Endostatin Stimulates Proliferation and Migration of Myofibroblasts Isolated from Myocardial Infarction Model Rats

Abstract
:
1. Introduction
2. Results
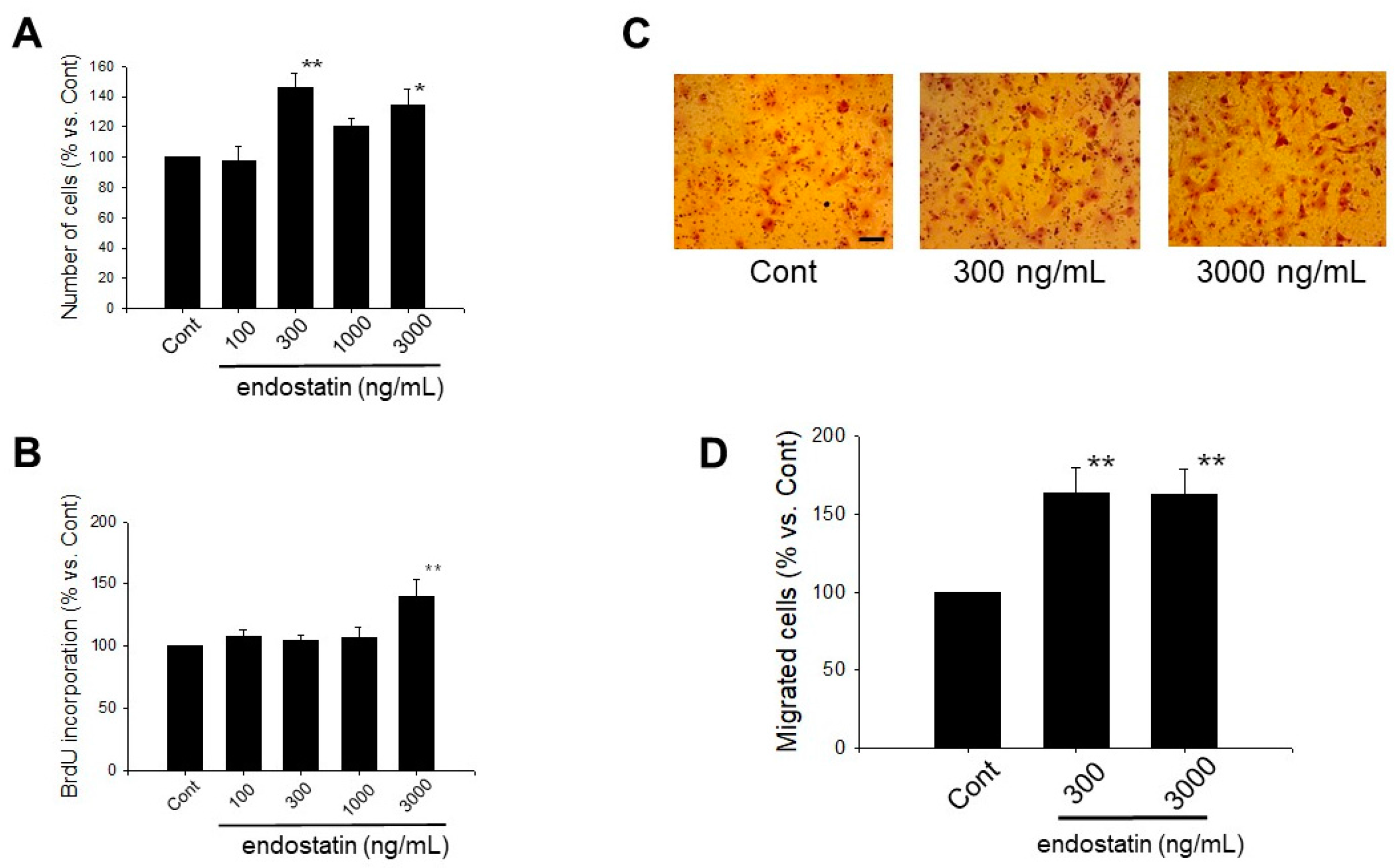
2.1. Effect of Endostatin on Proliferation of Myofibroblasts
2.2. Effect of Endostatin on Migration of Myofibroblasts
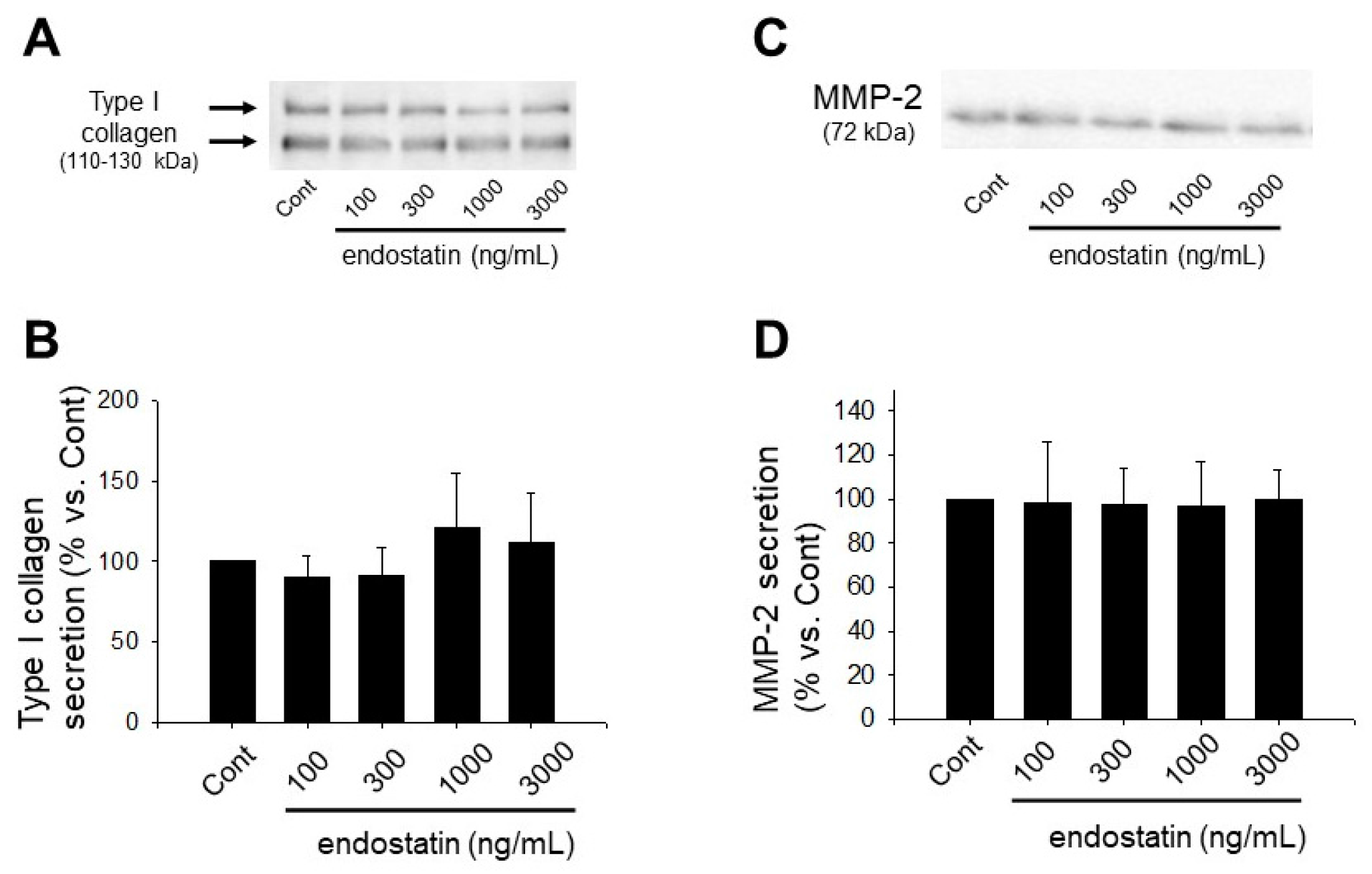
2.3. Effect of Endostatin on ECM Production and MMPs Secretion of Myofibroblasts
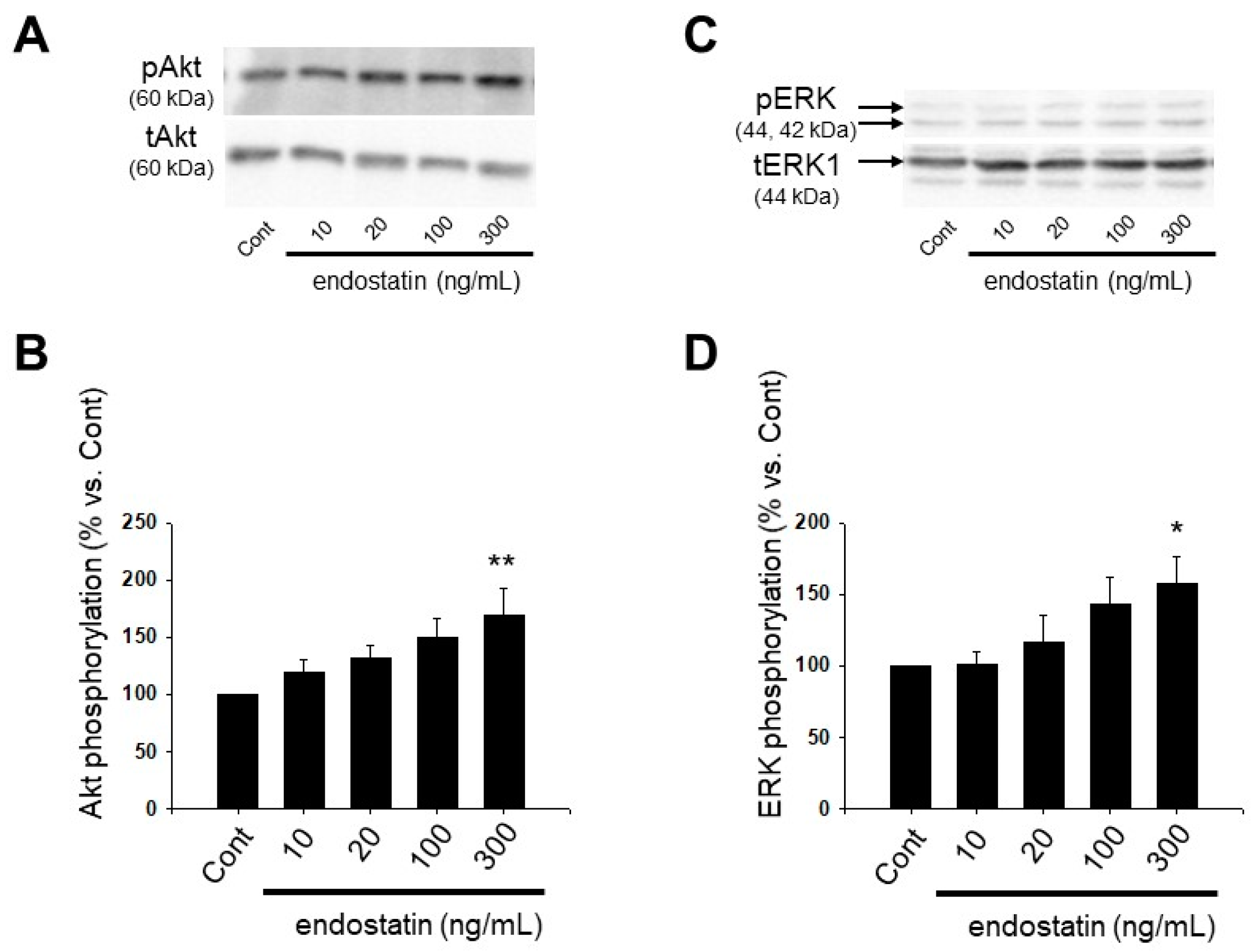
2.4. Effect of Endostatin on Activation of Akt and Extracellular Signal-Regulated Kinase (ERK) in Myofibroblasts
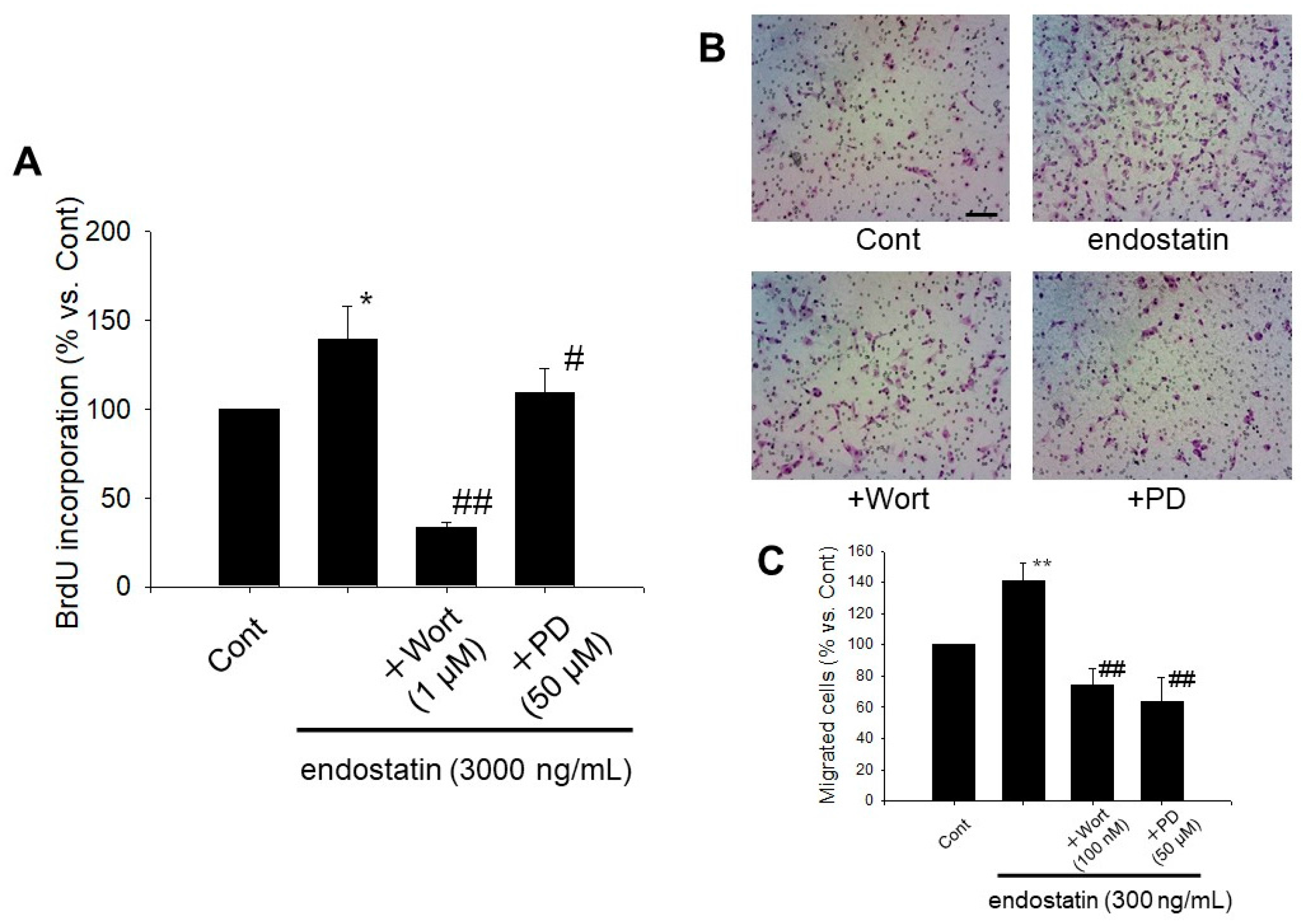
2.5. Endostatin Induced Proliferation and Migration of Myofibroblasts through Activation of Phosphoinositide 3-Kinase (PI3K)/Akt and Mitogen-Activated Protein Kinase (MAPK)/ERK Kinase (MEK)/ERK Pathway
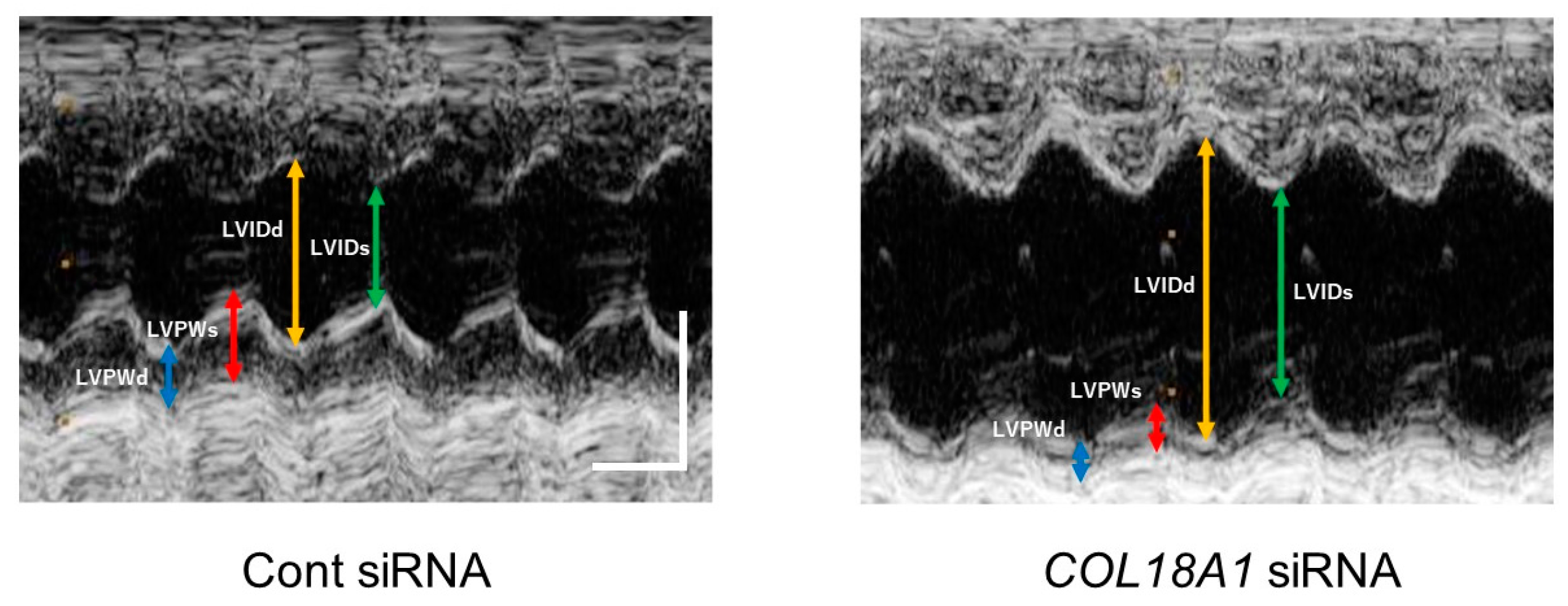
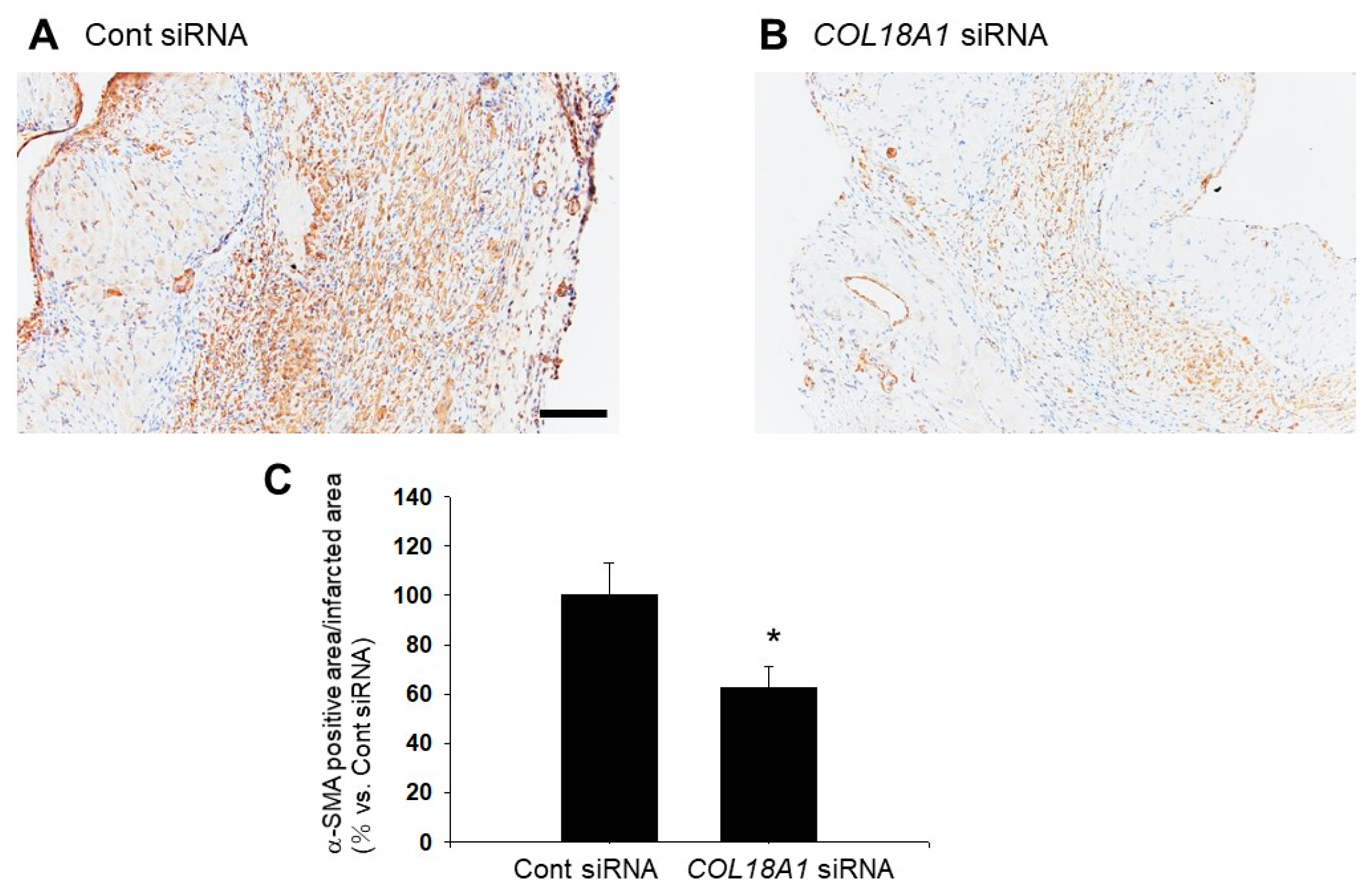
2.6. A Knockdown of the COL18A1 Gene in the Infarcted Area by COL18A1 Small Interference (si) RNA Worsened Cardiac Function and Decreased α-SMA-Positive Myofibroblasts
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Myocardial Infarction Model
4.3. Isolation of Myofibroblasts from the Areas of Myocardial Infarction
4.4. Cell Proliferation Assay
4.5. Boyden Chamber Assay
4.6. Western Blotting
4.7. Echocardiography
4.8. Immunohistochemical Staining
4.9. Azan Staining
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ECM | extracellular matrix |
| α-SMA | α-smooth muscle actin |
| MMPs | matrix metalloproteinases |
| IL | interleukin |
| S.E.M. | standard error of the mean |
| ERK | extracellular signal-regulated kinase |
| PI3K | phosphoinositide 3-kinase |
| MAPK | mitogen-activated protein kinase |
| MEK | MAPK/ERK kinase |
| siRNA | small interference RNA |
| LAD | left anterior descending artery |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| BW | body weight |
| LV | left ventricular |
| LVIDd | LV internal dimension at end-diastole |
| LVIDs | LV internal dimension at end-systole |
| IVSd | interventricular septum at end-diastole |
| IVSs | interventricular septum at end-systole |
| IVS% | percentage IVS thickness |
| LVPWd | LV posterior wall at end-diastole |
| LVPWs | LV posterior wall at end-systole |
| LVPW% | percentage LVPW thickness |
| EDV | end-diastolic volume |
| ESV | end-systolic volume |
| FS | fractional shortening |
| EF | ejection fraction |
| ROS | reactive oxygen species |
| NAC | N-acetylcysteine |
| TBS | tris buffered saline |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| FBS | fetal bovine serum |
| CC8 | cell counting kit-8 |
References
- Goldsmith, E.C.; Bradshaw, A.D.; Spinale, F.G. Cellular Mechanisms of Tissue Fibrosis. 2. Contributory pathways leading to myocardial fibrosis: Moving beyond collagen expression. Am. J. Physiol. Cell Physiol. 2013, 304, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Sproul, E.P.; Argraves, W.S. A cytokine axis regulates elastin formation and degradation. Matrix Biol. 2013, 32, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Aziz-Seible, R.S.; Casey, C.A. Fibronectin: Functional character and role in alcoholic liver disease. World J. Gastroenterol. 2011, 17, 2482–2499. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Brás, L.E.D.C.; Toba, H.; Iyer, R.P.; Lindsey, M.L. Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflügers Arch. Eur. J. Physiol. 2014, 446, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Creemers, E.E.; Pinto, Y.M. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc. Res. 2011, 89, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Miró, L.; Pérez-bosque, A.; Maijó, M.; Amat, C.; Naftalin, R.J.; Moretó, M. Aldosterone induces myofibroblast EGF secretion to regulate epithelial colonic permeability. Am. J. Physiol. Cell Physiol. 2017, 304, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, H.; Thomasy, S.M.; Russell, P.; Murphy, C.J. The role of hepatocyte growth factor in corneal wound healing. Exp. Eye Res. 2018, 166, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Neaud, V.; Faouzi, S.; Guirouilh, J.; Le Bail, B.; Balabaud, C.; Bioulac-Sage, P.; Rosenbaum, J. Human hepatic myofibroblasts increase invasiveness of hepatocellular carcinoma cells: Evidence for a role of hepatocyte growth factor. Hepatology 1997, 26, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Felbor, U.; Dreier, L.; Bryant, R.A.; Ploegh, H.L.; Olsen, B.R.; Mothes, W. Secreted cathepsin L generates endostatin from collagen XVIII. Eur. Mol. Biol. Organ. J. 2000, 19, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Heljasvaara, R.; Nyberg, P.; Luostarinen, J.; Parikka, M.; Heikkilä, P.; Rehn, M.; Sorsa, T.; Salo, T.; Pihlajaniemi, T. Generation of biologically active endostatin fragments from human collagen XVIII by distinct matrix metalloproteases. Exp. Cell Res. 2005, 307, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.H.K.; Yao, J.Y.; Kuo, M.T.; See, L.C.; Lin, K.Y.; Chen, S.C.; Chen, J.K.; Chao, A.S.; Wang, S.F.; Lin, K.K. Generation of endostatin by matrix metalloproteinase and cathepsin from human limbocorneal epithelial cells cultivated on amniotic membrane. Investig. Ophthalmol. Vis. Sci. 2007, 48, 644–651. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef]
- Wen, W.; Moses, M.A.; Wiederschain, D.; Arbiser, J.L.; Folkman, J. The generation of endostatin is mediated by elastase. Cancer Res. 1999, 59, 6052–6056. [Google Scholar] [PubMed]
- Dhanabal, M.; Ramchandran, R.; Volk, R.; Simons, M.; Sukhatme, V.P. Endostatin : Yeast Production, Mutants, and Antitumor Effect in Renal Cell Carcinoma. Cancer Res. 1999, 59, 189–197. [Google Scholar] [PubMed]
- Dhanabal, M.; Ramchandran, R.; Waterman, M.J.F.; Lu, H.; Knebelmann, B.; Segal, M.; Sukhatme, V.P. Endostatin Induces Endothelial Cell Apoptosis. J. Biol. Chem. 1999, 274, 11721–11726. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Duong, T.; Lee, G.; Seong, B.L.; El-Rifai, W.; Ruley, H.E.; Jo, D. The effect of intracellular protein delivery on the anti-tumor activity of recombinant human endostatin. Biomaterials 2013, 34, 6261–6271. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Tang, H.; Huang, Y.; Song, N.; Luo, Y. Unraveling the mysteries of endostatin. IUBMB Life 2009, 61, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, S.B.; Dallabrida, S.M.; Durand, E.; Ismail, N.S.; Bazinet, L.; Birsner, A.E.; Sohn, R.; Ikeda, S.; Pu, W.T.; Kulke, M.H.; et al. Endostatin lowers blood pressure via nitric oxide and prevents hypertension associated with VEGF inhibition. Proc. Natl. Acad. Sci. USA 2012, 109, 11306–11311. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takihara, T.; Chambers, R.A.; Veraldi, K.L.; Larregina, A.T.; Feghali-Bostwick, C.A. A Peptide Derived from Endostatin Ameliorates Organ Fibrosis. Sci. Transl. Med. 2012, 4, 136ra7. [Google Scholar] [CrossRef] [PubMed]
- Kurosaka, D.; Yoshida, K.; Yasuda, J.; Yokoyama, T.; Kingetsu, I.; Yamaguchi, N.; Joh, K.; Matsushima, M.; Saito, S.; Yamada, A. Inhibition of arthritis by systemic administration of endostatin in passive murine collagen induced arthritis. Ann. Rheum. Dis. 2003, 62, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Mitsuma, W.; Kodama, M.; Hanawa, H.; Ito, M.; Ramadan, M.M.; Hirono, S.; Obata, H.; Okada, S.; Sanada, F.; Yanagawa, T.; et al. Serum Endostatin in the Coronary Circulation of Patients With Coronary Heart Disease and Its Relation to Coronary Collateral Formation. Am. J. Cardiol. 2007, 99, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Gouya, G.; Siller-Matula, J.M.; Fritzer-Szekeres, M.; Neuhold, S.; Storka, A.; Neuhofer, L.M.; Clodi, M.; Hülsmann, M.; Pacher, R.; Wolzt, M. Association of endostatin with mortality in patients with chronic heart failure. Eur. J. Clin. Investig. 2014, 44, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Givvimani, S.; Munjal, C.; Gargoum, R.; Sen, U.; Tyagi, N.; Vacek, J.C.; Tyagi, S.C. Hydrogen sulfide mitigates transition from compensatory hypertrophy to heart failure. J. Appl. Physiol. 2011, 110, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, A.; Ablasser, K.; Ballmoos, M.C.W.; Von Poutias, D.; Kaza, E.; Mcgowan, F.X.; Moses, M.A.; Nido, P.J.; Friehs, I. Endogenous Angiogenesis Inhibitors Prevent Adaptive Capillary Growth in Left Ventricular Pressure Overload Hypertrophy. Ann. Thorac. Surg. 2012, 94, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Isobe, K.; Kuba, K.; Maejima, Y.; Szuki, J.; Kubota, S.; Isobe, M. Inhibition of Endostatin/Collagen XVIII Deteriorates Left Ventricular Remodeling and Heart Failure in Rat Myocardial Infarction Model. Circ. J. 2010, 74, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Qipshidze, N.; Metreveli, N.; Mishra, P.K.; Lominadze, D.; Tyagi, S.C. Hydrogen sulfide mitigates cardiac remodeling during myocardial infarction via improvement of angiogenesis. Int. J. Biol. Sci. 2012, 8, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Oba, Y.; Yamawaki, H. Endostatin stimulates proliferation and migration of adult rat cardiac fibroblasts through PI3K/Akt pathway. Eur. J. Pharmacol. 2015, 750, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, W.; Che, H.; Liu, J.; Sun, H. Adenosine-5Ј-triphosphate up-regulates proliferation of human cardiac fibroblasts. Br. J. Pharmacol. 2012, 16, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, J.; Fukui, K.; Okada, M. T3 peptide, a fragment of tumstatin, stimulates proliferation and migration of cardiac fibroblasts through activation of Akt signaling pathway. N.-S. Arch. Pharmacol. 2017, 390, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Imoto, K.; Kumatani, S.; Okada, M.; Yamawaki, H. Endostatin is protective against monocrotaline-induced right heart disease through the inhibition of T-type Ca2+channel. Pflügers Arch. Eur. J. Physiol. 2016, 468, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Aljinović, J.; Vukojević, K.; Košta, V.; Guić, M.M.; Saraga-Babić, M.; Grković, I. Histological differences in healing following experimental transmural infarction in rats. Histol. Histopathol. 2010, 25, 1507–1517. [Google Scholar] [PubMed]
- Kłubo-Gwieździnska, J.; Junik, R.; Kopczyńska, E. Serum endostatin levels in patients with metastatic and non-metastatic well-differentiated thyroid cancer. Endokrynol. Pol. 2010, 61, 7–12. [Google Scholar] [PubMed]
- Sudhakar, A.; Sugimoto, H.; Yang, C.; Lively, J.; Zeisberg, M.; Kalluri, R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc. Natl. Acad. Sci. USA 2003, 100, 4766–4771. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Hwang, S.; Kim, Y.M.; Pyun, B.J.; Kim, T.Y.; Lee, S.T.; Gho, Y.S.; Kwon, Y.G. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J. Biol. Chem. 2002, 277, 27872–27879. [Google Scholar] [CrossRef] [PubMed]
- Alahuhta, I.; Aikio, M.; Väyrynen, O.; Nurmenniemi, S.; Suojanen, J.; Teppo, S.; Pihlajaniemi, T.; Heljasvaara, R.; Salo, T.; Nyberg, P. Endostatin induces proliferation of oral carcinoma cells but its effect on invasion is modified by the tumor microenvironment. Exp. Cell Res. 2015, 336, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-Y.; Zhang, X.-M.; Chen, F.-H.; Zhou, L.-L.; Deng, X.-F.; Liu, Y.-J.; Li, X.-J. Anti-proliferative effect of recombinant human endostatin on synovial fibroblasts in rats with adjuvant arthritis. Eur. J. Pharmacol. 2014, 723, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Benelli, R.; Venè, R.; Minghelli, S.; Carlone, S.; Gatteschi, B.; Ferrari, N. Celecoxib induces proliferation and Amphiregulin production in colon subepithelial myofibroblasts, activating erk1-2 signaling in synergy with EGFR. Cancer Lett. 2013, 328, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Wang, C.N.; Lee, Y.L.; Tsai, Y.R.; Liu, J.J.; Yang, C. High mobility group box 1 induced human lung myofibroblasts differentiation and enhanced migration by activation of MMP-9. PLoS ONE 2015, 10, e0116393. [Google Scholar] [CrossRef] [PubMed]
- Uyama, N.; Iimuro, Y.; Kawada, N.; Reynaert, H.; Suzumura, K.; Hirano, T.; Kuroda, N.; Fujimoto, J. Fascin, a novel marker of human hepatic stellate cells, may regulate their proliferation, migration, and collagen gene expression through the FAK-PI3K-Akt pathway. Lab. Investig. 2012, 92, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, A.; Okada, M.; Yamawaki, H. Pathophysiological roles of canstatin on myofibroblasts after myocardial infarction in rats. Eur. J. Pharmacol. 2017, 807, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, D.; Schmidt, A.; Reimann, K.; Hescheler, J.; Pfitzer, G.; Bloch, W.; Fleischmann, B.K. Endostatin, the proteolytic fragment of collagen XVIII, induces vasorelaxation. Circ. Res. 2006, 98, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Y.; Teggatz, E.G.; Zou, A.; Campbell, W.B.; Li, P.-L. Endostatin uncouples NO and Ca2+ response to bradykinin through enhanced O2− production in the intact coronary endothelium. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Chang, S.C.; Wen, H.C.; Chen, C.Y.; Liao, J.F.; Chang, C.H. Plumbagin activates ERK1/2 and Akt via superoxide, Src and PI3-kinase in 3T3-L1 Cells. Eur. J. Pharmacol. 2010, 638, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Porter, K.E. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair 2013, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Liu, J.; Zhang, H.; Wang, J.; Li, H. Intramyocardial Injection of siRNAs Can Efficiently Establish Myocardial Tissue-Specific Renalase Knockdown Mouse Model. BioMed Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Sakatsume, T.; Nijima, R.; Otani, K.; Kazama, K.; Morita, T.; Kameshima, S.; Okada, M.; Yamawaki, H. Death-associated protein kinase 3 mediates vascular structural remodelling via stimulating smooth muscle cell proliferation and migration. Clin. Sci. (Lond.) 2014, 127, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Kameshima, S.; Kakuda, C.; Okamura, Y.; Kodama, T.; Okada, M.; Yamawaki, H. Visceral adipose tissue-derived serine protease inhibitor prevents the development of monocrotaline-induced pulmonary arterial hypertension in rats. Pflügers Arch. Eur. J. Physiol. 2017, 1425–1432. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Cont siRNA (n = 6) | COL18A1 siRNA (n = 6) |
|---|---|---|
| BW (before operation) | 265.8 ± 16.4 g | 270.9 ± 17.2 g |
| BW (2 weeks after operation) | 318.6 ± 10.0 g | 310.7 ± 13.2 g |
| ΔBW | 52.8 ± 6.9 g | 39.8 ± 5.9 g |
| Parameters | Unit | Cont siRNA(n = 6) | COL18A1 siRNA (n = 6) |
|---|---|---|---|
| IVSd | cm | 0.18 ± 0.01 | 0.17 ± 0.01 |
| LVIDd | cm | 0.72 ± 0.04 | 0.85 ± 0.04 |
| LVPWd | cm | 0.20 ± 0.01 | 0.15 ± 0.01 ** |
| IVSs | cm | 0.30 ± 0.01 | 0.29 ± 0.02 |
| LVIDs | cm | 0.40 ± 0.02 | 0.57 ± 0.04 ** |
| LVPWs | cm | 0.29 ± 0.02 | 0.19 ± 0.02 ** |
| EDV (MM-Teich) | mL | 0.89 ± 0.12 | 1.38 ± 0.21 |
| IVS/LVPW (MM) | 0.94 ± 0.07 | 1.17 ± 0.06 * | |
| LV Mass (Cubed) | g | 1.4 ± 0.1 | 1.4 ± 0.1 |
| IVS% (MM) | % | 69.0 ± 9.3 | 69.7 ± 7.6 |
| ESV (MM-Teich) | mL | 0.16 ± 0.02 | 0.49 ± 0.11 * |
| FS (MM-Teich) | % | 44.5 ± 2.7 | 32.9 ± 2.0 ** |
| EF (MM-Teich) | % | 80.0 ± 2.9 | 66.5 ± 2.7 ** |
| LVPW% (MM) | % | 51.7 ± 5.8 | 26.3 ± 8.3 * |
| Heart rate | bpm | 405.9 ± 9.7 | 376.2 ± 7.4 * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugiyama, A.; Hirano, Y.; Okada, M.; Yamawaki, H. Endostatin Stimulates Proliferation and Migration of Myofibroblasts Isolated from Myocardial Infarction Model Rats. Int. J. Mol. Sci. 2018, 19, 741. https://doi.org/10.3390/ijms19030741
Sugiyama A, Hirano Y, Okada M, Yamawaki H. Endostatin Stimulates Proliferation and Migration of Myofibroblasts Isolated from Myocardial Infarction Model Rats. International Journal of Molecular Sciences. 2018; 19(3):741. https://doi.org/10.3390/ijms19030741
Chicago/Turabian StyleSugiyama, Akira, Yuka Hirano, Muneyoshi Okada, and Hideyuki Yamawaki. 2018. "Endostatin Stimulates Proliferation and Migration of Myofibroblasts Isolated from Myocardial Infarction Model Rats" International Journal of Molecular Sciences 19, no. 3: 741. https://doi.org/10.3390/ijms19030741
APA StyleSugiyama, A., Hirano, Y., Okada, M., & Yamawaki, H. (2018). Endostatin Stimulates Proliferation and Migration of Myofibroblasts Isolated from Myocardial Infarction Model Rats. International Journal of Molecular Sciences, 19(3), 741. https://doi.org/10.3390/ijms19030741