Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle


{kind=link}
Abstract
:1. Setting the Stage: Estrogen, the Cardiovascular System and the Immune Response
2. Estrogen Receptors
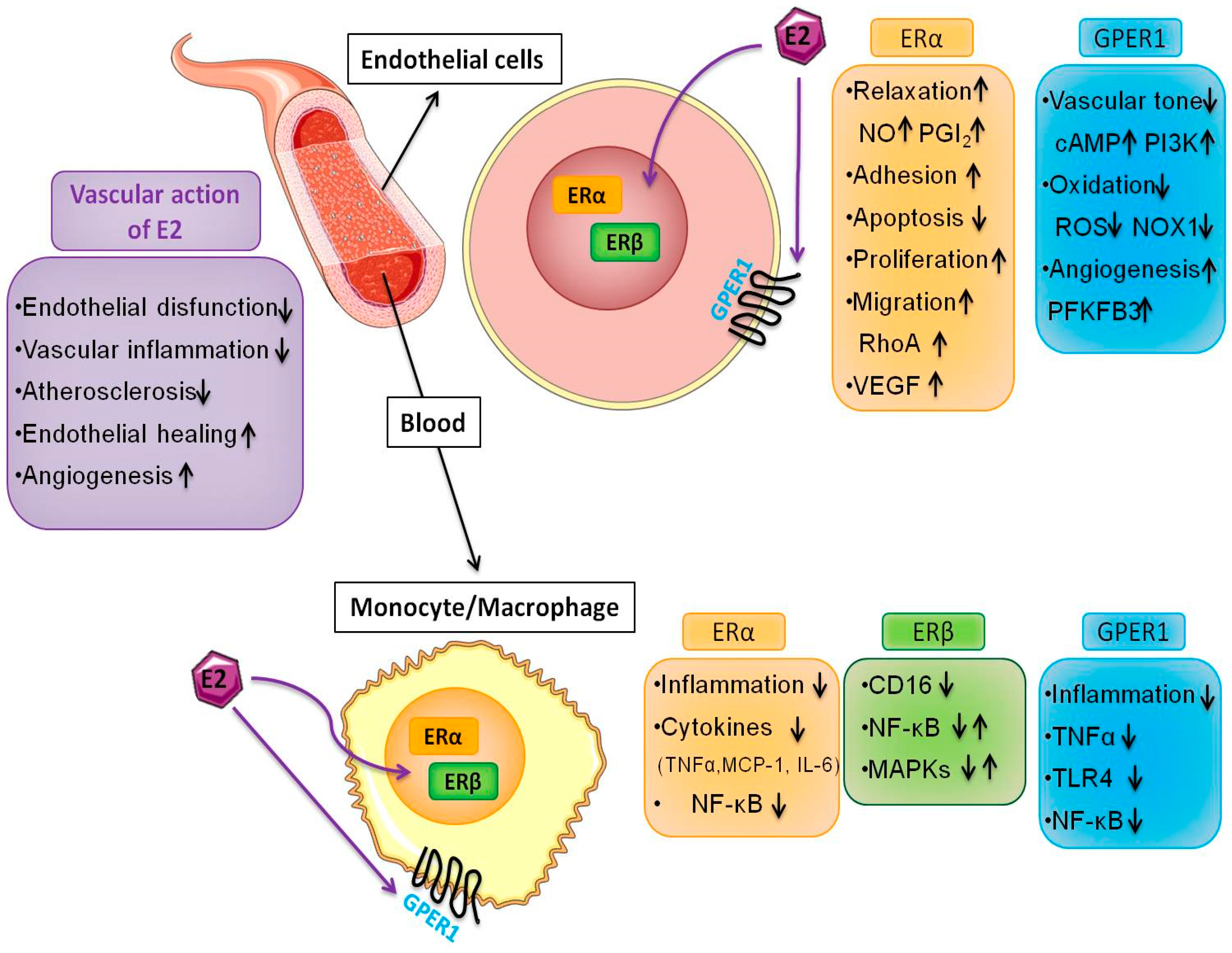
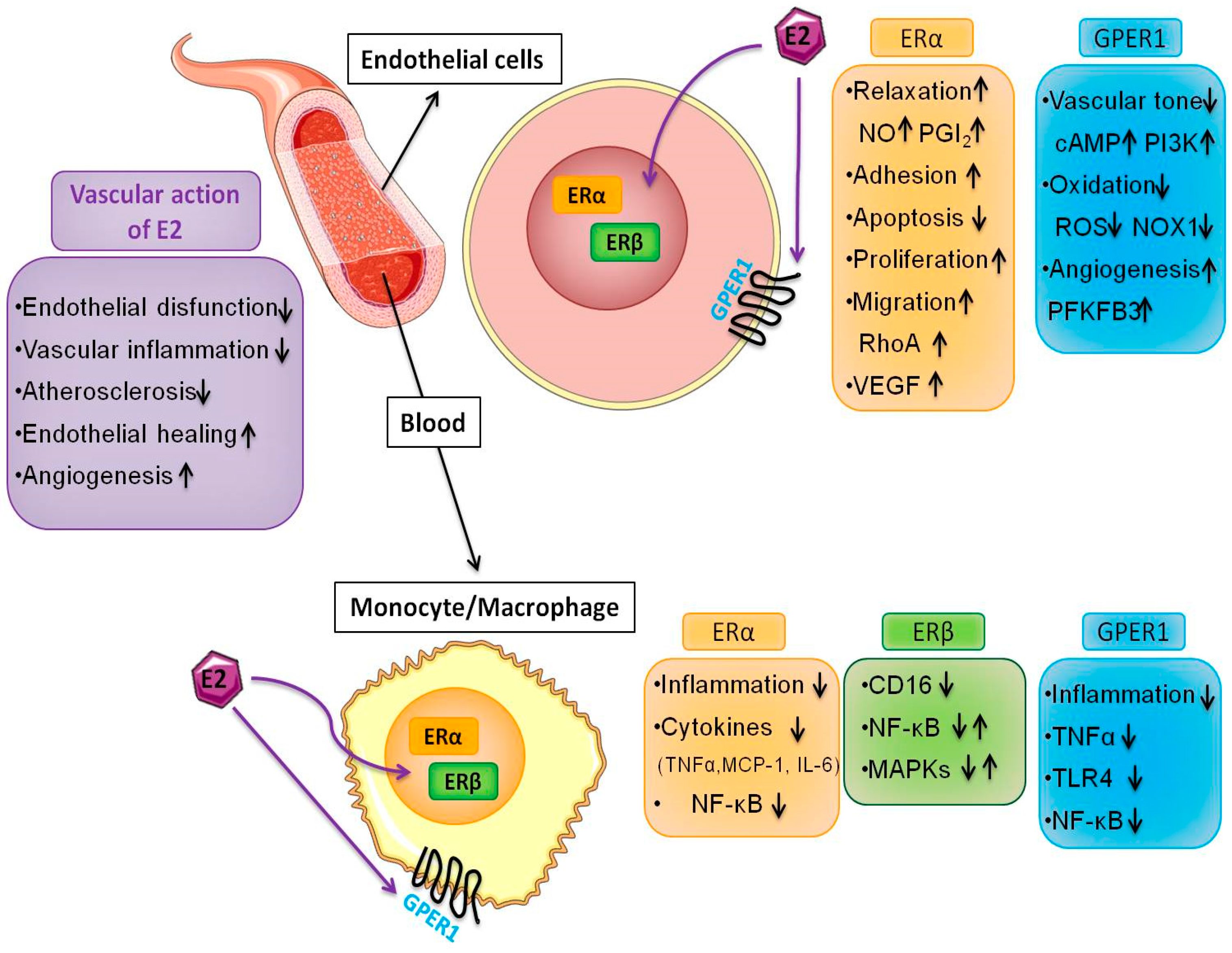
3. Estrogen Receptors and Endothelial Function
4. Estrogen, Angiogenesis and Metabolism
5. Estrogen and Macrophage Function
6. Estrogen Receptors in the Monocyte/Macrophage System
7. Estrogen in Women’s Health
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AF | activator factor |
| cAMP | cyclic adenosine monophosphate |
| E2 | 17β-estradiol |
| eNOS | endothelial nitric oxide synthase |
| ERE | estrogen response element |
| ER | estrogen receptor |
| FAK | focal adhesion kinase |
| G-1 | (±)-1-[(3aR*,4S*,9bS*)-4-(6-Bromo-1,3-benzodioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone |
| GPER1 | G-protein-coupled ER |
| HUVEC | human umbilical vein endothelial cells |
| IL | interleukin |
| IFNγ | interferon-γ |
| LBD | Ligand-Binding Domain |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemoattractant protein-1 |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | nitric oxide |
| PFKFB3 | phosphofructokinase-2/fructose-2,6-bisphosphatase-3 |
| PI3K | phosphatidylinositol-3-kinases |
| TNF-α | tumor necrosis factor α |
| VEGF | vascular endothelial growth factor |
References
- Prossnitz, E.R.; Arterburn, J.B. International Union of Basic and Clinical Pharmacology. XCVII. G protein-coupled estrogen receptor and its pharmacologic modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef] [PubMed]
- Pare, G.; Krust, A.; Karas, R.H.; Dupont, S.; Aronovitz, M.; Chambon, P.; Mendelsohn, M.E. Estrogen receptor-α mediates the protective effects of estrogen against vascular injury. Circ. Res. 2002, 90, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Bolego, C.; Rossoni, G.; Fadini, G.P.; Vegeto, E.; Pinna, C.; Albiero, M.; Boscaro, E.; Agostini, C.; Avogaro, A.; Gaion, R.M.; et al. Selective estrogen receptor-α agonist provides widespread heart and vascular protection with enhanced endothelial progenitor cell mobilization in the absence of uterotrophic action. FASEB J. 2010, 24, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Marcantoni, E.; Di Francesco, L.; Totani, L.; Piccoli, A.; Evangelista, V.; Tacconelli, S.; Patrignani, P. Effects of estrogen on endothelial prostanoid production and cyclooxygenase-2 and heme oxygenase-1 expression. Prostaglandins Other Lipid Mediat. 2012, 98, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Guyre, P.M.; Wira, C.R.; Pioli, P.A. Estradiol regulates expression of estrogen receptor ERα46 in human macrophages. PLoS ONE 2009, 4, e5539. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, P.; Sirois, M.G.; Bernatchez, P.N.; Tanguay, J.F. Estrogen regulation of endothelial and smooth muscle cell migration and proliferation: Role of p38 and p42/44 mitogen-activated protein kinase. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1585–1590. [Google Scholar] [CrossRef] [PubMed]
- Maggi, A.; Cignarella, A.; Brusadelli, A.; Bolego, C.; Pinna, C.; Puglisi, L. Diabetes undermines estrogen control of inducible nitric oxide synthase function in rat aortic smooth muscle cells through overexpression of estrogen receptor-β. Circulation 2003, 108, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.E.; McGowan, K.A.; Grant, D.S.; Maheshwari, S.; Bhartiya, D.; Cid, M.C.; Kleinman, H.K.; Schnaper, H.W. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation 1995, 91, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Concina, P.; Sordello, S.; Barbacanne, M.A.; Elhage, R.; Pieraggi, M.T.; Fournial, G.; Plouet, J.; Bayard, F.; Arnal, J.F. The mitogenic effect of 17β-estradiol on in vitro endothelial cell proliferation and on in vivo reendothelialization are both dependent on vascular endothelial growth factor. J. Vasc. Res. 2000, 37, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, T.; Scorticati, C.; Mannella, P.; Fadiel, A.; Giretti, M.S.; Fu, X.D.; Baldacci, C.; Garibaldi, S.; Caruso, A.; Fornari, L.; et al. Estrogen receptor α interacts with Gα13 to drive actin remodeling and endothelial cell migration via the RhoA/Rho kinase/moesin pathway. Mol. Endocrinol. 2006, 20, 1756–1771. [Google Scholar] [CrossRef] [PubMed]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Ferri, N.; Cignarella, A.; Trevisi, L.; Bolego, C. The glycolytic enzyme PFKFB3 is involved in estrogen-mediated angiogenesis via GPER1. J. Pharmacol. Exp. Ther. 2017, 361, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Bao, A.M.; Liu, R.Y.; van Someren, E.J.; Hofman, M.A.; Cao, Y.X.; Zhou, J.N. Diurnal rhythm of free estradiol during the menstrual cycle. Eur. J. Endocrinol. 2003, 148, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Wildman, R.P.; Colvin, A.B.; Powell, L.H.; Matthews, K.A.; Everson-Rose, S.A.; Hollenberg, S.; Johnston, J.M.; Sutton-Tyrrell, K. Associations of endogenous sex hormones with the vasculature in menopausal women: The Study of Women’s health Across the Nation (SWAN). Menopause 2008, 15, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Cignarella, A.; Kratz, M.; Bolego, C. Emerging role of estrogen in the control of cardiometabolic disease. Trends Pharmacol. Sci. 2010, 31, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S.; Benedusi, V.; Fontana, R.; Maggi, A. Energy metabolism and fertility: A balance preserved for female health. Nat. Rev. Endocrinol. 2014, 10, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Krasinski, K.; Spyridopoulos, I.; Asahara, T.; van der Zee, R.; Isner, J.M.; Losordo, D.W. Estradiol accelerates functional endothelial recovery after arterial injury. Circulation 1997, 95, 1768–1772. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, A.; Luedemann, C.; Shastry, S.; Hanley, A.; Kearney, M.; Aikawa, R.; Isner, J.M.; Asahara, T.; Losordo, D.W. Estrogen-mediated, endothelial nitric oxide synthase-dependent mobilization of bone marrow-derived endothelial progenitor cells contributes to reendothelialization after arterial injury. Circulation 2003, 108, 3115–3121. [Google Scholar] [CrossRef] [PubMed]
- Strehlow, K.; Werner, N.; Berweiler, J.; Link, A.; Dirnagl, U.; Priller, J.; Laufs, K.; Ghaeni, L.; Milosevic, M.; Bohm, M.; et al. Estrogen increases bone marrow-derived endothelial progenitor cell production and diminishes neointima formation. Circulation 2003, 107, 3059–3065. [Google Scholar] [CrossRef] [PubMed]
- Toutain, C.E.; Filipe, C.; Billon, A.; Fontaine, C.; Brouchet, L.; Guery, J.C.; Gourdy, P.; Arnal, J.F.; Lenfant, F. Estrogen receptor α expression in both endothelium and hematopoietic cells is required for the accelerative effect of estradiol on reendothelialization. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Rubanyi, G.M.; Johns, A.; Kauser, K. Effect of estrogen on endothelial function and angiogenesis. Vasc. Pharmacol. 2002, 38, 89–98. [Google Scholar] [CrossRef]
- Filipe, C.; Lam Shang Leen, L.; Brouchet, L.; Billon, A.; Benouaich, V.; Fontaine, V.; Gourdy, P.; Lenfant, F.; Arnal, J.F.; Gadeau, A.P.; et al. Estradiol accelerates endothelial healing through the retrograde commitment of uninjured endothelium. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2822–H2830. [Google Scholar] [CrossRef] [PubMed]
- Brouchet, L.; Krust, A.; Dupont, S.; Chambon, P.; Bayard, F.; Arnal, J.F. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-α but not estrogen receptor-β. Circulation 2001, 103, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Hardman, M.J.; Ashcroft, G.S. Estrogen, not intrinsic aging, is the major regulator of delayed human wound healing in the elderly. Genome Biol. 2008, 9, R80. [Google Scholar] [CrossRef] [PubMed]
- Libert, C.; Dejager, L.; Pinheiro, I. The X chromosome in immune functions: When a chromosome makes the difference. Nat. Rev. Immunol. 2010, 10, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Gubbels Bupp, M.R. Sex, the aging immune system, and chronic disease. Cell. Immunol. 2015, 294, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Amadori, A.; Zamarchi, R.; De Silvestro, G.; Forza, G.; Cavatton, G.; Danieli, G.A.; Clementi, M.; Chieco-Bianchi, L. Genetic control of the CD4/CD8 T-cell ratio in humans. Nat. Med. 1995, 1, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Scotland, R.S.; Stables, M.J.; Madalli, S.; Watson, P.; Gilroy, D.W. Sex differences in resident immune cell phenotype underlie more efficient acute inflammatory responses in female mice. Blood 2011, 118, 5918–5927. [Google Scholar] [CrossRef] [PubMed]
- Benedek, G.; Zhang, J.; Nguyen, H.; Kent, G.; Seifert, H.; Vandenbark, A.A.; Offner, H. Novel feedback loop between M2 macrophages/microglia and regulatory B cells in estrogen-protected EAE mice. J. Neuroimmunol. 2017, 305, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, S.; McArthur, S. Oestrogen and immunomodulation: New mechanisms that impact on peripheral and central immunity. Curr. Opin. Pharmacol. 2013, 13, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S.; Maggi, A. Sex differences: A resultant of an evolutionary pressure? Cell. Metab. 2017, 25, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Bolego, C.; Vegeto, E.; Pinna, C.; Maggi, A.; Cignarella, A. Selective agonists of estrogen receptor isoforms: New perspectives for cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2192–2199. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Davidge, S.T. G-protein coupled receptor 30 (GPR30): A novel regulator of endothelial inflammation. PLoS ONE 2012, 7, e52357. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The dynamic structure of the estrogen receptor. J. Amino Acids 2011, 2011, 812540. [Google Scholar] [CrossRef] [PubMed]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Strom, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed]
- Leitman, D.C.; Paruthiyil, S.; Vivar, O.I.; Saunier, E.F.; Herber, C.B.; Cohen, I.; Tagliaferri, M.; Speed, T.P. Regulation of specific target genes and biological responses by estrogen receptor subtype agonists. Curr. Opin. Pharmacol. 2010, 10, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Bender, J.R. Membrane-initiated actions of estrogen on the endothelium. Mol. Cell. Endocrinol. 2009, 308, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-dependent estrogen receptor α membrane localization: Regulation by 17β-estradiol. Mol. Biol. Cell 2005, 16, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Billon-Gales, A.; Krust, A.; Fontaine, C.; Abot, A.; Flouriot, G.; Toutain, C.; Berges, H.; Gadeau, A.P.; Lenfant, F.; Gourdy, P.; et al. Activation function 2 (AF2) of estrogen receptor-α is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc. Natl. Acad. Sci. USA 2011, 108, 13311–13316. [Google Scholar] [CrossRef] [PubMed]
- Adlanmerini, M.; Solinhac, R.; Abot, A.; Fabre, A.; Raymond-Letron, I.; Guihot, A.L.; Boudou, F.; Sautier, L.; Vessieres, E.; Kim, S.H.; et al. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. USA 2014, 111, E283–E290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filardo, E.J.; Thomas, P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: Its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology 2012, 153, 2953–2962. [Google Scholar] [CrossRef] [PubMed]
- Pelekanou, V.; Kampa, M.; Kiagiadaki, F.; Deli, A.; Theodoropoulos, P.; Agrogiannis, G.; Patsouris, E.; Tsapis, A.; Castanas, E.; Notas, G. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPER1. J. Leukoc. Biol. 2016, 99, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Albanito, L.; Madeo, A.; Lappano, R.; Vivacqua, A.; Rago, V.; Carpino, A.; Oprea, T.I.; Prossnitz, E.R.; Musti, A.M.; Ando, S.; et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007, 67, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Ma, X.; Ostmann, A.B.; Das, S.K. GPR30 activation opposes estrogen-dependent uterine growth via inhibition of stromal ERK1/2 and estrogen receptor alpha (ERα) phosphorylation signals. Endocrinology 2011, 152, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Traupe, T.; Stettler, C.D.; Li, H.; Haas, E.; Bhattacharya, I.; Minotti, R.; Barton, M. Distinct roles of estrogen receptors α and β mediating acute vasodilation of epicardial coronary arteries. Hypertension 2007, 49, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.N.; Gorelick, D.A. Crosstalk between nuclear and G protein-coupled estrogen receptors. Gen. Comp. Endocrinol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Venkov, C.D.; Rankin, A.B.; Vaughan, D.E. Identification of authentic estrogen receptor in cultured endothelial cells—A potential mechanism for steroid hormone regulation of endothelial function. Circulation 1996, 94, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Karas, R.H.; Patterson, B.L.; Mendelsohn, M.E. Human vascular smooth muscle cells contain functional estrogen receptor. Circulation 1994, 89, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, T.; Maffei, S.; Basta, G.; Barsacchi, G.; Genazzani, A.R.; Liao, J.K.; De Caterina, R. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ. Res. 2000, 87, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.A.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Characterization of the biological roles of the estrogen receptors, ERα and ERβ, in estrogen target tissues in vivo through the use of an ERα-selective ligand. Endocrinology 2002, 143, 4172–4177. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, M.E.; Karas, R.H. Molecular and cellular basis of cardiovascular gender differences. Science 2005, 308, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Ospina, J.A.; Krause, D.N.; Duckles, S.P. 17β-estradiol increases rat cerebrovascular prostacyclin synthesis by elevating cyclooxygenase-1 and prostacyclin synthase. Stroke 2002, 33, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.M.; Lawson, J.A.; Fries, S.; Koller, B.; Rader, D.J.; Smyth, E.M.; Fitzgerald, G.A. COX-2-derived prostacyclin confers atheroprotection on female mice. Science 2004, 306, 1954–1957. [Google Scholar] [CrossRef] [PubMed]
- Nevzati, E.; Shafighi, M.; Bakhtian, K.D.; Treiber, H.; Fandino, J.; Fathi, A.R. Estrogen induces nitric oxide production via nitric oxide synthase activation in endothelial cells. Acta Neurochir. Suppl. 2015, 120, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Garcia-Cardena, G.; Madri, J.A.; Sessa, W.C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Investig. 1997, 100, 3131–3139. [Google Scholar] [CrossRef] [PubMed]
- Ziche, M.; Morbidelli, L.; Choudhuri, R.; Zhang, H.T.; Donnini, S.; Granger, H.J.; Bicknell, R. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J. Clin. Investig. 1997, 99, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Novensa, L.; Novella, S.; Medina, P.; Segarra, G.; Castillo, N.; Heras, M.; Hermenegildo, C.; Dantas, A.P. Aging negatively affects estrogens-mediated effects on nitric oxide bioavailability by shifting ERα/ERβ balance in female mice. PLoS ONE 2011, 6, e25335. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, D.J.; Chapple, S.; Siow, R.C.; Mann, G.E. Equol-stimulated mitochondrial reactive oxygen species activate endothelial nitric oxide synthase and redox signaling in endothelial cells: Roles for F-actin and GPR30. Hypertension 2011, 57, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.R.; Fredette, N.C.; Daniel, C.; Sharma, G.; Amann, K.; Arterburn, J.B.; Barton, M.; Prossnitz, E.R. Obligatory role for GPER in cardiovascular aging and disease. Sci. Signal. 2016, 9, ra105. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.P.; Killilea, S.D.; Redmer, D.A. Angiogenesis in the female reproductive system. FASEB J. 1992, 6, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.J.; Gips, S.J.; Moldovan, N.; Wilhide, C.C.; Milliken, E.E.; Hoang, A.T.; Hruban, R.H.; Silverman, H.S.; Dang, C.V.; Goldschmidt-Clermont, P.J. 17β-estradiol inhibits apoptosis of endothelial cells. Biochem. Biophys. Res. Commun. 1997, 237, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Spyridopoulos, I.; Sullivan, A.B.; Kearney, M.; Isner, J.M.; Losordo, D.W. Estrogen-receptor-mediated inhibition of human endothelial cell apoptosis. Estradiol as a survival factor. Circulation 1997, 95, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Flamini, M.I.; Zullino, S.; Gopal, S.; Genazzani, A.R.; Simoncini, T. Estrogen receptor-α promotes endothelial cell motility through focal adhesion kinase. Mol. Hum. Reprod. 2011, 17, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Johns, A.; Freay, A.D.; Fraser, W.; Korach, K.S.; Rubanyi, G.M. Disruption of estrogen receptor gene prevents 17 beta estradiol-induced angiogenesis in transgenic mice. Endocrinology 1996, 137, 4511–4513. [Google Scholar] [CrossRef] [PubMed]
- Oviedo, P.J.; Hermenegildo, C.; Tarin, J.J.; Cano, A. Raloxifene increases proliferation of human endothelial cells in association with increased gene expression of cyclins A and B1. Fertil. Steril. 2007, 88, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Oviedo, P.J.; Sobrino, A.; Laguna-Fernandez, A.; Novella, S.; Tarin, J.J.; Garcia-Perez, M.A.; Sanchis, J.; Cano, A.; Hermenegildo, C. Estradiol induces endothelial cell migration and proliferation through estrogen receptor-enhanced RhoA/ROCK pathway. Mol. Cell. Endocrinol. 2011, 335, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Karas, R.H.; Gauer, E.A.; Bieber, H.E.; Baur, W.E.; Mendelsohn, M.E. Growth factor activation of the estrogen receptor in vascular cells occurs via a mitogen-activated protein kinase-independent pathway. J. Clin. Investig. 1998, 101, 2851–2861. [Google Scholar] [CrossRef] [PubMed]
- Shifren, J.L.; Tseng, J.F.; Zaloudek, C.J.; Ryan, I.P.; Meng, Y.G.; Ferrara, N.; Jaffe, R.B.; Taylor, R.N. Ovarian steroid regulation of vascular endothelial growth factor in the human endometrium: Implications for angiogenesis during the menstrual cycle and in the pathogenesis of endometriosis. J. Clin. Endocrinol. Metab. 1996, 81, 3112–3118. [Google Scholar] [PubMed]
- Gargett, C.E.; Zaitseva, M.; Bucak, K.; Chu, S.; Fuller, P.J.; Rogers, P.A. 17β-estradiol up-regulates vascular endothelial growth factor receptor-2 expression in human myometrial microvascular endothelial cells: Role of estrogen receptor-α and -β. J. Clin. Endocrinol. Metab. 2002, 87, 4341–4349. [Google Scholar] [CrossRef] [PubMed]
- Cid, M.C.; Esparza, J.; Schnaper, H.W.; Juan, M.; Yague, J.; Grant, D.S.; Urbano-Marquez, A.; Hoffman, G.S.; Kleinman, H.K. Estradiol enhances endothelial cell interactions with extracellular matrix proteins via an increase in integrin expression and function. Angiogenesis 1999, 3, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Haran, E.F.; Maretzek, A.F.; Goldberg, I.; Horowitz, A.; Degani, H. Tamoxifen enhances cell death in implanted MCF7 breast cancer by inhibiting endothelium growth. Cancer Res. 1994, 54, 5511–5514. [Google Scholar] [PubMed]
- Dadiani, M.; Seger, D.; Kreizman, T.; Badikhi, D.; Margalit, R.; Eilam, R.; Degani, H. Estrogen regulation of vascular endothelial growth factor in breast cancer in vitro and in vivo: The role of estrogen receptor α and c-Myc. Endocr. Relat. Cancer 2009, 16, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed]
- Pedram, A.; Razandi, M.; O’Mahony, F.; Harvey, H.; Harvey, B.J.; Levin, E.R. Estrogen reduces lipid content in the liver exclusively from membrane receptor signaling. Sci. Signal. 2013, 6, ra36. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, M.; Montagner, A.; Fontaine, C.; Lenfant, F.; Arnal, J.F.; Gourdy, P. Nuclear and membrane actions of estrogen receptor alpha: Contribution to the regulation of energy and glucose homeostasis. Adv. Exp. Med. Biol. 2017, 1043, 401–426. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Imbert-Fernandez, Y.; Clem, B.F.; O’Neal, J.; Kerr, D.A.; Spaulding, R.; Lanceta, L.; Clem, A.L.; Telang, S.; Chesney, J. Estradiol stimulates glucose metabolism via 6-phosphofructo-2-kinase (PFKFB3). J. Biol. Chem. 2014, 289, 9440–9448. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sotgia, F.; Clarke, R.B.; Lisanti, M.P.; Maggiolini, M. G protein-coupled receptors at the crossroad between physiologic and pathologic angiogenesis: Old paradigms and emerging concepts. Int J. Mol. Sci. 2017, 18, 2713. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Brecht, K.; Weigert, A.; Hu, J.; Popp, R.; Fisslthaler, B.; Korff, T.; Fleming, I.; Geisslinger, G.; Brune, B. Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J. 2011, 25, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Bruemmer, D. Targeting angiogenesis as treatment for obesity. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 161–162. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.P.; Feng, W.; Xing, D.; Weathington, N.M.; Blalock, J.E.; Chen, Y.F.; Oparil, S. Estrogen modulates inflammatory mediator expression and neutrophil chemotaxis in injured arteries. Circulation 2004, 110, 1664–1669. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.C.; Frink, M.; Hsieh, C.H.; Choudhry, M.A.; Schwacha, M.G.; Bland, K.I.; Chaudry, I.H. Downregulation of migration inhibitory factor is critical for estrogen-mediated attenuation of lung tissue damage following trauma-hemorrhage. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1227–L1232. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Guignard, L.; Knaup Reymond, M.; Perreau, M.; Roth-Kleiner, M.; Calandra, T.; Roger, T. Estradiol and progesterone strongly inhibit the innate immune response of mononuclear cells in newborns. Infect. Immun. 2011, 79, 2690–2698. [Google Scholar] [CrossRef] [PubMed]
- Campesi, I.; Sanna, M.; Zinellu, A.; Carru, C.; Rubattu, L.; Bulzomi, P.; Seghieri, G.; Tonolo, G.; Palermo, M.; Rosano, G.; et al. Oral contraceptives modify DNA methylation and monocyte-derived macrophage function. Biol. Sex. Differ. 2012, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pechenino, A.S.; Lin, L.; Mbai, F.N.; Lee, A.R.; He, X.M.; Stallone, J.N.; Knowlton, A.A. Impact of aging vs. estrogen loss on cardiac gene expression: Estrogen replacement and inflammation. Physiol. Genom. 2011, 43, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, A.; Fadini, G.P.; Tedesco, S.; Cappellari, R.; Vegeto, E.; Maggi, A.; Avogaro, A.; Bolego, C.; Cignarella, A. Alternative activation of human macrophages is rescued by estrogen treatment in vitro and impaired by menopausal status. J. Clin. Endocrinol. Metab. 2015, 100, E50–E58. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Oparil, S.; Yu, H.; Gong, K.; Feng, W.; Black, J.; Chen, Y.F.; Nozell, S. Estrogen modulates NFκB signaling by enhancing IκBα levels and blocking p65 binding at the promoters of inflammatory genes via estrogen receptor-β. PLoS ONE 2012, 7, e36890. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Guyre, P.M.; Pioli, P.A. Estradiol suppresses NF-κB activation through coordinated regulation of let-7a and miR-125b in primary human macrophages. J. Immunol. 2010, 184, 5029–5037. [Google Scholar] [CrossRef] [PubMed]
- Lambert, K.C.; Curran, E.M.; Judy, B.M.; Lubahn, D.B.; Estes, D.M. Estrogen receptor-α deficiency promotes increased TNF-α secretion and bacterial killing by murine macrophages in response to microbial stimuli in vitro. J. Leukoc. Biol. 2004, 75, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Calippe, B.; Douin-Echinard, V.; Delpy, L.; Laffargue, M.; Lelu, K.; Krust, A.; Pipy, B.; Bayard, F.; Arnal, J.F.; Guery, J.C.; et al. 17β-estradiol promotes TLR4-triggered proinflammatory mediator production through direct estrogen receptor α signaling in macrophages in vivo. J. Immunol. 2010, 185, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H. The complex role of estrogens in inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Spiller, K.L.; Anfang, R.R.; Spiller, K.J.; Ng, J.; Nakazawa, K.R.; Daulton, J.W.; Vunjak-Novakovic, G. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 2014, 35, 4477–4488. [Google Scholar] [CrossRef] [PubMed]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.; Emmerson, E.; Williams, H.; Saville, C.R.; Krust, A.; Chambon, P.; Mace, K.A.; Hardman, M.J. Estrogen receptor-alpha promotes alternative macrophage activation during cutaneous repair. J. Investig. Dermatol. 2014, 134, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Rizzi, N.; Vegeto, E.; Ciana, P.; Maggi, A. Estrogen accelerates the resolution of inflammation in macrophagic cells. Sci. Rep. 2015, 5, 15224. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.-Y.; Mauro, C. Similarities in the metabolic reprogramming of immune system and endothelium. Front. Immunol. 2017, 8, 837. [Google Scholar] [CrossRef] [PubMed]
- Blasko, E.; Haskell, C.A.; Leung, S.; Gualtieri, G.; Halks-Miller, M.; Mahmoudi, M.; Dennis, M.K.; Prossnitz, E.R.; Karpus, W.J.; Horuk, R. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J. Neuroimmunol. 2009, 214, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Rettew, J.A.; McCall, S.H.T.; Marriott, I. GPR30/GPER-1 mediates rapid decreases in TLR4 expression on murine macrophages. Mol. Cell. Endocrinol. 2010, 328, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.R.; Winger, V.; Kramer, S.F. 17β-estradiol utilizes the estrogen receptor to regulate CD16 expression in monocytes. Mol. Cell. Endocrinol. 2007, 279, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Campesi, I.; Marino, M.; Montella, A.; Pais, S.; Franconi, F. Sex differences in estrogen receptor α and β levels and activation status in LPS-stimulated human macrophages. J. Cell. Physiol. 2017, 232, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Pepe, G.; Braga, D.; Renzi, T.A.; Villa, A.; Bolego, C.; D’Avila, F.; Barlassina, C.; Maggi, A.; Locati, M.; Vegeto, E. Self-renewal and phenotypic conversion are the main physiological responses of macrophages to the endogenous estrogen surge. Sci. Rep. 2017, 7, 44270. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; Drew, B.G.; Le, J.A.; Soleymani, T.; Daraei, P.; Sitz, D.; Mohammad, L.; Henstridge, D.C.; Febbraio, M.A.; Hewitt, S.C.; et al. Myeloid-specific estrogen receptor α deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc. Natl. Acad. Sci. USA 2011, 108, 16457–16462. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S.; Mitro, N.; Fontana, R.; Gomaraschi, M.; Favari, E.; Recordati, C.; Lolli, F.; Quagliarini, F.; Meda, C.; Ohlsson, C.; et al. An essential role for liver ERα in coupling hepatic metabolism to the reproductive cycle. Cell Rep. 2016, 15, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Bowling, M.R.; Xing, D.; Kapadia, A.; Chen, Y.F.; Szalai, A.J.; Oparil, S.; Hage, F.G. Estrogen effects on vascular inflammation are age dependent: Role of estrogen receptors. Arterioscler. Thromb. Vasc. Biol.. 2014, 34, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. The sexual dimorphism of obesity. Mol. Cell. Endocrinol. 2015, 402, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Aragaki, A.K.; Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Chlebowski, R.T.; Howard, B.V.; Thomson, C.A.; Margolis, K.L.; et al. Menopausal hormone therapy and long-term all-cause and cause-specific mortality: The Women’s Health Initiative randomized trials. JAMA 2017, 318, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Schierbeck, L.L.; Rejnmark, L.; Tofteng, C.L.; Stilgren, L.; Eiken, P.; Mosekilde, L.; Kober, L.; Jensen, J.E. Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: Randomised trial. BMJ 2012, 345, e6409. [Google Scholar] [CrossRef] [PubMed]
- Stopinska-Gluszak, U.; Waligora, J.; Grzela, T.; Gluszak, M.; Jozwiak, J.; Radomski, D.; Roszkowski, P.I.; Malejczyk, J. Effect of estrogen/progesterone hormone replacement therapy on natural killer cell cytotoxicity and immunoregulatory cytokine release by peripheral blood mononuclear cells of postmenopausal women. J. Reprod. Immunol. 2006, 69, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, M.P.; Meydani, M.; Lichtenstein, A.H.; Schaefer, E.J.; Dillard, A.; Lamon-Fava, S. Sex hormone modulation of proinflammatory cytokine and C-reactive protein expression in macrophages from older men and postmenopausal women. J. Endocrinol. 2010, 206, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Campesi, I.; Carru, C.; Zinellu, A.; Occhioni, S.; Sanna, M.; Palermo, M.; Tonolo, G.; Mercuro, G.; Franconi, F. Regular cigarette smoking influences the transsulfuration pathway, endothelial function, and inflammation biomarkers in a sex-gender specific manner in healthy young humans. Am. J. Transl. Res. 2013, 5, 497–509. [Google Scholar] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trenti, A.; Tedesco, S.; Boscaro, C.; Trevisi, L.; Bolego, C.; Cignarella, A. Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. Int. J. Mol. Sci. 2018, 19, 859. https://doi.org/10.3390/ijms19030859
Trenti A, Tedesco S, Boscaro C, Trevisi L, Bolego C, Cignarella A. Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. International Journal of Molecular Sciences. 2018; 19(3):859. https://doi.org/10.3390/ijms19030859
Chicago/Turabian StyleTrenti, Annalisa, Serena Tedesco, Carlotta Boscaro, Lucia Trevisi, Chiara Bolego, and Andrea Cignarella. 2018. "Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle" International Journal of Molecular Sciences 19, no. 3: 859. https://doi.org/10.3390/ijms19030859
APA StyleTrenti, A., Tedesco, S., Boscaro, C., Trevisi, L., Bolego, C., & Cignarella, A. (2018). Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. International Journal of Molecular Sciences, 19(3), 859. https://doi.org/10.3390/ijms19030859