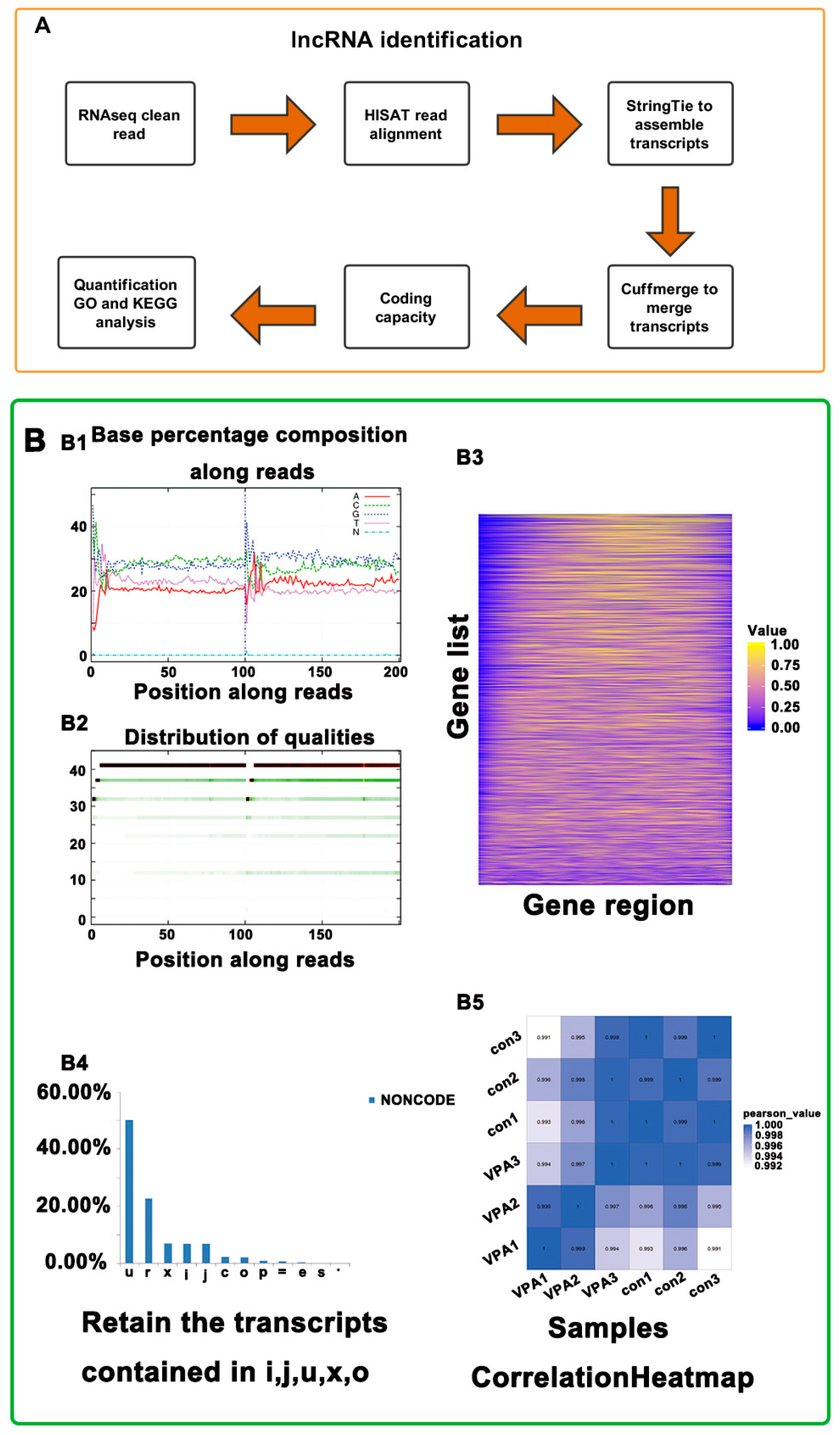
2.2. Overview of RNA-Seq in hHSCs Myofibroblasts
2.3. Identification of lncRNA and mRNA in hHSC Myofibroblasts
2.4. Profiling of Differentially Expressed (DE) lncRNA and mRNA in Activated hHSCs and Quiescent hHSCs
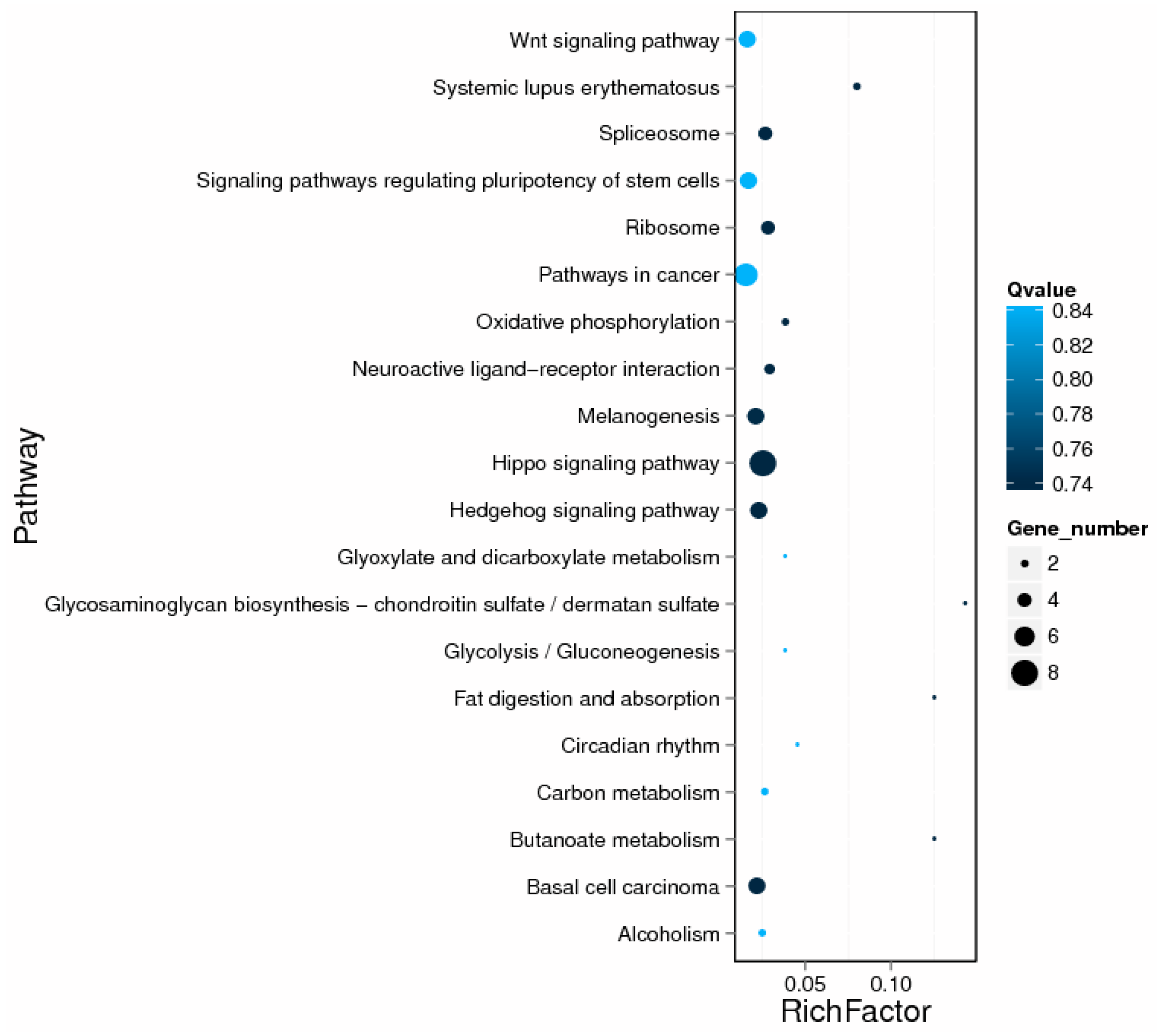
2.5. GO and KEGG Enrichment Analysis of DE lncRNA Target Genes
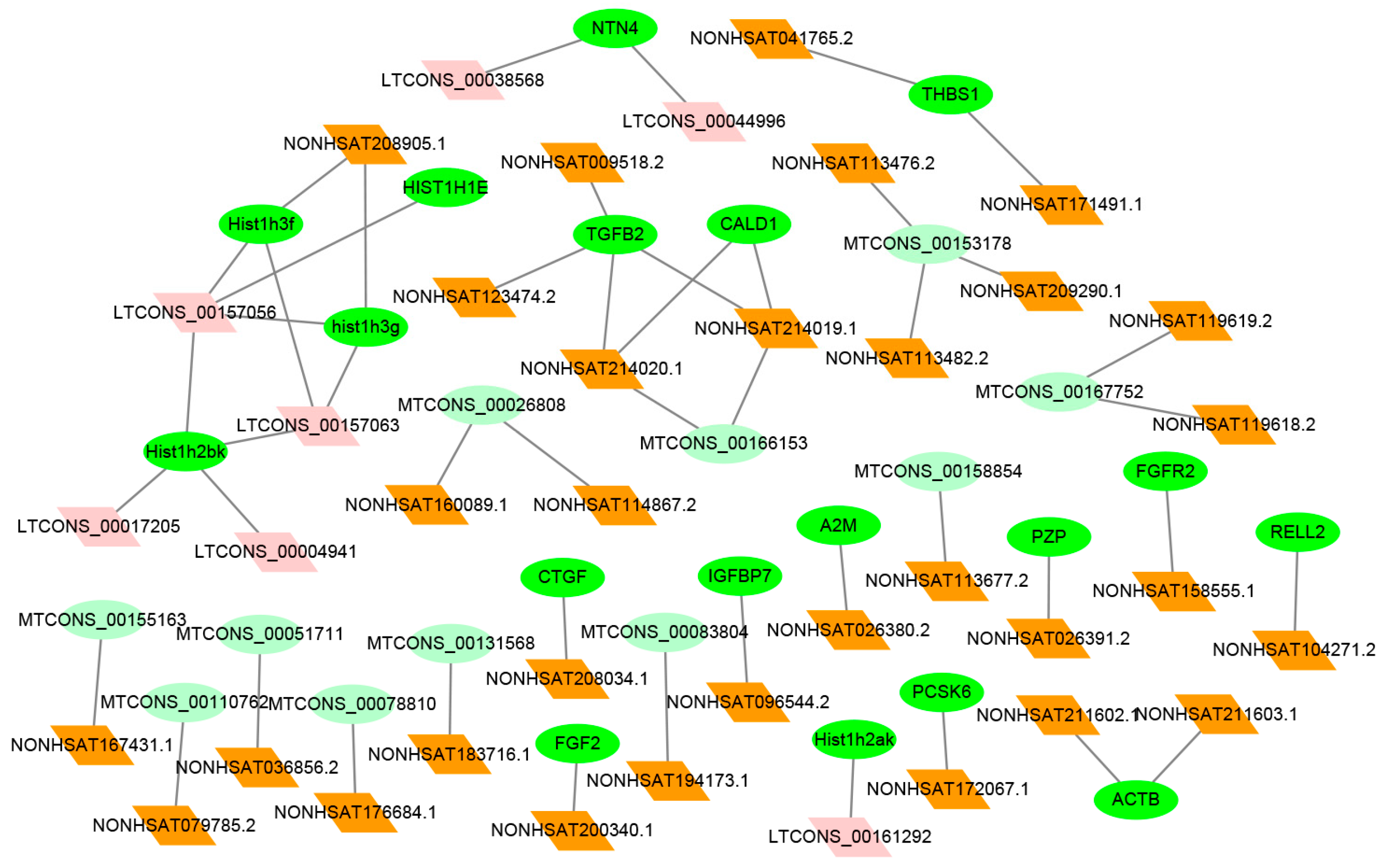
2.6. Co-Expression Analysis between Differentially Expression (DE) lncRNAs–mRNAs
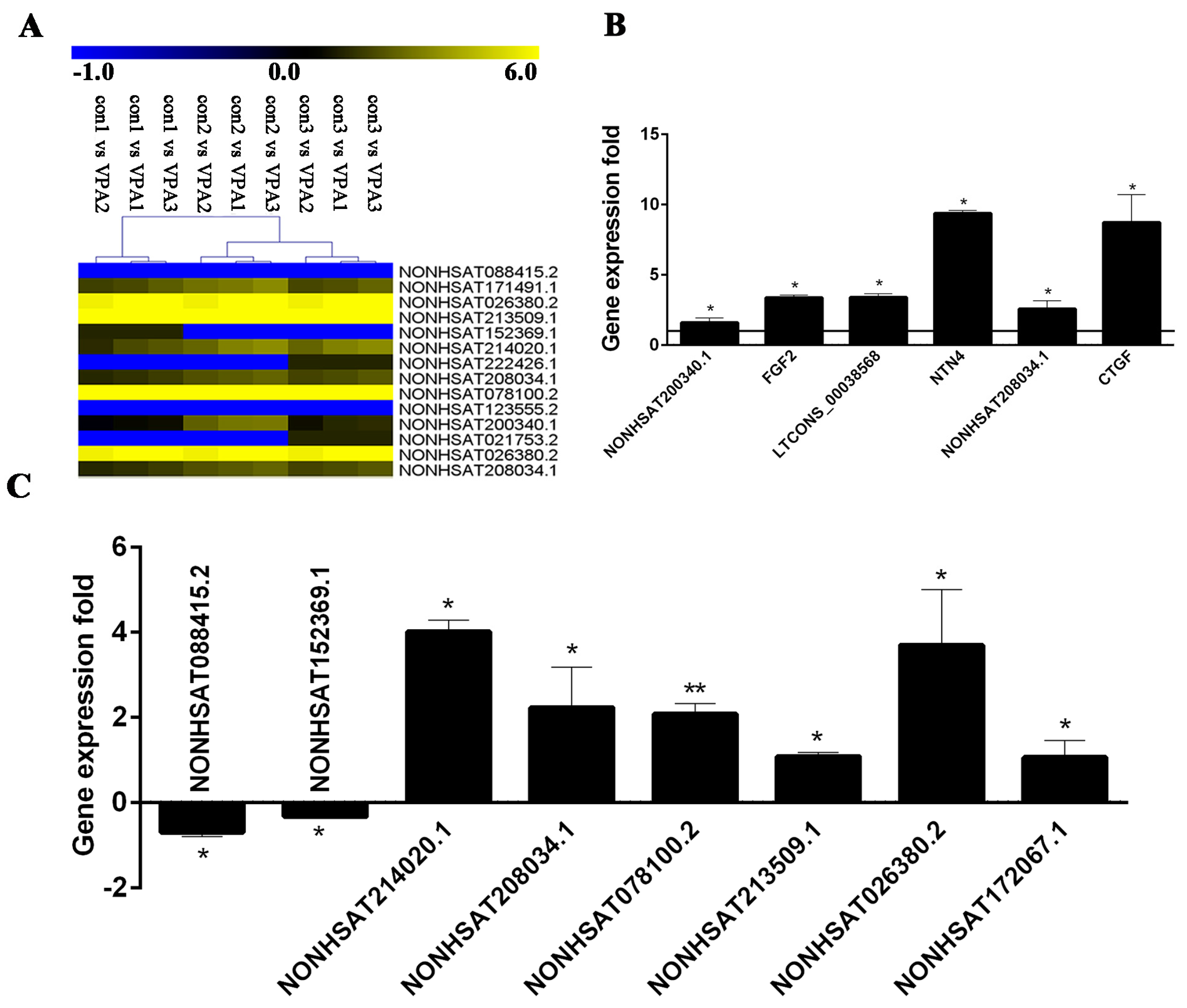
2.7. Verification of DE lncRNA and Co-Expression mRNA
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Immunofluorescence
4.3. Quantitative Real-Time PCR
4.4. cDNA Library Construction and Sequencing
4.5. Alignment and Transcript Assembly
- Reads were splice aligned to the genome.
- A graph representing alternative splicing events was constructed.
- The graph were aligned transversely to assemble.
- Isoforms were assembled.
4.6. Identification of lncRNAs and mRNAs
4.7. Differential Gene Expression Analyses
4.8. Gene Ontology (GO) Terms and Kyoto Encyclopaedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis
4.9. Co-Expression Network Analysis and Prediction of the Target Genes of DE lncRNAs
4.10. Statistical Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| hHSC | human hepatic stellate cell |
| lncRNA | Long noncoding RNA |
| RNA-seq | RNA sequencing |
| DE | differentially expressed |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopaedia of Genes & Genomes |
| CTGF | connective tissue growth factor |
| FGF2 | fibroblast growth factor |
| NTN4 | netrin-4 |
| ACTA2 | actin, alpha2 smooth muscle |
| COL1A1 | collagen type I alpha 1 chain |
| LOX | lysyl oxidase |
| LOXL2 | lysyl oxidase like 2 |
| FGFR2 | fibroblast growth factor receptor 2 |
| TGF-β | transforming growth factor beta |
| PPAR | peroxisome proliferator-activated receptors |
| ChIP-seq | Chromatin immunoprecipitation sequencing |
| α-SMA | actin, aortic smooth muscle |
| HDAC | Histone Deacetylase |
| IGFBP7 | insulin like growth factor binding protein 7 |
| FPKM | fragments per kilobase of exon per million fragments mapped |
| RPKM | Reads Per Kilobase per Million mapped reads |
References
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Deng, X.; Liang, J. Modulation of hepatic stellate cells and reversibility of hepatic fibrosis. Exp. Cell Res. 2017, 352, 4204–4226. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Graff, A.; Kruger, S.; Bochard, F.; Gabbiani, G.; Denk, H. Modulation of alpha smooth muscle actin and desmin expression in perisinusoidal cells of normal and diseased human livers. Am. J. Pathol. 1991, 138, 1233–1242. [Google Scholar]
- Yum, M.J.; Koppula, S.; Kim, J.S.; Shin, G.M.; Chae, Y.J.; Yoon, T.; Chun, C.S.; Lee, J.D.; Song, M. Protective effects of ampelopsis brevipedunculata against in vitro hepatic stellate cells system and thioacetamide-induced liver fibrosis rat model. Pharm. Biol. 2017, 55, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Perepelyuk, M.; Terajima, M.; Wang, A.Y.; Georges, P.C.; Janmey, P.A.; Yamauchi, M.; Wells, R.G. Hepatic stellate cells and portal fibroblasts are the major cellular sources of collagens and lysyl oxidases in normal liver and early after injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G605–G614. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, J.; Brenner, D.A.; Kisseleva, T. Reversibility of liver fibrosis and inactivation of fibrogenic myofibroblasts. Curr. Pathobiol. Rep. 2013, 1, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Ganai, A.A.; Husain, M. Genistein attenuates d-galn induced liver fibrosis/chronic liver damage in rats by blocking the TGF-β/smad signaling pathways. Chem. Biol. Interact. 2017, 261, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Sato-Matsubara, M.; Matsubara, T.; Daikoku, A.; Okina, Y.; Longato, L.; Rombouts, K.; Thuy, L.T.T.; Adachi, J.; Tomonaga, T.; Ikeda, K.; et al. Fibroblast growth factor 2 (FGF2) regulates cytoglobin expression and activation of human hepatic stellate cells via JNK signaling. J. Biol. Chem. 2017, 292, 18961–18972. [Google Scholar] [CrossRef] [PubMed]
- Bohm, F.; Speicher, T.; Hellerbrand, C.; Dickson, C.; Partanen, J.M.; Ornitz, D.M.; Werner, S. FGF receptors 1 and 2 control chemically induced injury and compound detoxification in regenerating livers of mice. Gastroenterology 2010, 139, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- LaQuaglia, M.J.; Grijalva, J.L.; Mueller, K.A.; Perez-Atayde, A.R.; Kim, H.B.; Sadri-Vakili, G.; Vakili, K. Yap subcellular localization and hippo pathway transcriptome analysis in pediatric hepatocellular carcinoma. Sci. Rep. 2016, 6, 30238. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.H.; Camargo, F.D.; Yimlamai, D. Hippo signaling in the liver regulates organ size, cell fate, and carcinogenesis. Gastroenterology 2017, 152, 5335–5345. [Google Scholar] [CrossRef] [PubMed]
- Zhubanchaliyev, A.; Temirbekuly, A.; Kongrtay, K.; Wanshura, L.C.; Kunz, J. Targeting mechanotransduction at the transcriptional level: Yap and brd4 are novel therapeutic targets for the reversal of liver fibrosis. Front. Pharmacol. 2016, 7, 462. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Krzyzanowski, P.M.; Muro, E.M.; Andrade-Navarro, M.A. Computational approaches to discovering noncoding RNA. RNA 2012, 3, 5675–5679. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.L.; Li, C.J.; Xu, Y.; Li, L.Y.; Zhou, X.; Li, D.D.; Chen, S.X.; Wang, F.G.; Zhang, X.Y.; Zheng, L.W. Role of lncRNAs as novel biomarkers and therapeutic targets in ovarian cancer. Crit. Rev. Eukaryotic Gene Express. 2017, 27, 1831–1895. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.; Baird, A.M.; Brady, L.; Lim, M.; Gray, S.G.; McDermott, R.; Finn, S.P. Circular RNAs: Biogenesis, function and role in human diseases. Front. Mol. Biosci. 2017, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Lavorgna, G.; Vago, R.; Sarmini, M.; Montorsi, F.; Salonia, A.; Bellone, M. Long non-coding RNAs as novel therapeutic targets in cancer. Pharmacol. Res. 2016, 110, 1311–1338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, H.; Han, L.; Li, F.; Zhang, T.; Pang, J.; Feng, X.; Ren, C.; Mao, S.; Wang, F. Long noncoding RNA expression profile changes associated with dietary energy in the sheep testis during sexual maturation. Sci. Rep. 2017, 7, 5180. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, C.; Owen, N.; Lees, J.; Tagalakis, A.; Hart, S.; Webster, A.; Orengo, C.; Khaw, P. Genome-wide RNA-sequencing analysis identifies a distinct fibrosis gene signature in the conjunctiva after glaucoma surgery. Sci Rep 2017, 7, 5644. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.J.; Xiao, X.; Sheng, L.; Chen, L.; Zhong, W.; Li, H.; Hua, J.; Ma, X. RNA sequencing and bioinformatics analysis implicate the regulatory role of a long noncoding RNA-mRNA network in hepatic stellate cell activation. Cell Physiol. Biochem. 2017, 42, 20302–203042. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; York, S.R.; Chen, J.Y.; Pondick, J.V.; Motola, D.L.; Chung, R.T.; Mullen, A.C. Long noncoding RNAs expressed in human hepatic stellate cells form networks with extracellular matrix proteins. Genome Med. 2016, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Jiang, Z.; Chen, B.; Dong, P.; Zheng, J. Neat1 accelerates the progression of liver fibrosis via regulation of microRNA-122 and kruppel-like factor 6. J. Mol. Med. 2017, 95, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Han, X.; Zhang, Z.; Zheng, L.; Hu, Z.; Yao, Q.; Cui, H.; Shu, G.; Si, M.; Li, C.; et al. The liver-enriched Lnc-LFAR1 promotes liver fibrosis by activating tgfβ and notch pathways. Nat. Commun. 2017, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Shen, X.Z.; Jiang, F.; Wu, Y.; Han, C. DNA-guided genome editing using the Natronobacterium gregoryi Argonaute. Nat. Biotechnol. 2016, 34, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Coco, F.L.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 69696–69978. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xue, Y.; Han, Y.; Lin, L.; Wu, C.; Xu, S.; Jiang, Z.; Xu, J.; Liu, Q.; Cao, X. The STAT3-binding long noncoding RNA LNC-DC controls human dendritic cell differentiation. Science 2014, 344, 3103–3113. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The hippo pathway effector yap controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Chen, S.; Pan, W.; Xu, L.; Hu, K.; Xu, R. Np603, a novel and potent inhibitor of FGFR1 tyrosine kinase, inhibits hepatic stellate cell proliferation and ameliorates hepatic fibrosis in rats. Am. J. Physiol. Cell Physiol. 2011, 301, C469–C477. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.F.; Liu, L.X.; Zhang, H.Y. Effect of tanshinone ii on hepatic fibrosis in mice. Zhongguo Zhong Xi Yi Jie He Za Zhi 2009, 29, 1012–1017. [Google Scholar]
- Xu, X.; Yan, Q.; Wang, Y.; Dong, X. NTN4 is associated with breast cancer metastasis via regulation of EMT-related biomarkers. Oncol. Rep. 2017, 37, 449–457. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Tsutsumi, M.; Tsuchishima, M. MMP-13 deletion decreases profibrogenic molecules and attenuates N-nitrosodimethylamine-induced liver injury and fibrosis in mice. J. Cell Mol. Med. 2017, 21, 3821–3835. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; He, Z.; Kong, L.L.; Chen, Q.; Yuan, Q.; Zhang, S.; Ye, J.; Liu, H.; Sun, X.; Geng, J.; et al. Pharmacological targeting of kinases MST1 and MST2 augments tissue repair and regeneration. Sci. Transl. Med. 2016, 8, 352ra108. [Google Scholar] [CrossRef] [PubMed]
- Geng, Z.-M.; Li, Q.-H.; Li, W.-Z.; Zheng, J.-B.; Shah, V. Activated human hepatic stellate cells promote growth of human hepatocellular carcinoma in a subcutaneous xenograft nude mouse model. Cell Biochem. Biophys. 2014, 70, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. Stringtie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 2902–2995. [Google Scholar] [CrossRef] [PubMed]
- Janosky, J.E. Pearson correlation coefficients vs. reliability coefficients. J. Am. Diet. Assoc. 1991, 91, 912–913. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Ontology Term | Cluster Frequency | Genome Frequency of Use | Corrected p-Value |
|---|---|---|---|
| Nucleosome | 6/147 | 107/34,590 | 0.00108 |
| DNA bending complex | 6/147 | 107/34,590 | 0.00108 |
| DNA packaging complex | 6/147 | 124/34,590 | 0.00253 |
| Actin cap | 3/147 | 13/34,590 | 0.00335 |
| Growth factor binding | 8/135 | 258/31,397 | 0.00334 |
| Gene Symbol | Up-/Downregulated | Chromosomal Location | Co-Expressed lncRNA | Up-/Downregulated | Chromosomal Location | COR | p-Value |
|---|---|---|---|---|---|---|---|
| CTGF | up | chr6 | NONHSAT208034.1 | up | chr6 | 0.8812 | 6.19 × 10−136 |
| FGF2 | up | chr6 | NONHSAT200340.1 | up | Chr4 | 0.9053 | 1.11 × 10−12 |
| NTN4 | up | chr12 | LTCONS_00038568 | up | Chr12 | 0.8797 | 1.15 × 10−14 |
| LTCONS_00044996 | up | chr12 | 0.9053 | 1.43 × 10−51 | |||
| FGFR2 | up | chr10 | NONHSAT158555.1 | up | Chr10 | 0.7655 | 0.000175775 |
| IGFBP7 | up | chr4 | NONHSAT096544.2 | up | Chr4 | 0.956 | 0.00334 |
| PCSK6 | up | chr15 | NONHSAT172067.1 | up | Chr15 | 0.6457 | 2.36 × 10−5 |
| Hist1h2ak | down | chr6 | LTCONS_00161292 | up | chr6 | −0.6806 | 0.000102578 |
| Hist1h3f | down | chr6 | LTCONS_00157063 | down | chr6 | 0.9388 | 1.04 × 10−158 |
| CALD1 | down | chr7 | NONHSAT214019.1 | up | chr7 | −0.6686 | 5.21 × 10−7 |
| hist1h3g | down | chr6 | LTCONS_00157063 | down | chr6 | 0.8589 | 1.04 × 10−158 |
| Gene Symbol | Primer Sequence | Product Size (bp) |
|---|---|---|
| 18S-F | AGGTGGAACGTGTGATCACC | 146 |
| 18S-R | CAGGTCTTCACGGAGCTTGT | |
| ACTA2-F | ACTGGGACGACATGGAAAAG | 129 |
| ACTA2-R | GAGTCATTTTCTCCCGGTTG | |
| COL1A1-F | TTCTGCAACATGGAGACTGG | 134 |
| COL1A1-R | AATCCATCGGTCATGCTCTC | |
| LOX-F | GACCTGCTTGATGCCAACAC | 126 |
| LOX-R | TCCCTGTGTGTGTGCAGTAC | |
| LOXL2-F | ATGGGCTTGCAGAAGAAGCT | 116 |
| LOXL2-R | GTTTTGGCCACACACCATCC | |
| GAPDH-F | AATCCCATCACCATCTTCCA | 120 |
| GAPDH-R | TGGACTCCACGACGTACTCA | |
| CTGF-F | CAGCATGGACGTTCGTCTG | 115 |
| CTGF-R | AACCACGGTTTGGTCCTTGG | |
| FGF2-F | CCGTTACCTGGCTATGAAGG | 144 |
| FGF2-R | TTCAGTGCCACATACCAACTG | |
| NTN4-F | ATGCTTGCAAACCGTGTTCC | 137 |
| NTN4-R | CATGCACCTGTCACAACGTC | |
| NHSAT088415.2-F | GTTCGGACAAGAGCCAGGA | 120 |
| NHSAT088415.2-R | CCAACTGCCAAGTTCCTTCC | |
| TCONS_00038568-F | GTGTCCAGAGCAGTGCTTCT | 149 |
| TCONS_00038568-R | TAATGCAGAGACCCAGGCC | |
| NONHSAT208034.1-F | CCCACAGGTCTTGGAACAGG | 132 |
| NONHSAT208034.1-R | AAAGCGGTGTTTGTGTGCTC | |
| NONHSAT152369.1-F | CAGGTGAATGCCACACAGGT | 132 |
| NONHSAT152369.1-R | TGCTGACTGAGGATGAGATGG | |
| NONHSAT214020.1-F | GTGATCAGGAGGAGCTCTTGT | 113 |
| NONHSAT214020.1-R | CAGGTAGCATTGACTCCCGT | |
| NONHSAT078100.2-F | TCTCCAGGCTCCACAATACC | 119 |
| NONHSAT078100.2-R | TGTTCATTGCCTGCTCTCAC | |
| NONHSAT213509.1-F | ATGAGGGAATCCCCAATTTC | 120 |
| NONHSAT213509.1-R | AGCTGTGGATGCTTTCTGCT | |
| NONHSAT026380.2-F | TCTCCAGGCTCCACAATACC | 120 |
| NONHSAT026380.2-R | GACACTCCAAATCCCAGGAA | |
| NONHSAT172067.1-F | ACAGATGCTCAGCGATGTTG | 127 |
| NONHSAT172067.1-R | AGGCAGGAGGATCTGGATTT | |
| NONHSAT200340.1-F | CCCAGATGATGGCTACTGGT | 142 |
| NONHSAT200340.1-R | CCCACCTCAACACCAAAGAT |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.-Q.; Ren, Z.-X.; Li, K.; Huang, J.-J.; Huang, Z.-T.; Zhou, T.-R.; Cao, H.-Y.; Zhang, F.-X.; Tan, B. Key Anti-Fibrosis Associated Long Noncoding RNAs Identified in Human Hepatic Stellate Cell via Transcriptome Sequencing Analysis. Int. J. Mol. Sci. 2018, 19, 675. https://doi.org/10.3390/ijms19030675
Li X-Q, Ren Z-X, Li K, Huang J-J, Huang Z-T, Zhou T-R, Cao H-Y, Zhang F-X, Tan B. Key Anti-Fibrosis Associated Long Noncoding RNAs Identified in Human Hepatic Stellate Cell via Transcriptome Sequencing Analysis. International Journal of Molecular Sciences. 2018; 19(3):675. https://doi.org/10.3390/ijms19030675
Chicago/Turabian StyleLi, Xian-Qian, Zhen-Xing Ren, Ke Li, Jing-Jing Huang, Zi-Tong Huang, Tian-Ran Zhou, Hong-Ying Cao, Feng-Xue Zhang, and Bo Tan. 2018. "Key Anti-Fibrosis Associated Long Noncoding RNAs Identified in Human Hepatic Stellate Cell via Transcriptome Sequencing Analysis" International Journal of Molecular Sciences 19, no. 3: 675. https://doi.org/10.3390/ijms19030675
APA StyleLi, X.-Q., Ren, Z.-X., Li, K., Huang, J.-J., Huang, Z.-T., Zhou, T.-R., Cao, H.-Y., Zhang, F.-X., & Tan, B. (2018). Key Anti-Fibrosis Associated Long Noncoding RNAs Identified in Human Hepatic Stellate Cell via Transcriptome Sequencing Analysis. International Journal of Molecular Sciences, 19(3), 675. https://doi.org/10.3390/ijms19030675