Transcriptome Analysis Identifies a 140 kb Region of Chromosome 3B Containing Genes Specific to Fusarium Head Blight Resistance in Wheat


Abstract
1. Introduction
2. Results
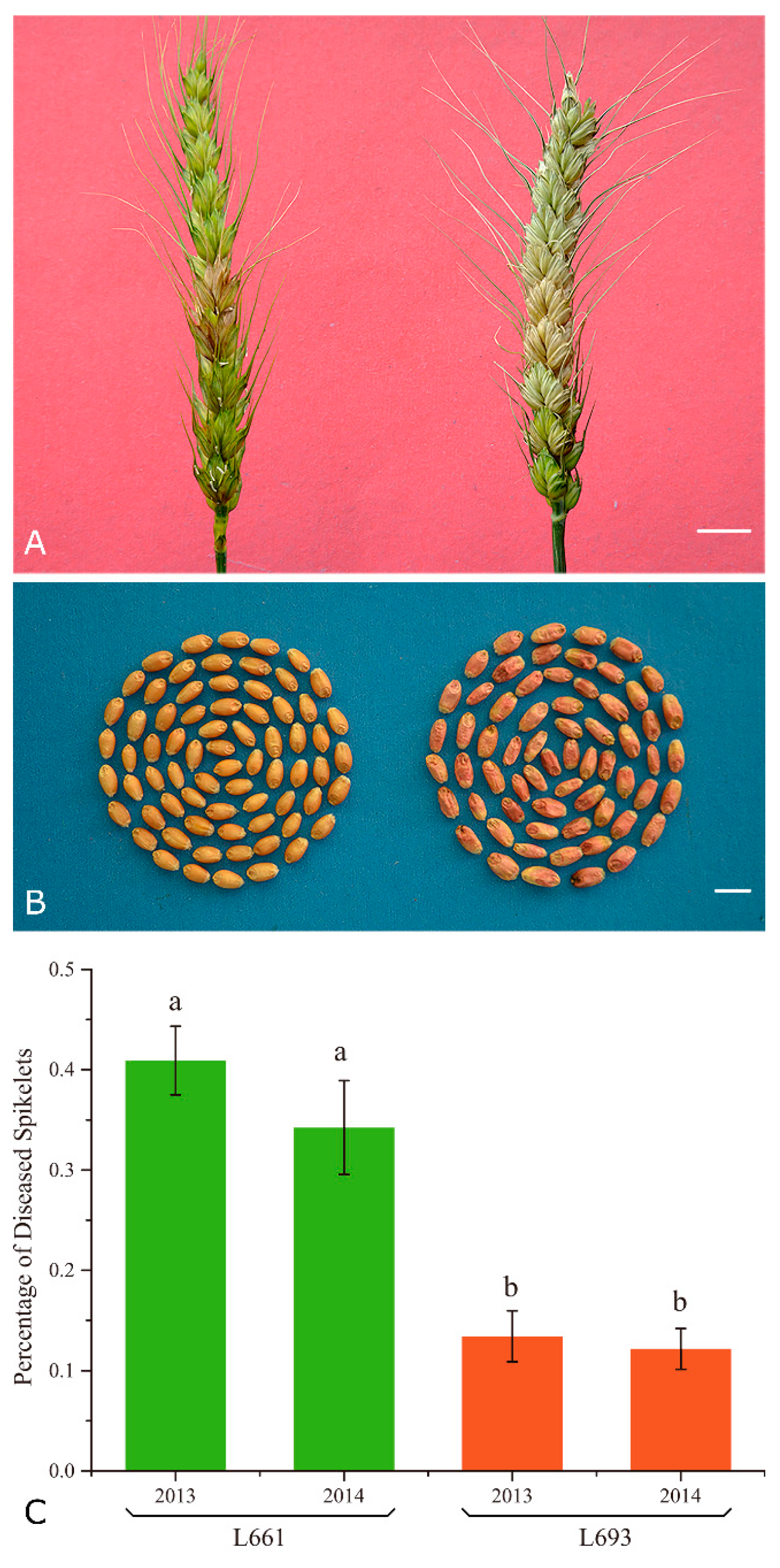
2.1. L693 and L661 Exhibit Different FHB Resistance Level, but Similar Transcriptome Sizes
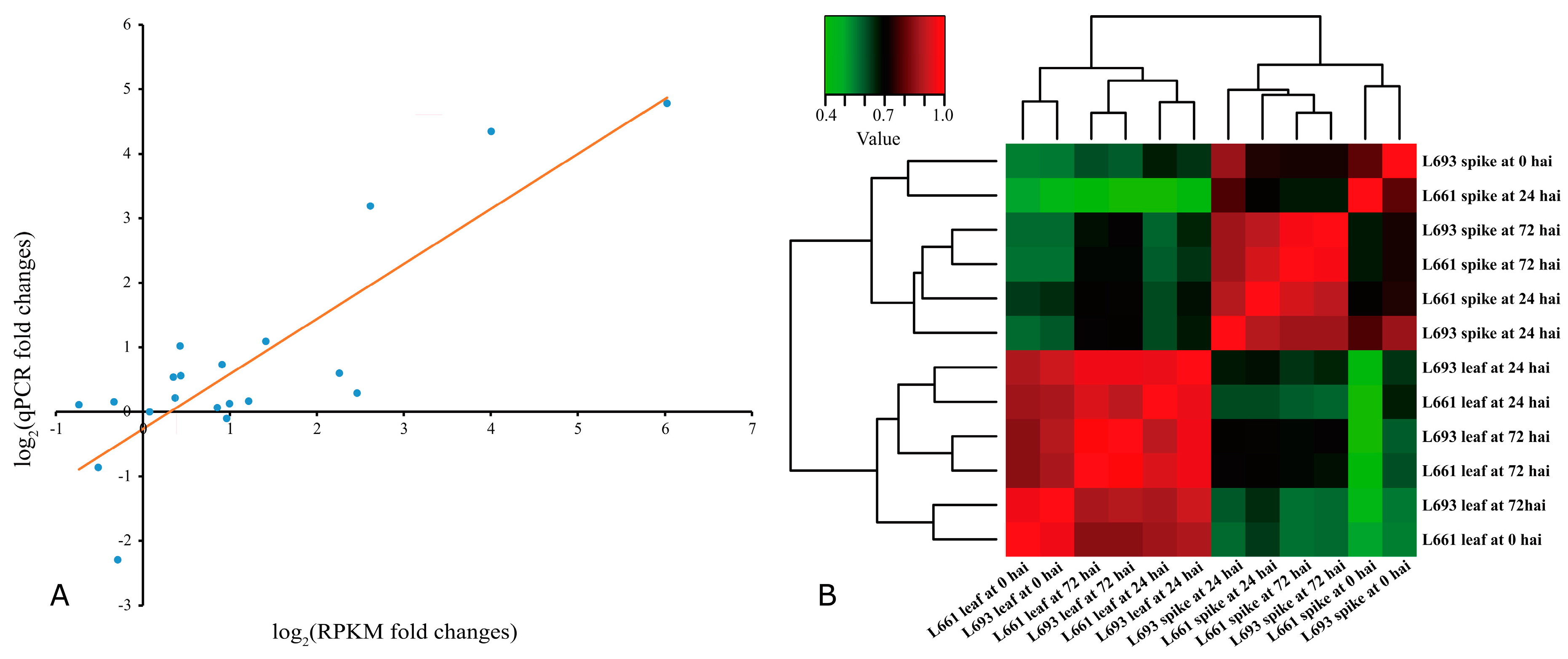
2.2. Validation of the Differences in Gene Expression by RT-qPCR and Clustering Analysis
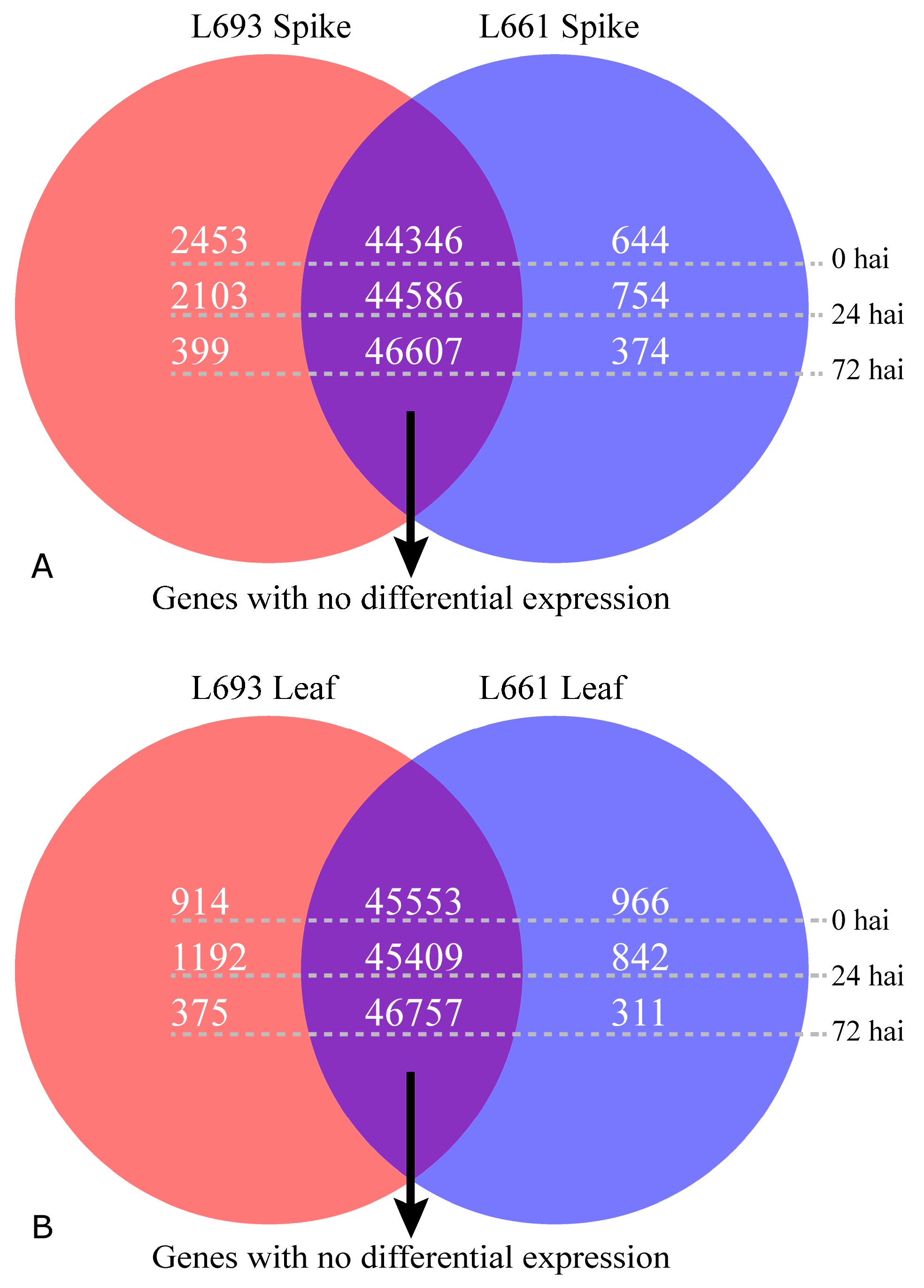
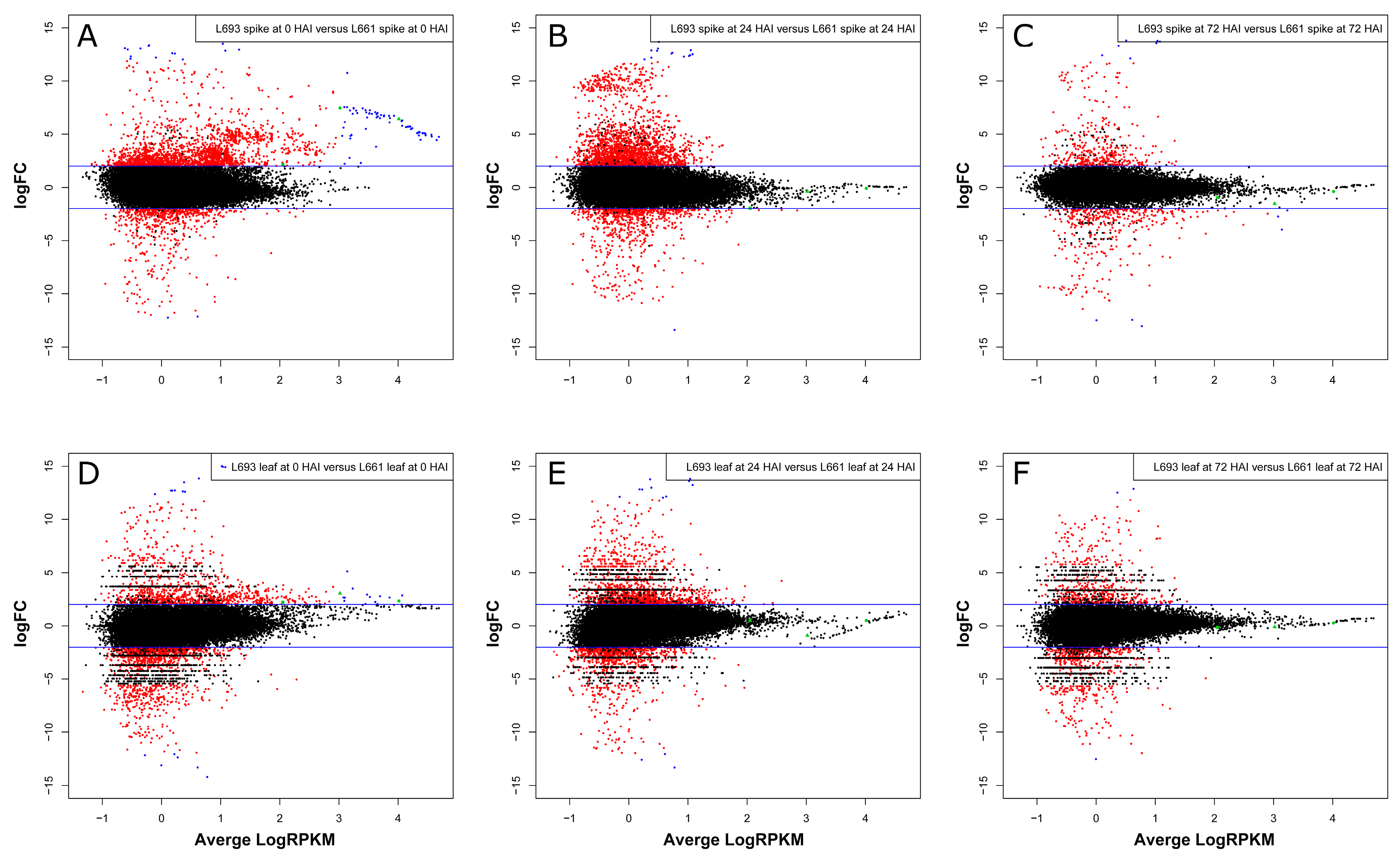
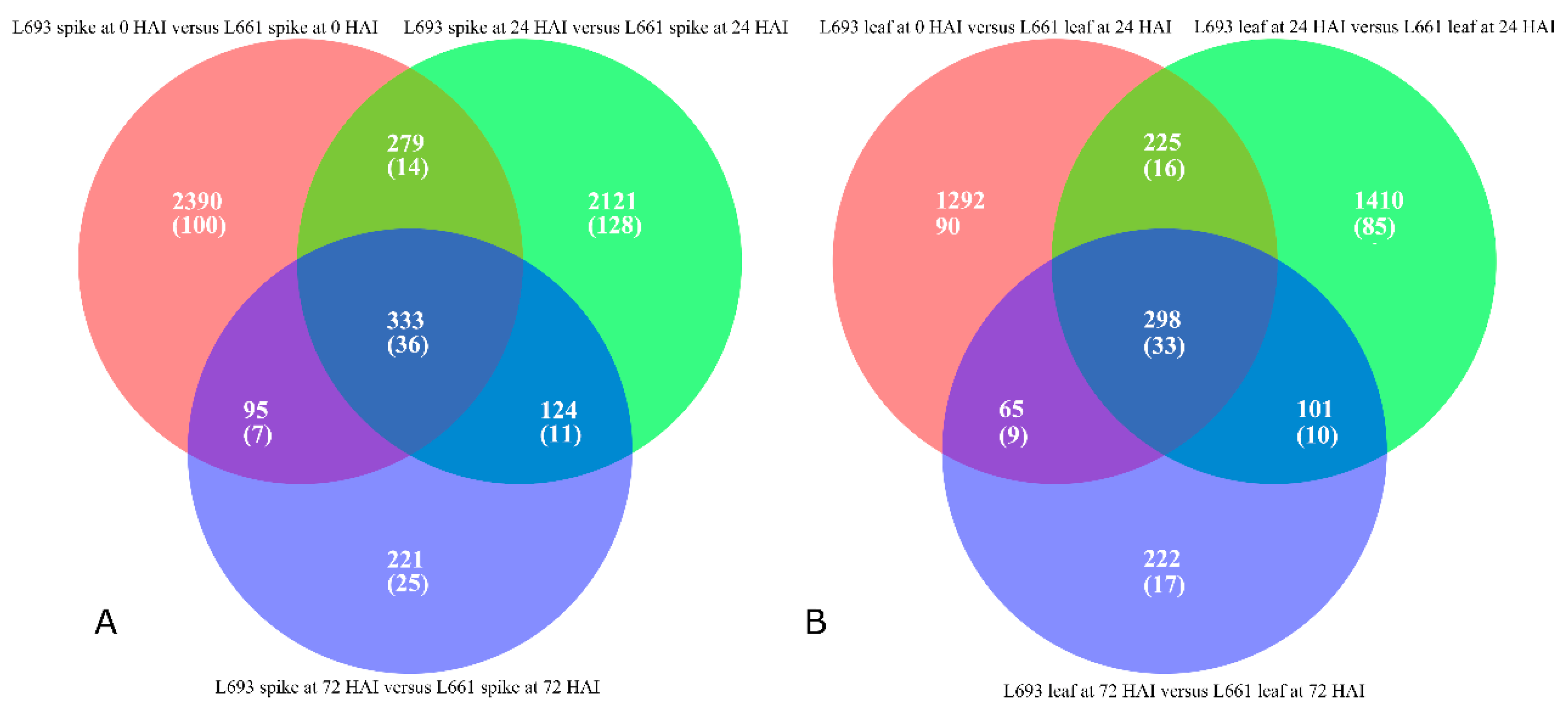
2.3. Differential Expression Analysis Revealed Tissue-Specific Expression Tendencies
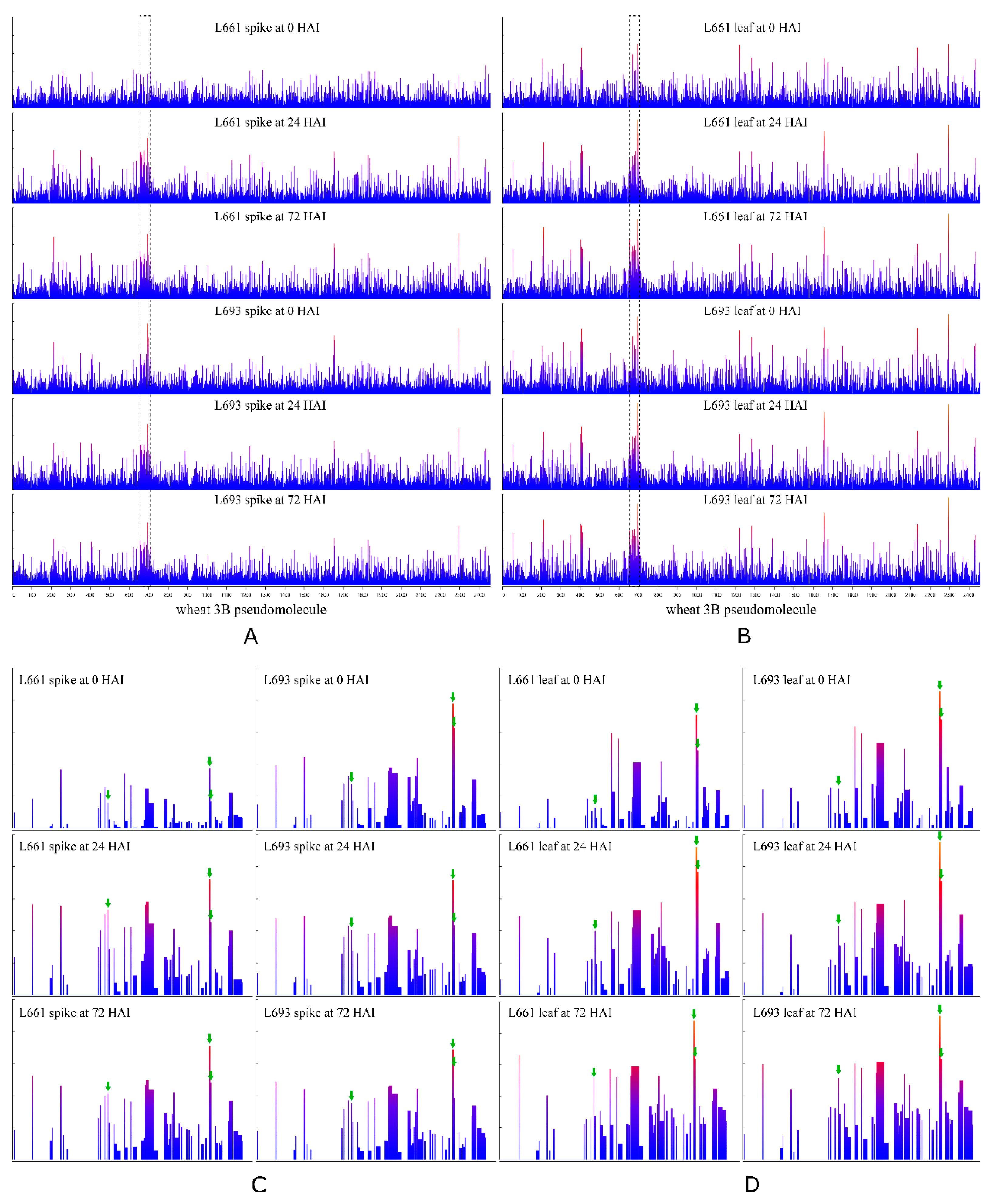
2.4. DEGs Have a Biased Chromosomal Distribution
2.5. A 140 kb Differential Expression Island Exists on Chromosome 3B
2.6. Genes in the 140 kb Expression Island Exhibit Higher Constitutive Expression in the FHB Resistance Genotype
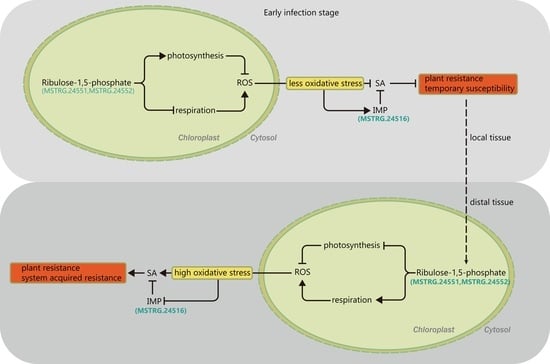
2.7. DEGs in the 140 kb Expression Island Are Mainly Involved in Chloroplast Function
3. Discussion
3.1. The Reliability of Original Data Was Important for Comparing Transcriptome Analyses
3.2. DEGs in Both the Leaf and Spike Play Key Roles in FHB Resistance Establishment
3.3. The Gene Island and 140 kb Differential Gene Expression Island on Chromosome 3B Were Highly Associated with FHB Resistance
3.4. Constitutive Differential Expression of Genes in the 181.40 to 181.54 Mb Region of Chromosome 3B Plays a Key Role in FHB Resistance
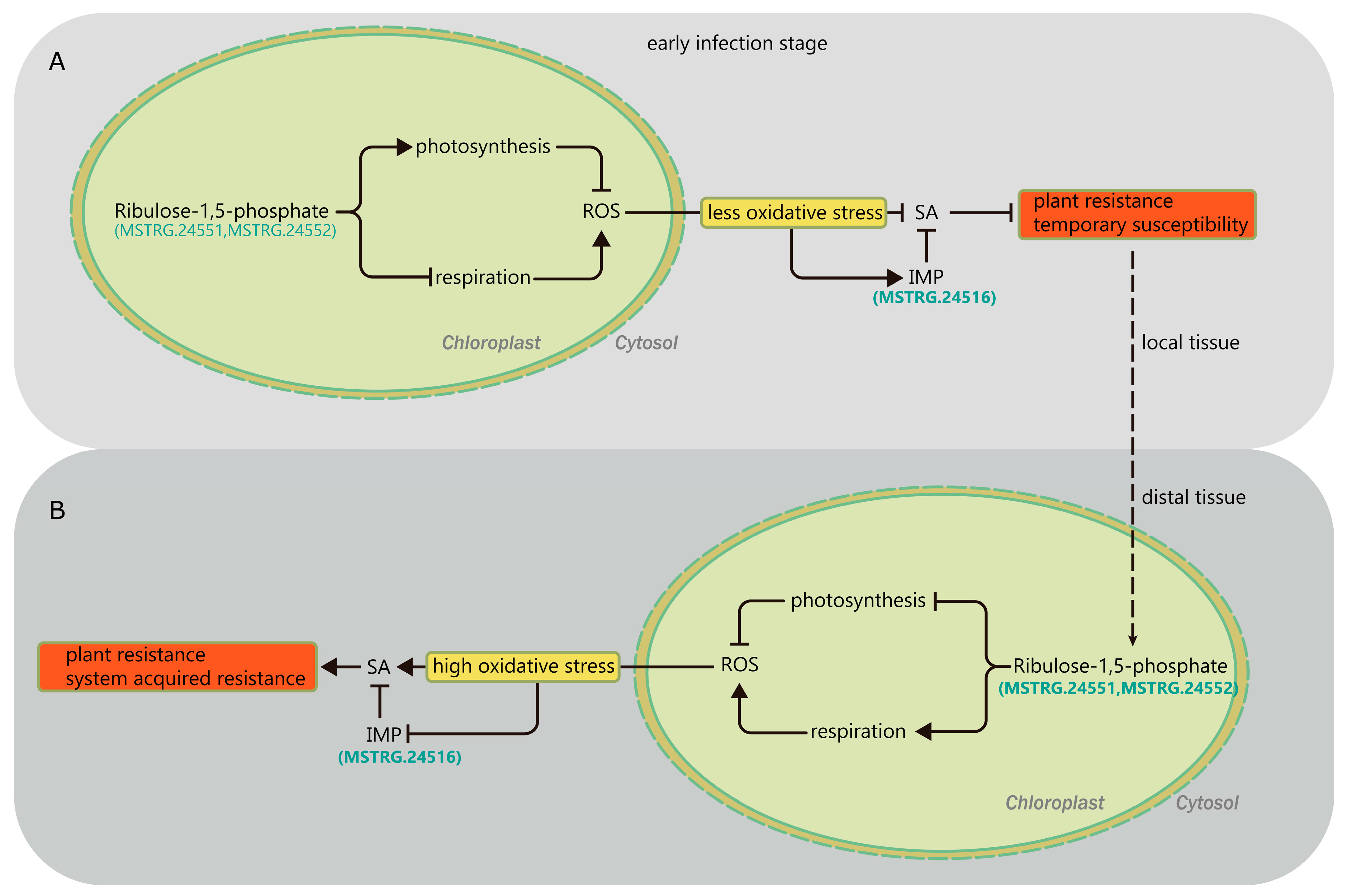
3.5. Photosynthesis Is Possibly Involved in FHB Resistance
4. Materials and Methods
4.1. Plant and Pathogen Materials
4.2. F. graminearum Spore Production and Inoculation
4.3. Experimental Design and Sample Harvesting
4.4. RNA Extraction and RNA-Sequencing
4.5. RNA-Seq Data Quality Control and Alignment Statistics
4.6. Reliable Analysis of Sequencing Data and Quantitative Real-Time PCR
4.7. Differential Gene Expression Analysis and Identification of Hotspot Expression Polymorphism
4.8. Bioinformatic Analysis of Pathway-Specific Gene Function in the FhbL693b Region
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nigro, D.; Giove, S.; Fortunato, S.; Incerti, O.; Zacheo, S.; Blanco, A.; Gadaleta, A. Allelic variation of wheat flour allergens in a collection of wheat genotypes. J. Chem. 2014, 2014. [Google Scholar] [CrossRef]
- McMullen, M.; Jones, R.; Gallenberg, D. Scab of wheat and barley: A re-emerging disease of devastating impact. Plant Dis. 1997, 81, 1340–1348. [Google Scholar] [CrossRef]
- Zwart, R.S.; Muylle, H.; Van Bockstaele, E.; Roldán-Ruiz, I. Evaluation of genetic diversity of fusarium head blight resistance in European winter wheat. Theor. Appl. Genet. 2008, 117, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Haber, S. Overview of some recent research developments in fusarium head blight of wheat. Can. J. Plant Pathol. 2013, 35, 149–174. [Google Scholar] [CrossRef]
- Bai, G.; Shaner, G. Management and resistance in wheat and barley to fusarium head blight 1. Annu. Rev. Phytopathol. 2004, 42, 135–161. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Nicholson, P.; Doohan, F.M. Action and reaction of host and pathogen during fusarium head blight disease. New Phytol. 2010, 185, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Oliver, R.; Cai, X.; Xu, S.; Chen, X.; Stack, R. Wheat-alien species derivatives: A novel source of resistance to fusarium head blight in wheat. Crop Sci. 2005, 45, 1353–1360. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, R.; Yang, J.; Luo, P. Identification of a new QTL for fusarium head blight resistance in the wheat genotype “wang shui-bai”. Mol. Biol. Rep. 2010, 37, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Karre, S.; Dhokane, D.; Kage, U.; Hukkeri, S.; Kushalappa, A.C. Real-time quantitative PCR based method for the quantification of fungal biomass to discriminate quantitative resistance in barley and wheat genotypes to fusarium head blight. J. Cereal Sci. 2015, 64, 16–22. [Google Scholar] [CrossRef]
- Dhokane, D.; Karre, S.; Kushalappa, A.C.; McCartney, C. Integrated metabolo-transcriptomics reveals fusarium head blight candidate resistance genes in wheat QTL-FHB2. PLoS ONE 2016, 11, e0155851. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Zhang, H.; Shu, K.; Wu, X.; Zhang, H.; Ren, Z. The physiological genetic effects of 1bl/1rs translocated chromosome in “stay green” wheat cultivar cn17. Can. J. Plant Sci. 2009, 89, 1–10. [Google Scholar] [CrossRef]
- Luo, P.; Deng, K.; Hu, X.; Li, L.; Li, X.; Chen, J.; Zhang, H.; Tang, Z.; Zhang, Y.; Sun, Q. Chloroplast ultrastructure regeneration with protection of photosystem ii is responsible for the functional ‘stay-green’ trait in wheat. Plant Cell Environ. 2013, 36, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhong, S.; Liu, N.; Chen, W.; Liu, T.; Li, X.; Zhang, M.; Ren, Z.; Yang, J.; Luo, P. Gene expression profile and physiological and biochemical characterization of hexaploid wheat inoculated with blumeria graminis f. Sp. Tritici. Physiol. Mol. Plant Pathol. 2015, 90, 39–48. [Google Scholar] [CrossRef]
- Luo, P.-G.; Hu, X.-Y.; Chang, Z.-J.; Zhang, M.; Zhang, H.-Q.; Ren, Z.-L. A new stripe rust resistance gene transferred from Thinopyrum intermedium to hexaploid wheat (Triticum aestivum). Phytoprotection 2009, 90, 57–63. [Google Scholar] [CrossRef][Green Version]
- Luo, P.; Luo, H.; Chang, Z.; Zhang, H.; Zhang, M.; Ren, Z. Characterization and chromosomal location of pm40 in common wheat: A new gene for resistance to powdery mildew derived from elytrigia intermedium. Theor. Appl. Genet. 2009, 118, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, X.; Chen, W.; Xiang, Z.; Zhong, S.; Chang, Z.; Zhang, M.; Zhang, H.; Tan, F.; Ren, Z. Genetic mapping of a putative thinopyrum intermedium-derived stripe rust resistance gene on wheat chromosome 1b. Theor. Appl. Genet. 2014, 127, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, M.; Xiang, Z.; Li, X.; Chen, W.; Luo, P. Registration of the novel wheat lines l658, l693, l696, and l699, with resistance to fusarium head blight, stripe rust, and powdery mildew. J. Plant Regist. 2015, 9, 121–124. [Google Scholar] [CrossRef]
- Zhang, L.; Chang, Z.; Li, X.; Zhang, H.; Ren, Z.; Luo, P. Screen and identification of wheat new resistant germplasms to fusarium head blight. Zhi Wu Bao Hu XueBao 2011, 38, 569–570. [Google Scholar]
- Yang, S.; Li, X.; Chen, W.; Liu, T.; Zhong, S.; Ma, L.; Zhang, M.; Zhang, H.; Yu, D.; Luo, P. Wheat resistance to fusarium head blight is associated with changes in photosynthetic parameters. Plant Dis. 2016, 100, 847–852. [Google Scholar] [CrossRef]
- Li, X.; Xiang, Z.; Chen, W.; Huang, Q.; Liu, T.; Li, Q.; Zhong, S.; Zhang, M.; Guo, J.; Lei, L.; et al. Reevaluation of two quantitative trait loci for type ii resistance to fusarium head blight in wheat germplasm pi 672538. Phytopathology 2016, 107, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Lovell, J.T.; Shakirov, E.V.; Schwartz, S.; Lowry, D.B.; Aspinwall, M.J.; Taylor, S.H.; Bonnette, J.; Palacio-Mejia, J.D.; Hawkes, C.V.; Fay, P.A. Promises and challenges of eco-physiological genomics in the field: Tests of drought responses in switchgrass. Plant Physiol. 2016, 172, 734–748. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, T.; Chen, W.; Zhong, S.; Zhang, H.; Tang, Z.; Chang, Z.; Wang, L.; Zhang, M.; Li, L.; et al. Wheat wcbp1 encodes a putative copper-binding protein involved in stripe rust resistance and inhibition of leaf senescence. BMC Plant Biol. 2015, 15, 239. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Sinha, A.K.; Roitsch, T. Plant physiology meets phytopathology: Plant primary metabolism and plant–pathogen interactions. J. Exp. Bot. 2007, 58, 4019–4026. [Google Scholar] [CrossRef] [PubMed]
- Rawat, N.; Pumphrey, M.O.; Liu, S.; Zhang, X.; Tiwari, V.K.; Ando, K.; Trick, H.N.; Bockus, W.W.; Akhunov, E.; Anderson, J.A. Wheat fhb1 encodes a chimeric lectin with agglutinin domains and a pore-forming toxin-like domain conferring resistance to fusarium head blight. Nat. Genet. 2016, 48, 1576. [Google Scholar] [CrossRef] [PubMed]
- Rustenholz, C.; Choulet, F.; Laugier, C.; Šafář, J.; Šimková, H.; Doležel, J.; Magni, F.; Scalabrin, S.; Cattonaro, F.; Vautrin, S. A 3000-loci transcription map of chromosome 3b unravels the structural and functional features of gene islands in hexaploid wheat. Plant Physiol. 2011, 157, 1596–1608. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, D.; Gill, K.S. Gene-containing regions of wheat and the other grass genomes. Plant Physiol. 2002, 128, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Zimmermann, W.; Perovic, D.; Paterson, A.H.; Ganal, M.; Graner, A.; Stein, N. A detailed look at 7 million years of genome evolution in a 439 kb contiguous sequence at the barley hv-eif4e locus: Recombination, rearrangements and repeats. Plant J. 2005, 41, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Rustenholz, C.; Hedley, P.E.; Morris, J.; Choulet, F.; Feuillet, C.; Waugh, R.; Paux, E. Specific patterns of gene space organisation revealed in wheat by using the combination of barley and wheat genomic resources. BMC Genom. 2010, 11, 714. [Google Scholar] [CrossRef] [PubMed]
- Serrano, I.; Audran, C.; Rivas, S. Chloroplasts at work during plant innate immunity. J. Exp. Bot. 2016, 67, 3845–3854. [Google Scholar] [CrossRef] [PubMed]
- Hill-Ambroz, K.; Webb, C.A.; Matthews, A.R.; Li, W.; Gill, B.S.; Fellers, J.P. Expression analysis and physical mapping of a CDNA library of fusarium head blight infected wheat spikes. Crop Sci. 2006, 46, S-15–S-26. [Google Scholar] [CrossRef]
- Bauriegel, E.; Herppich, W.B. Hyperspectral and chlorophyll fluorescence imaging for early detection of plant diseases, with special reference to fusarium spec. Infections on wheat. Agriculture 2014, 4, 32–57. [Google Scholar] [CrossRef]
- Ajigboye, O.O.; Bousquet, L.; Murchie, E.H.; Ray, R.V. Chlorophyll fluorescence parameters allow the rapid detection and differentiation of plant responses in three different wheat pathosystems. Funct. Plant Biol. 2016, 43, 356–369. [Google Scholar] [CrossRef]
- Berridge, M.J.; Irvine, R.F. Inositol phosphates and cell signalling. Nature 1989, 341, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Nourbakhsh, A.; Collakova, E.; Gillaspy, G.E. Characterization of the inositol monophosphatase gene family in arabidopsis. Front. Plant Sci. 2014, 5, 725. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, S.; Noctor, G. Myo-inositol abolishes salicylic acid-dependent cell death and pathogen defence responses triggered by peroxisomal hydrogen peroxide. New Phytol. 2010, 188, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Coram, T.E.; Huang, X.; Zhan, G.; Settles, M.L.; Chen, X. Meta-analysis of transcripts associated with race-specific resistance to stripe rust in wheat demonstrates common induction of blue copper-binding protein, heat-stress transcription factor, pathogen-induced wir1a protein, and ent-kaurene synthase transcripts. Funct. Integr. Genom. 2010, 10, 383–392. [Google Scholar]
- Asai, S.; Yoshioka, H. Nitric oxide as a partner of reactive oxygen species participates in disease resistance to necrotrophic pathogen botrytis cinerea in nicotiana benthamiana. Mol. Plant-Microbe Interact. 2009, 22, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Cárdenas, M.L.; Narváez-Vásquez, J.; Ryan, C.A. Hydrogen peroxide acts as a second messenger for the induction of defense genes in tomato plants in response to wounding, systemin, and methyl jasmonate. Plant Cell 2001, 13, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Barna, B.; Fodor, J.; Harrach, B.; Pogány, M.; Király, Z. The janus face of reactive oxygen species in resistance and susceptibility of plants to necrotrophic and biotrophic pathogens. Plant Physiol. Biochem. 2012, 59, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Caplan, J.L.; Mamillapalli, P.; Burch-Smith, T.M.; Czymmek, K.; Dinesh-Kumar, S. Chloroplastic protein nrip1 mediates innate immune receptor recognition of a viral effector. Cell 2008, 132, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Belhaj, K.; Lin, B.; Mauch, F. The chloroplast protein rph1 plays a role in the immune response of arabidopsis to phytophthora brassicae. Plant J. 2009, 58, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Gou, J.-Y.; Li, K.; Wu, K.; Wang, X.; Lin, H.; Cantu, D.; Uauy, C.; Dobon-Alonso, A.; Midorikawa, T.; Inoue, K. Wheat stripe rust resistance protein wks1 reduces the ability of the thylakoid-associated ascorbate peroxidase to detoxify reactive oxygen species. Plant Cell 2015, 27, 1755–1770. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar]
- Clavijo, B.J.; Venturini, L.; Schudoma, C.; Accinelli, G.G.; Kaithakottil, G.; Wright, J.; Borrill, P.; Kettleborough, G.; Heavens, D.; Chapman, H.; et al. An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 2017, 27, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef] [PubMed]
- Phanstiel, D.H. Sushi: Tools for Visualizing Genomics Data. Available online: http://bioconductor.org/packages/release/bioc/html/Sushi.html (accessed on 13 March 2018).
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Samples | Raw Data | Clean Reads | Mapped | Mapped % | Multiple | Multiple % | Unique | Unique % |
|---|---|---|---|---|---|---|---|---|---|
| spike | L661 spike at 0 HAI | 48,619,708 | 44,064,636 | 31,413,442 | 71.3% | 7,948,442 | 25.3% | 23,472,782 | 74.7% |
| L693 spike at 0 HAI | 54,638,926 | 50,729,857 | 37,433,618 | 73.8% | 15,223,441 | 40.7% | 22,210,177 | 59.3% | |
| L661 spike at 24 HAI | 80,401,044 | 14,261,236 | 6,799,856 | 47.7% | 2,277,018 | 33.5% | 4,522,838 | 66.5% | |
| L693 spike at 24 HAI | 56,001,617 | 37,706,168 | 24,971,426 | 66.2% | 7,770,208 | 31.1% | 17,201,218 | 68.9% | |
| L661 spike at 72 HAI | 64,589,048 | 47,444,396 | 29,577,848 | 62.3% | 9,209,005 | 31.1% | 20,368,843 | 68.9% | |
| L693 spike at 72 HAI | 73,276,052 | 52,378,107 | 33,229,807 | 63.4% | 9,061,381 | 27.3% | 24,168,426 | 72.7% | |
| leaf | L661 leaf at 0 HAI | 74,642,980 | 40,067,567 | 28,798,631 | 71.9% | 9,266,948 | 32.2% | 19,983,610 | 69.4% |
| L693 leaf at 0 HAI | 53,896,907 | 37,849,318 | 18,909,880 | 50.0% | 8,498,338 | 44.9% | 10,411,542 | 55.1% | |
| L661 leaf at 24 HAI | 52,460,707 | 45,960,283 | 35,981,574 | 78.3% | 20,792,164 | 57.8% | 15,189,410 | 42.2% | |
| L693 leaf at 24 HAI | 54,132,118 | 45,690,464 | 32,199,399 | 70.5% | 20,182,030 | 62.7% | 12,017,369 | 37.3% | |
| L661 leaf at 72 HAI | 50,556,369 | 26,746,381 | 18,315,598 | 68.5% | 9,430,647 | 51.5% | 6,319,347 | 34.5% | |
| L693 leaf at 72 HAI | 49,560,263 | 26,728,939 | 18,240,700 | 68.2% | 10,006,376 | 54.9% | 8,234,324 | 45.1% |
| Pathways | Enzyme | Definition | Seq Nu. | Sequence |
|---|---|---|---|---|
| Oxidative phosphorylation | ec:1.10.2.2—reductase | ubiquinol-cytochrome c reductase cytochrome b/c1 subunit | 7 | MSTRG.24508 |
| ec:1.6.99.3—dehydrogenase | NADH dehydrogenase | MSTRG.24509, MSTRG.24524, MSTRG.24557, MSTRG.24558, MSTRG.24527, TRAES3BF007300290CFD_g | ||
| ec:1.9.3.1—oxidase | cytochrome c oxidase cbb3-type subunit I | MSTRG.24509 | ||
| ec:1.6.5.3—reductase (H+-translocating) | NADH:ubiquinone reductase (H+-translocating) | MSTRG.24509, MSTRG.24524, MSTRG.24557, MSTRG.24558, MSTRG.24527, TRAES3BF007300290CFD_g | ||
| Purine metabolism | ec:3.6.1.3—adenylpyrophosphatase | adenosinetriphosphatase | 3 | MSTRG.24512, MSTRG.24554 |
| ec:3.6.1.15—phosphatase | nucleoside-triphosphatase | MSTRG.24512, MSTRG.24554 | ||
| ec:2.7.7.6—RNA polymerase | DNA-directed RNA polymerase subunit alpha | MSTRG.24544 | ||
| Glyoxylate and dicarboxylate metabolism | ec:4.1.1.39—carboxylase | 2 | MSTRG.24551, MSTRG.24552 | |
| Biosythesis of antibiotics | ec:4.1.1.39—carboxylase | 2 | MSTRG.24551, MSTRG.24552 | |
| Carbon fixation in photosynthetic organisms | ec:4.1.1.39—carboxylase | 2 | MSTRG.24551, MSTRG.24552 | |
| Thiamine metabolism | ec:3.6.1.15—phosphatase | 2 | MSTRG.24512, MSTRG.24554 | |
| Pyrimidine metabolism | ec:2.7.7.6—RNA polymerase | 1 | MSTRG.24544 | |
| Galactose metabolism | ec:3.2.1.26—invertase | 1 | MSTRG.24512 | |
| Phenylpropanoid biosynthesis | ec:1.11.1.7—lactoperoxidase | 1 | MSTRG.24516 | |
| Starch and sucrose metabolism | ec:3.2.1.26—invertase | 1 | MSTRG.24512 | |
| ec:3.2.1.48—α-glucosidase | 1 | MSTRG.24512 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhong, S.; Chen, W.; Fatima, S.A.; Huang, Q.; Li, Q.; Tan, F.; Luo, P. Transcriptome Analysis Identifies a 140 kb Region of Chromosome 3B Containing Genes Specific to Fusarium Head Blight Resistance in Wheat. Int. J. Mol. Sci. 2018, 19, 852. https://doi.org/10.3390/ijms19030852
Li X, Zhong S, Chen W, Fatima SA, Huang Q, Li Q, Tan F, Luo P. Transcriptome Analysis Identifies a 140 kb Region of Chromosome 3B Containing Genes Specific to Fusarium Head Blight Resistance in Wheat. International Journal of Molecular Sciences. 2018; 19(3):852. https://doi.org/10.3390/ijms19030852
Chicago/Turabian StyleLi, Xin, Shengfu Zhong, Wanquan Chen, Syeda Akash Fatima, Qianglan Huang, Qing Li, Feiquan Tan, and Peigao Luo. 2018. "Transcriptome Analysis Identifies a 140 kb Region of Chromosome 3B Containing Genes Specific to Fusarium Head Blight Resistance in Wheat" International Journal of Molecular Sciences 19, no. 3: 852. https://doi.org/10.3390/ijms19030852
APA StyleLi, X., Zhong, S., Chen, W., Fatima, S. A., Huang, Q., Li, Q., Tan, F., & Luo, P. (2018). Transcriptome Analysis Identifies a 140 kb Region of Chromosome 3B Containing Genes Specific to Fusarium Head Blight Resistance in Wheat. International Journal of Molecular Sciences, 19(3), 852. https://doi.org/10.3390/ijms19030852