Epigenetic Programming of Synthesis, Release, and/or Receptor Expression of Common Mediators Participating in the Risk/Resilience for Comorbid Stress-Related Disorders and Coronary Artery Disease


Abstract
:1. Introduction
2. Similar Underlying Neuroendocrine Function in Psychiatric and Cardiometabolic Diseases
3. Psychiatric and Cardiometabolic Mediators That Could Be under Early Programming Regulation
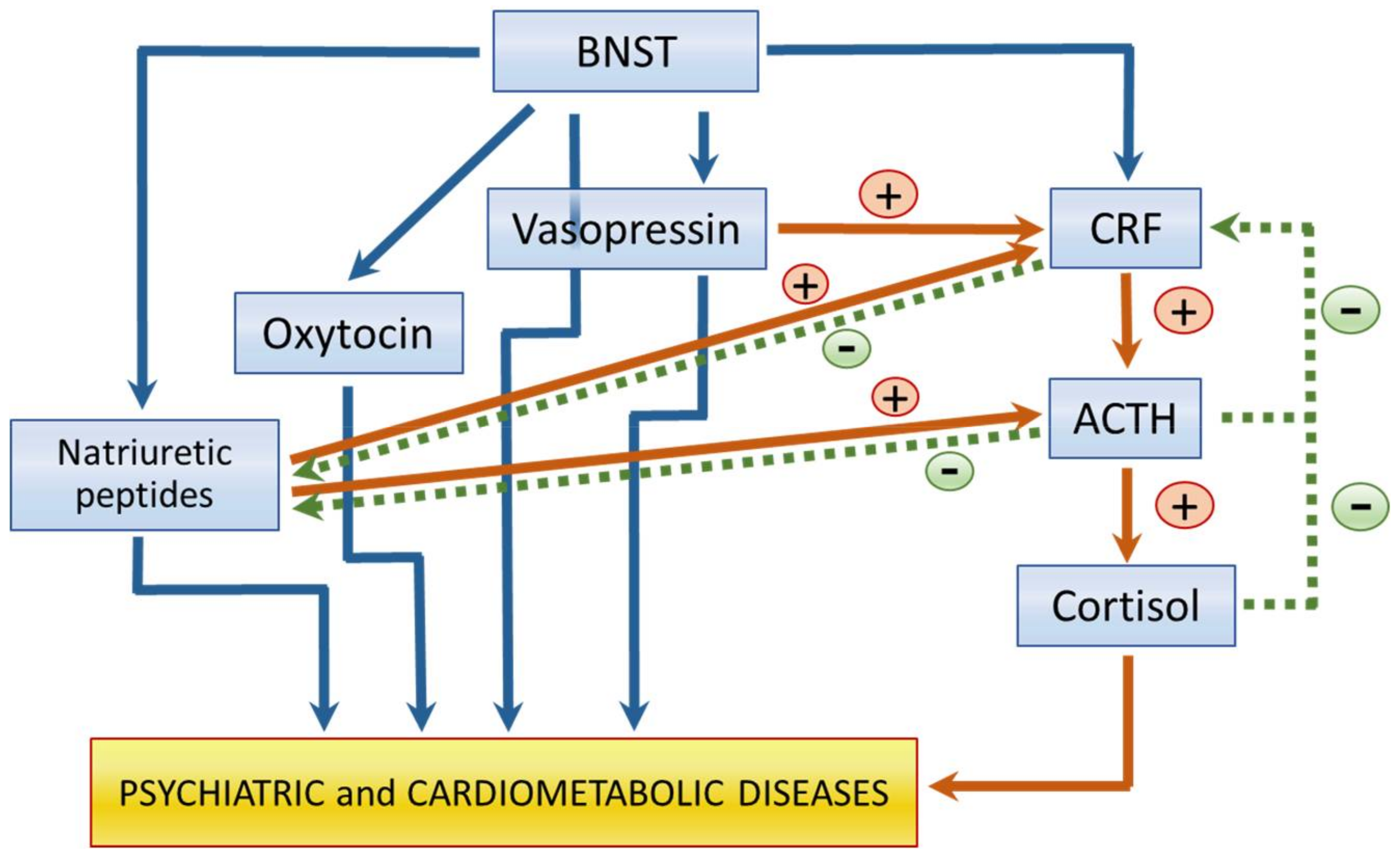
3.1. Hypothalamus-Hypophysis-Adrenal Gland (HHA) Axis Mediators
3.2. Oxytocin and Vasopressin
3.3. Natriuretic Peptides (NPs)
3.4. Renin-Angiotensin-Aldosterone System (RAAS)
3.5. Neuregulins (NRGs)
3.6. Purinergic Neuromediators
3.7. Inflammatory Mediators
4. Possible Re-Programming of Expression of Cardio and Neuroactive Substances Participating in the Comorbidity of Neuropsychiatric and Cardiometabolic Diseases
5. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| ACE | angiotensin converting enzyme |
| ACTH | adrenocorticotrophic hormone |
| ADPR | ADP ribose |
| AMP | adenosine mono phosphate |
| Ang II | angiotensin II |
| ANP | atrial natriuretic peptide |
| ANS | autonomous nervous system |
| ATP | adenosine triphosphate |
| AVP | arginine vasopressin gene, |
| AvpR1 | vasopressin receptor |
| BNP | brain natriuretic peptide |
| BNST | bed nucleus of the stria terminalis |
| Ca2+ | calcium |
| CAD | coronary artery disease |
| CMD | cardiometabolic diseases |
| CNP | C-type natriuretic |
| CNS | central nervous system |
| CpG | cytosine-phosphate-guanine |
| CRF | corticotrophin releasing factor |
| CV | cardiovascular |
| DNA | deoxyribonucleic acid |
| EGF | epithelial growth factor |
| ErbB receptors | receptors pertaining to EGF |
| FKBP5 | gene coding for chaperones |
| GABA | gamma amino butyric acid |
| GR | glucocorticoid receptors |
| HAT | histone acetyltransferases |
| HDAC | histone deacetylases |
| HHA | hypothalamus-hypophysis-adrenal |
| HMT | histone methyltransferases |
| IL | interleukin |
| ICNS | intrinsic cardiac nervous system |
| K+ | potassium |
| MAP | mitogen activated protein |
| MR | mineralocorticoid receptors |
| mRNA | micro ribonucleic acid |
| Na+ | sodium |
| NAD+ | β-nicotinamide adenine dinucleotide |
| NMDA | N-methyl-d-aspartate |
| NPR-A, NPR-B and NPR-C | natriuretic peptide receptors |
| NPs | natriuretic peptides |
| NR3C1 | steroid receptor gene |
| NRGs | neuregulins |
| NRSF | neuron restrictive silencing factor |
| PFC | prefrontal cortex |
| PI-3 | phosphatidylinositol-3 |
| PKC | protein kinase C |
| pMeCP2 | phosphorylated protein related to methylation of histones |
| PVN | paraventricular nucleus |
| RAAS | renin–angiotensin–aldosterone system |
| RNA | ribonucleic acid |
| ROS | reactive oxygen species |
| SNS | sympathetic nervous system |
| SRD | stress related disorders |
| TNF-α | tumor necrosis factor alpha |
References
- Pasipoularides, A. Greek underpinnings to his methodology in unraveling De Motu Cordis and what Harvey has to teach us still today. Int. J. Cardiol. 2013, 168, 3173–3182. [Google Scholar] [CrossRef] [PubMed]
- Rumsfeld, S.J.; Ho, P.M. Depression and cardiovascular disease. A call for recognition. Circulation 2005, 111, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Nusair, M.; Al-dadah, A.; Kumar, A. The tale of mind and heart: Psychiatric disorders & coronary heart disease. Mol. Med. 2012, 109, 199–203. [Google Scholar]
- Malzberg, B. Mortality among patients with involutional melancholia. Am. J. Psychiatry 1937, 93, 1231–1238. [Google Scholar] [CrossRef]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Murray, C. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Whiteford, H.A.; Ferrari, A.J.; Degenhardt, L.; Feigin, V.; Vos, T. The global burden of mental, neurological and substance use disorders: An analysis from the Global Burden of Disease Study 2010. PLoS ONE 2015, 10, e0116820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duric, V.; Clayton, S.; Leong, M.L.; Yuan, L.-L. Comorbidity Factors and Brain Mechanisms Linking Chronic Stress and Systemic Illness. Neural Plast. 2016, 5460732. [Google Scholar] [CrossRef] [PubMed]
- Bankier, B.; Januzzi, J.L.; Littman, A.B. The high prevalence of multiple psychiatric disorders in stable outpatients with coronary heart disease. Psychosom. Med. 2004, 66, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Roest, A.M.; Martens, E.J.; de Jonge, P.; Denollet, J. Anxiety and risk of incident coronary heart disease: A meta-analysis. J. Am. Coll. Cardiol. 2010, 56, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Barger, S.D.; Sydeman, S.J. Does generalized anxiety disorder predict coronary heart disease risk factors independently of major depressive disorder? J. Affect. Disord. 2005, 88, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Albert, C.M.; Chae, C.U.; Rexrode, K.M.; Manson, J.E.; Kawachi, I. Phobic anxiety and risk of coronary heart disease and sudden cardiac death among women. Circulation 2005, 111, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Caminero, A.; Blumentals, W.A.; Russo, L.J.; Brown, R.R.; Castilla-Puentes, R. Does panic disorder increase the risk of coronary heart disease? A cohort study of a national managed care database. Psychosom. Med. 2005, 67, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Weissman, M.M.; Markowitz, J.S.; Ouellette, R.; Greenwald, S.; Kahn, J. Panic disorders and cardiovascular/cerebrovascular problems: Results from a community survey. Am. J. Psychiatry 1990, 147, 1504–1508. [Google Scholar] [PubMed]
- Kubzansky, L.D.; Koenen, K.C.; Spiro, A., 3rd; Vokonas, P.S.; Sparrow, D. Prospective study of posttraumatic stress disorder symptoms and coronary heart disease in the Normative Aging Study. Arch. Gen. Psychiatry 2007, 64, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Hayward, C. Psychiatric illness and cardiovascular disease risk. Epidemiol. Rev. 1995, 17, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Todaro, J.F.; Shen, B.J.; Raffa, S.D.; Tilkemeier, P.L.; Niaura, R. Prevalence of anxiety disorders in men and women with established coronary heart disease. J. Cardiopulm. Rehabil. Prev. 2007, 27, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. The inheritance of epigenetic defects. Science 1987, 238, 63–170. [Google Scholar] [CrossRef]
- Meagher, R.B.; Müssar, J.K. The influence of DNA sequence on epigenome induced pathologies. Epigenet. Chromatin 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. Der epigenotypus. Endeavour 1942, 1, 18–20. [Google Scholar]
- Lewis, A.J.; Austin, E.; Knapp, R.; Vaiano, T.; Galbally, M. Perinatal Maternal Mental Health, Fetal Programming and Child Development. Healthcare 2015, 3, 1212–1227. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.C.; Polvani, G.; Pesce, M. Epigenetic programming and risk: The birthplace of cardiovascular disease? Stem Cell Rev. 2013, 9, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.L.; Walker, C.L. Molecular pathways: Environmental estrogens activate nongenomic signaling to developmentally reprogram the epigenome. Clin. Cancer Res. 2013, 19, 3732–3737. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Barker, D.J.; Eriksson, J.G.; Forsen, T.; Osmond, C. Fetal origins of adult disease: Strength of effects and biological basis. Int. J. Epidemiol. 2002, 31, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.J.; Galbally, M.; Gannon, T.; Symeonides, C. Early life programming as a target for prevention of child and adolescent mental disorders. BMC Med. 2014, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Post, W.S.; Goldschmidt-Clermont, P.J.; Wilhide, C.C.; Heldman, A.W.; Sussman, M.S.; Ouyang, P.; Milliken, E.E.; Issa, J.P. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc. Res. 1999, 43, 985–991. [Google Scholar] [CrossRef]
- Reynolds, R.M. Glucocorticoid excess and the developmental origins of disease: Two decades of testing the hypothesis—2012 Curt Richter Award Winner. Psychoneuroendocrinology 2013, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Pearson, R.M.; Goodman, S.H.; Rapa, E.; Rahman, A.; McCallum, M.; Howard, L.M.; Pariante, C.M. Effects of perinatal mental disorders on the fetus and child. Lancet 2014, 384, 1800–1819. [Google Scholar] [CrossRef]
- Srinivasan, M.; Laychock, S.G.; Hill, D.J.; Patel, M.S. Neonatal Nutrition: Metabolic programming of pancreatic islets and obesity. Exp. Biol. Med. 2003, 228, 15–23. [Google Scholar] [CrossRef]
- Aalinkeel, R.; Srinivasan, M.; Song, F.; Patel, M.S. Programming into adulthood of islet adaptations induced by early nutritional intervention in the rat. Am. J. Physiol. Endocr. Metab. 2001, 281, E640–E648. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, M.; Monteggia, L.M. Epigenetics and Psychiatry. Neurotherapeutics 2013, 10, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C. Lifestyle Modulators of Neuroplasticity: How Physical Activity, Mental Engagement, and Diet Promote Cognitive Health during. Aging Neural Plast. 2017, 2017, 3589271. [Google Scholar] [CrossRef] [PubMed]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.J.; Stein, D.J. A systematic review of the neural bases of psychotherapy for anxiety and related disorders. Dialogues Clin. Neurosci. 2015, 17, 261–279. [Google Scholar] [PubMed]
- Armour, J.A. Cardiac neuronal hierarchy in health and disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R262–R271. [Google Scholar] [CrossRef] [PubMed]
- Stepniakowski, K.; Budzikowski, A.; Lon, S.; Szczepanska-Sadowska, E. Central ANP attenuates pressor responses to central AVP in WKY and SHR. Brain Res. Bull. 1991, 27, 247–249. [Google Scholar] [CrossRef]
- Zannas, A.S.; West, A.E. Epigenetics and the regulation of stress vulnerability and resilience. Neuroscience 2014, 264, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.M. Nick Hales Award Lecture 2011: Glucocorticoids and early life programming of cardiometabolic disease. J. Dev. Orig. Health Dis. 2012, 3, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Galbally, M.; Lewis, A.J.; Ijzendoorn, M.; Permezel, M. The role of oxytocin in mother-infant relations: A systematic review of human studies. Harv. Rev. Psychiatry 2011, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Berry, E.M.; De Geest, S. Tell me what you eat and I will tell you your sociotype: Coping with diabesity. Rambam Maimonides Med. J. 2012, 3, e0010. [Google Scholar] [CrossRef] [PubMed]
- Lesse, A.; Rether, K.; Gröger, N.; Braun, K.; Bock, J. Chronic Postnatal Stress Induces Depressive-like Behavior in Male Mice and Programs Second-Hit Stress-Induced Gene Expression Patterns of OxtR and AvpR1a in Adulthood. Mol. Neurobiol. 2017, 54, 4813–4819. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.G.; Desai, M.; Guerra, C.; Wang, S. Prenatal programming of hypernatremia and hypertension in neonatal lambs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R97–R103. [Google Scholar] [CrossRef] [PubMed]
- Mathiyalagan, P.; Chang, L.; Du, X.J.; El-Osta, A. Cardiac ventricular chambers are epigenetically distinguishable. Cell Cycle 2010, 9, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.T. The Impact of Nutritional Insults during Fetal Life on Blood pressure. J. Nutr. Sci. Vitaminol. 2015, 61, S5–S6. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Abe, Y.; Sotoyama, H.; Kakita, A.; Kominami, R.; Hirokawa, S.; Ozaki, M.; Takahashi, H.; Nawa, H. Transient exposure of neonatal mice to neuregulin-1 results in hyperdopaminergic states in adulthood: Implication in neurodevelopmental hypothesis for schizophrenia. Mol. Psychiatry 2011, 16, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Feron, O.; Dessy, C.; Han, X.; Marchionni, M.A.; Kelly, R.A. Neuregulin signaling in the heart. Dynamic targeting of erbB4 to caveolarmicrodomains in cardiac myocytes. Circ. Res. 1999, 84, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H. Nucleotide signaling in nervous system development. Pflugers Arch. 2006, 452, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Rivkees, S.A.; Wendler, C.C. Regulation of cardiovascular development by adenosine and adenosine-mediated embryo protection. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Buscariollo, D.L.; Fang, X.; Greenwood, V.; Xue, H.; Rivkees, S.A.; Wendler, C.C. Embryonic caffeine exposure acts via A1 adenosine receptors to alter adult cardiac function and DNA methylation in mice. PLoS ONE 2014, 9, e87547. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Bilbo, S.D. Developmental programming of brain and behavior by perinatal diet: Focus oninflammatory mechanisms. Dialogues Clin. Neurosci. 2014, 16, 307–320. [Google Scholar] [PubMed]
- Spencer, S.J.; Meyer, U. Perinatal programming by inflammation. Brain Behav. Immun. 2017, 63, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bale, T.L.; Vale, W.W. CRF and CRF receptors: Role in stress responsivity and other behaviors. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 525–557. [Google Scholar] [CrossRef] [PubMed]
- Coste, S.C.; Quintos, R.F.; Stenzel-Poore, M.P. Corticotropin releasing hormone-related peptides and receptors: Emergentregulators of cardiovascular adaptations to stress. Trends Cardiovasc. Med. 2002, 12, 176–182. [Google Scholar] [CrossRef]
- Inoue, K.; Valdez, G.R.; Reyes, T.M.; Reinhardt, L.E.; Tabarin, A.; Rivier, J.; Vale, W.W.; Sawchenko, P.E.; Koob, G.F.; Zorrilla, E.P. Human urocortin II. A selective agonist for the type 2 corticotropin releasing factor receptor, decreases feeding and drinking in the rat. J. Pharmacol. Exp. Ther. 2003, 305, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Stengel, A.; Tache, Y. Corticotropin-releasing factor signaling and visceral response to stress. Exp. Biol. Med. 2010, 235, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Thorsell, A. Brain neuropeptide Y and corticotropin-releasing hormone in mediating stress and anxiety. Exp. Biol. Med. 2010, 235, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Hauger, R.L.; Grigoriadis, D.E.; Dallman, M.F.; Plotsky, P.M.; Vale, W.W.; Dautzenberg, F.M. International Union of Pharmacology. XXXVI. Current status of the nomenclature for receptors for corticotropin-releasing factor and their ligands. Pharmacol. Rev. 2003, 55, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Andres, A.L.; Regev, L.; Phi, L.; Seese, R.R.; Chen, Y.; Gall, C.M.; Baram, T.Z. NMDA Receptor Activation and Calpain Contribute to Disruption of Dendritic Spines by the Stress Neuropeptide CRH. J. Neurosci. 2013, 33, 16945–16960. [Google Scholar] [CrossRef] [PubMed]
- Jänig, W. Sympathetic nervous system and inflammation: A conceptual view. Auton. Neurosci. 2014, 182, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Jänig, W. Autonomic nervous system and inflammation. Auton. Neurosci. 2014, 182, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Buijs, R.M.; Escobar, C.; Swaab, D.F. The circadian system and the balance of the autonomic nervous system. Handb. Clin. Neurol. 2013, 117, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Ferenczi, S.; Kovács, K.J.; Garafova, A.; Jezova, D.; Zelena, D. Comparison of Stress-Induced Changes in Adults and Pups: Is Aldosterone the Main Adrenocortical Stress Hormone during the Perinatal Period in Rats? PLoS ONE 2013, 8, e72313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, C.E.; Hanson, M.A. Prenatal programming of postnatal endocrine responses by glucocorticoids. Reproduction 2002, 124, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Ter Heegde, F.; De Rijk, R.H.; Vinkers, C.H. The brain mineralocorticoid receptor and stress resilience. Psychoneuroendocrinology 2015, 52, 92–110. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F.; Heinrichs, S.C. A role for corticotropin releasing factor and urocortin in behavioral responses to stressors. Brain Res. 1999, 848, 141–152. [Google Scholar] [CrossRef]
- Griebel, G.; Stemmelin, J.; Gal, C.S.; Soubrié, P. Non-peptide vasopressin V1b receptor antagonists as potential drugs for the treatment of stress-related disorders. Curr. Pharm. Des. 2005, 11, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Nishiyama, M.; Tanaka, Y.; Noguchi, T.; Asaba, K.; Hossein, P.N.; Nishioka, T.; Makino, S. Urocortins and corticotropin releasing factor type 2 receptors in the hypothalamus and the cardiovascular system. Peptides 2004, 25, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Veloso, G.F.; Ohad, D.G.; Francis, A.J.; Vaughan, J.M.; Brownstein, D.G.; Culshaw, G.J.; Vale, W.W.; French, A.T.; Jamieson, P.M. Expression of urocortin peptides in canine myocardium and plasma. Vet. J. 2011, 188, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Sánchez, E.M.; Ruiz-Hurtado, G.; Smani, T.; Delgado, C.; Benitah, J.P.; Gómez, A.M.; Ordóñez, A. Cardioprotective action of urocortin in postconditioning involves recovery of intracellular calcium handling. Cell Calcium 2011, 50, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Brar, B.K.; Jonassen, A.K.; Egorina, E.M.; Chen, A.; Negro, A.; Perrin, M.H.; Mjøs, O.D.; Latchman, D.S.; Lee, K.F.; Vale, W. Urocortin-II and urocortin-III are cardioprotective against ischemia reperfusion injury: An essential endogenous cardioprotective role for corticotropin releasing factor receptor type 2 in the murine heart. Endocrinology 2004, 145, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Adão, R.; Santos-Ribeiro, D.; Rademaker, M.T.; Leite-Moreira, A.F.; Brás-Silva, C. Urocortin 2 in cardiovascular health and disease. Drug Discov. Today 2015, 20, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, K.M.; Kabir, A.M.; Bellahcene, M.; Davidson, S.; Cao, X.B.; McCormick, J.; Mesquita, R.A.; Carroll, C.J.; Chanalaris, A.; Townsend, P.A.; et al. Cardioprotection mediated by urocortin is dependent on PKCepsilon activation. FASEB J. 2005, 19, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Chanalaris, A.; Lawrence, K.M.; Stephanou, A.; Knight, R.D.; Hsu, S.Y.; Hsueh, A.J.; Latchman, D.S. Protective effects of the urocortin homologues stresscopin (SCP) and stresscopin-related peptide (SRP) against hypoxia/reoxygenation injury in rat neonatal cardiomyocytes. J. Mol. Cell. Cardiol. 2003, 35, 1295–1305. [Google Scholar] [CrossRef]
- Fekete, E.M.; Zorrilla, E.P. Physiology, pharmacology, and therapeutic relevance of urocortins in mammals: Ancient CRF paralogs. Front. Neuroendocrinol. 2007, 28, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Chong, A.C.N.; Merly, C.; Vogt, M.C.; Hill, A.S.; Jens, C.; Brüning, J.C.; Lori, M.; Zeltser, L.M. Central insulin signaling modulates hypothalamus-pituitary-adrenal axis responsiveness. Mol. Metab. 2015, 4, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.B.; Landgraf, R.; Keck, M.E. Vasopressin, major depression, and hypothalamic-pituitary-adrenocortical desensitization. Biol. Psychiatry 2000, 48, 330–333. [Google Scholar] [CrossRef]
- Dong, H.W.; Swanson, L.W. Projections from bed nuclei of the stria terminalis, antero medial area: Cerebral hemisphere integration of neuroendocrine, autonomic, and behavioral aspects of energy balance. J. Comp. Neurol. 2006, 494, 142–178. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.C.; Furay, A.R.; Evanson, N.K.; Ulrich-Lai, Y.M.; Nguyen, M.M.; Ostrander, M.M.; Herman, J.P. The role of the posterior medial bed nucleus of the stria terminalis in modulating hypothalamic-pituitary-adrenocortical axis responsiveness to acuteand chronic stress. Psychoneuroendocrinology 2008, 33, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.C.; Evanson, N.K.; Furay, A.R.; Ulrich-Lai, Y.M.; Ostrander, M.M.; Herman, J.P. The anteroventral bed n ucleus of the stria terminalis differentially regulates hypothalamic-pituitary adrenocortical axis responses to acute and chronic stress. Endocrinology 2008, 149, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Conradt, E.; Lester, B.M.; Appleton, A.A.; Armstrong, D.A.; Marsit, C.J. The roles of DNA methylation of NR3C1 and 11β-HSD2 and exposure to maternal mood disorder in utero on newborn neurobehavior. Epigenetics 2013, 8, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Meaney, M.J. Epigenetics and the biological definition of gene × environment interactions. Child Dev. 2010, 81, 41–79. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, J.C., III; Zhang, B.; Koren, J., III; Blair, L.; Dickey, C.A. The role of FKBP5 in mood disorders: Action of FKBP5 on steroid hormone receptors leads to questions about its evolutionary importance CNS. Neurol. Disord. Drug Targets 2013, 12, 1157–1162. [Google Scholar]
- Swaab, D.F.; Nijveldt, F.; Pool, C.W. Distribution of oxytocin and vasopressin in the rat supraoptic and paraventricular nucleus. J. Endocrinol. 1975, 67, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Leng, G.; Brown, C.H.; Russell, J.A. Physiological pathways regulating the activity of magnocellular neurosecretory cells. Prog. Neurobiol. 1999, 57, 625–655. [Google Scholar] [CrossRef]
- Gimpl, G.; Fahrenholz, F. The oxytocin receptor system: Structure, function, and regulation. Physiol. Rev. 2001, 81, 629–683. [Google Scholar] [CrossRef] [PubMed]
- Gutkowska, J.; Jankowski, M. Oxytocin revisited: Its role in cardiovascular regulation. J. Neuroendocrinol. 2012, 24, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Tom, N.; Assinder, S.J. Oxytocin in health and disease. Int. J. Biochem. Cell Biol. 2010, 42, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.L.; Landry, D.W.; Granton, J.T. Science Review: Vasopressin and the cardiovascular system part 2—Clinicalphysiology. Crit. Care 2004, 8, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treschan, T.A.; Peters, J. The vasopressin system: Physiology and clinical strategies. Anesthesiology 2006, 105, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Uvnas-Moberg, K.; Ahlenius, S.; Hillgaart, V.; Alster, P. High doses of oxytocin cause sedation and low doses cause an anxiolytic-like effect in male rats. Pharmacol. Biochem. Behav. 1994, 9, 101–106. [Google Scholar] [CrossRef]
- Windle, R.J.; Shanks, N.; Lightman, S.L.; Ingram, C.D. Central oxytocin administration reduces stress -induced corticosterone release and anxiety behavior in rats. Endocrinology 1997, 138, 2829–2834. [Google Scholar] [CrossRef] [PubMed]
- Bale, T.L.; Davis, A.M.; Auger, A.P.; Dorsa, D.M.; McCarthy, M.M. CNS region-specific oxytocin receptor expression: Importance in regulation of anxiety and sex behavior. J. Neurosci. 2001, 21, 2546–2552. [Google Scholar] [CrossRef] [PubMed]
- Erskine, M.S.; Barfield, R.J.; Goldman, B.D. Intraspecific fighting during late pregnancy and lactation in rats and effects of litter removal. Behav. Rev. 1978, 23, 206–213. [Google Scholar] [CrossRef]
- Liu, J.J.; Lou, F.; Lavebratt, C.; Forsell, Y. Impact of Childhood Adversity and Vasopressin receptor 1a Variation on Social Interaction in Adulthood: A Cross-Sectional Study. PLoS ONE 2015, 10, e0136436. [Google Scholar] [CrossRef] [PubMed]
- Wigger, A.; Sánchez, M.M.; Mathys, K.C.; Ebner, K.; Frank, E.; Liu, D.; Kresse, A.; Neumann, I.D.; Holsboer, F.; Plotsky, P.M.; et al. Alterations in central neuropeptide expression, release, and receptor binding in rats bred for high anxiety: Critical role of vasopressin. Neuropsychopharmacology 2004, 29, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Melander, O. Vasopressin, from Regulator toDisease Predictor for Diabetes and Cardiometabolic Risk. Ann. Nutr. Metab. 2016, 68 (Suppl. 2), 24–28. [Google Scholar] [CrossRef] [PubMed]
- Keck, M.E.; Wigger, A.; Welt, T.; Gesing, A.; Muller, M.B.; Reul, J.M.H.M.; Holsboer, F.; Landgraf, R.; Neumann, I.D. Pathological outcome of the combined dexamethasone/CRH test in hyperanxious rats: Involvement of endogenous vasopressin. Neuropsychopharmacology 2002, 26, 94–105. [Google Scholar] [CrossRef]
- Gutkowska, J.; Jankowski, M.; Antunes-Rodrigues, J. The role of oxytocin in cardiovascular regulation. Braz. J. Med. Biol. Res. 2014, 47, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Doris, P.A. Central cardiovascular regulation and the role of vasopressin: A review. Clin. Exp. Hypertens. Part A Theory Pract. 1984, 6, 2197–2217. [Google Scholar] [CrossRef]
- Altura, B.M.; Altura, B.T. Actions of vasopressin, oxytocin, and synthetic analogs on vascular smooth muscle. Fed. Proc. 1984, 43, 80–86. [Google Scholar] [PubMed]
- Fernandes, K.B.; Crippa, G.E.; Tavares, R.F.; Antunes-Rodrigues, J.; Correa, F.M. Mechanisms involved in the pressor response to noradrenaline injection into the cingulate cortex of unanesthetized rats. Neuropharmacology 2003, 44, 757–763. [Google Scholar] [CrossRef]
- Szczpanska-Sadaowska, E.; Cudnoch-Jedrzejewska, A.; Ufnal, M.; Zera, T. Brain and cardiovascular disease: Common neurogenic background of cardiovascular, metabolic and inflammatory diseases. J. Physiol. Pharmacol. 2010, 61, 509–521. [Google Scholar]
- Indrambarya, T.; John, H.; Wang, B.Y.; McConechy, M.; Walley, K.R. Low-dose vasopressin infusion results in increased mortality and cardiac dysfunction following ischemia-reperfusion injury in mice. Crit. Care 2009, 13, R98. [Google Scholar] [CrossRef] [PubMed]
- Crestani, C.C.; Alves, F.H.; Resstel, L.B.; Correa, F.M. Cardiovascular effects of noradrenaline microinjection in the bed nucleus of the stria terminalis of the rat brain. J. Neurosci. Res. 2007, 85, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Hatam, M.; Kharazmi, F.; Nasimi, A. Vasopressin and sympathetic systems mediate the cardiovascular effects of the GABAergic system in the bed nucleus of the stria terminalis. Neurosci. Res. 2009, 65, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.W.; Swanson, L.W. Organization of axonal projections from the anterolateral area of the bed nuclei of the stria terminalis. J. Comp. Neurol. 2004, 468, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J. The natriuretic peptides and fat metabolism. N. Engl. J. Med. 2012, 367, 377–378. [Google Scholar] [CrossRef] [PubMed]
- De Bold, A.J.; Borenstein, H.B.; Veress, A.T.; Sonnenberg, H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981, 28, 89–94. [Google Scholar] [CrossRef]
- Hodes, A.; Lichtstein, D. Natriuretic hormones in brain function. Front. Endocrinol. 2014, 28, 201. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Malvin, R.L.; Claybaugh, J.R.; Huang, B.S. Atrial natriuretic factor inhibits vasopressin secretion in conscious sheep. Proc. Soc. Exp. Biol. Med. 1987, 185, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Al-Barazanji, K.A.; Balment, R.J. The renal and vascular effects of central angiotensin II and atrial natriuretic factor in the anaesthetized rat. J. Physiol. 1990, 423, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Nakao, K.; Itoh, H.; Shirakami, G.; Kangawa, K.; Minamino, N.; Matsuo, H.; Imura, H. Intracerebroventricular injection of brain natriuretic peptide inhibits vaso-pressin secretion in conscious rats. Neurosci. Lett. 1988, 95, 223–228. [Google Scholar] [CrossRef]
- Samson, W.K.; Aguila, M.C.; Martinovic, J.; Antunes-Rodrigues, J.; Norris, M. Hypothalamic action of atrial natriuretic factor to inhibit vasopressin secretion. Peptides 1987, 8, 449–454. [Google Scholar] [CrossRef]
- Yasue, H.; Yoshimura, M.; Sumida, H.; Kikuta, K.; Kugiyama, K.; Jougasaki, M.; Ogawa, H.; Okumura, K.; Mukoyama, M.; Nakao, K. Localization and mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic peptide in normal subjects and patients with heart failure. Circulation 1994, 90, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, M.; Yasue, H.; Ogawa, H. Pathophysiological significance and clinical application of ANP and BNP in patients with heart failure. Can. J. Physiol. Pharmacol. 2001, 79, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R.; Gardner, D.G.; Samson, W.K. Natriuretic peptides. N. Engl. J. Med. 1998, 339, 321–328. [Google Scholar] [PubMed]
- Ogawa, Y.; Itoh, H.; Yoshitake, Y.; Inoue, M.; Yoshimasa, T.; Serikawa, T.; Nakao, K. Molecular cloning and chromosomal assignment of the mouse C-type natriuretic peptide (CNP) gene (Nppc): Comparison with the human CNP gene (NPPC). Genomics 1994, 24, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.G.; Chen, S.; Glenn, D.J.; Grigsby, C.L. Molecular biology of the natriuretic peptide system: Implications for physiology and hypertension. Hypertension 2007, 49, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Kasama, S.; Furuya, M.; Toyama, T.; Ichikawa, S.; Kurabayashi, M. Effect of atrial natriuretic peptide on left ventricular remodelling in patients with acute myocardial infarction. Eur. Heart J. 2008, 29, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Delgado Pastor, V.; Carbó, R.; Guarner, V. Participation of glucose transporters on atrial natriuretic peptide-induced glucose uptake by adult and neonatal cardiomyocytes under oxygenation and hypoxia. Eur. J. Pharmacol. 2007, 568, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Strohle, A.; Jahn, H.; Montkowski, A.; Liebsch, G.; Boll, E.; Landgraf, R.; Holsboer, F.; Wiedemann, K. Central and peripheral administration of atriopeptin is anxiolytic in rats. Neuroendocrinology 1997, 65, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Arlt, J.; Jahn, H.; Kellner, M.; Strohle, A.; Yassouridis, A.; Wiedemann, K. Modulation of sympathetic activity by corticotropin-releasing hormone and atrial natriuretic peptide. Neuropeptides 2003, 37, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Kellner, M.; Knaudt, K.; Jahn, H.; Holsboer, F.; Wiedemann, K. Atrial natriuretic hormone in lactate-induced panic attacks: Mode of release and endocrine and pathophysiological consequences. J. Psychiatr. Res. 1998, 32, 37–48. [Google Scholar] [CrossRef]
- Levinson, D.F.; Zubenko, G.S.; Crowe, R.R.; Depaulo, R.J.; Scheftner, W.S.; Weissman, M.M.; Holmans, P.; Zubenko, W.N.; Boutelle, S.; Murphy-Eberenz, K.; et al. Genetics of recurrent early-onset depression (GenRED): Design and preliminary clinical characteristics of are pository sample for genetic link-age studies. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 119B, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Herrmann-Lingen, C.; Binder, L.; Klinge, M.; Sander, J.; Schenker, W.; Beyermann, B.; Von Lewinski, D.; Pieske, B. High plasma levels of N-terminal pro-atrial natriuretic peptide associ-ated with low anxiety in severe heart failure. Psychosom. Med. 2003, 65, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Biro, E.; Toth, G.; Telegdy, G. Effect of receptor blockers on brain natriuretic peptide and C-type natriuretic peptide caused anxiolytic state in rats. Neuropeptides 1996, 30, 59–65. [Google Scholar] [CrossRef]
- Charles, C.J.; Espiner, E.A.; Richards, A.M.; Donald, R.A. Central C-type natriuretic peptide augments the hormone response to hemorrhage in conscious sheep. Peptides 1995, 16, 129–132. [Google Scholar] [CrossRef]
- Montkowski, A.; Jahn, H.; Strohle, A.; Poettig, M.; Holsboer, F.; Wiedemann, K. C-type natriuretic peptide exerts effects opposing those of atrial natriuretic peptide on anxiety-related behaviour in rats. Brain Res. 1998, 792, 358–360. [Google Scholar] [CrossRef]
- Jahn, H.; Montkowski, A.; Knaudt, K.; Strohle, A.; Kiefer, F.; Schick, M.; Wiedemann, K. Alpha-helical-corticotropin-releasing hormone reverses anxiogenic effects of C-type natriuretic peptide in rats. Brain Res. 2001, 893, 21–28. [Google Scholar] [CrossRef]
- Kellner, M.; Yassouridis, A.; Hua, Y.; Wendrich, M.; Jahn, H.; Wiedemann, K. Intra-venous C-type natriuretic peptide augments behavioral and endocrine effects of cholecystokinin tetrapeptide in healthy men. J. Psychiatr. Res. 2002, 36, 1–6. [Google Scholar] [CrossRef]
- Kellner, M.; Diehl, I.; Knaudt, K.; Schule, C.; Jahn, H.; Wiedemann, K. C-type natriuretic peptide exerts stimulatory effects on the corticotropin-releasing hormone-induced secretion of hormones in normal man. Eur. J. Endocrinol. 1997, 136, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Kawata, M.; Nakao, K.; Morii, N.; Kiso, Y.; Yamashita, H.; Imura, H.; Sano, Y. Atrial natriuretic polypeptide: Topographical distribution in the rat brain by radioimmunoassay and immunohistochemistry. Neuroscience 1985, 16, 521–546. [Google Scholar] [CrossRef]
- Li, N.; Zheng, D.; Sun, L.; Shi, H.; Zhu, X.; Xu, G.; Wang, Q.; Zhu, C.; Shao, G. Hypermethylation of brain natriuretic peptide gene is associated with the risk of rheumatic heart disease. Biosci. Rep. 2017, 37, BSR20160408. [Google Scholar] [CrossRef] [PubMed]
- Duygu, B.; Poels, E.M.; da Costa Martins, P.A. Genetics and epigenetics of arrhythmia and heart failure. Front. Genet. 2013, 4, 219. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, A.; Sessa, M.; Scavone, C.; De Angelis, A.; Vitale, C.; Berrino, L.; Rossi, F.; Rosano, G.; Capuano, A. New and old roles of the peripheral and brain renin-angiotensin-aldosterone system (RAAS): Focus on cardiovascular and neurological diseases. Int. J. Cardiol. 2017, 227, 734–742. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, A.D.; Wang, L.; Pitra, S.; Hiller, H.; Smith, J.A.; Tan, Y.; Nguyen, D.; Cahill, K.M.; Sumners, C.; Stern, J.E.; et al. A Unique “Angiotensin-Sensitive” Neuronal Population Coordinates Neuroendocrine, Cardiovascular, and Behavioral Responses to Stress. J. Neurosci. 2017, 37, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Mansur, S.J.; Hage, F.G.; Oparil, S. Have the renin-angiotensin-aldosterone system perturbations in cardiovascular disease been exhausted? Curr. Cardiol. Rep. 2010, 12, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Atlas, S.A. The renin-angiotensin aldosterone system: Pathophysiological role and pharmacologic inhibition. J. Manag. Care Pharm. 2007, 13, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Sokol, S.I.; Portnay, E.L.; Curtis, J.P.; Nelson, M.A.; Hebert, P.R.; Setaro, J.F.; Foody, J.M. Modulation of the renin-angiotensin-aldosterone system for the secondary prevention of stroke. Neurology 2004, 63, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Bondy, B.; Baghai, T.C.; Zill, P.; Bottlender, R.; Jaeger, M.; Minov, C.; Schule, C.; Zwanzger, P.; Rupprecht, R.; Engel, R.R. Combined action of the ACE D- and the G-protein beta3 T-allele in major depression: A possible link to cardiovascular disease? Mol. Psychiatry 2002, 7, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Santos, A.F.; Kangussu, L.M.; Moreira, F.A.; Santos, R.A.; Aguiar, D.C.; Campagnole-Santos, M.J. Anxiolytic- and antidepressant-like effects of angiotensin-(1–7) in hypertensive transgenic (mRen2)27 rats. Clin. Sci. 2016, 30, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Britsch, S. The neuregulin-I/ErbB signaling system in development and disease. Adv. Anat. Embryol. Cell Biol. 2007, 190, 1–65. [Google Scholar] [PubMed]
- Russell, K.S.; Stern, D.F.; Polverini, P.J.; Bender, J.R. Neuregulin activation of ErbB receptors in vascular endothelium leads to angiogenesis. Am. J. Physiol. 1999, 277, H2205–H2211. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.B.; Taylor, A.R.; Koenig, J.I. The interaction of disrupted type II neuregulin 1 and chronic adolescent stress on adult anxiety- and fear-related behaviors. Neuroscience 2013, 249, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.B.; Taylor, A.R.; Markham, J.A.; Geurts, A.M.; Kanaskie, B.Z.; Koenig, J.I. Disruption of the neuregulin 1 gene in the rat alters HPA axis activity and behavioral responses to environmental stimuli. Physiol. Behav. 2011, 104, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Zhang, J.; Wu, J.L.; Zou, W.J.; Liang, Z.P.; Bi, L.L.; Liu, J.H.; Kong, Y.; Huang, C.Q.; Li, X.W.; et al. Neuregulin 1-ErbB4 signaling in the bed nucleus of the stria terminalis regulates anxiety-like behavior. Neuroscience 2016, 329, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.L.; Sun, X.D.; Zhang, J.; Lu, Y.S.; Chen, Y.H.; Wang, J.; Geng, F.; Liu, F.; Zhang, M.; Liu, J.H.; et al. Amygdala NRG1-ErbB4 is critical for the modulation of anxiety-like behaviors. Neuropsychopharmacology 2015, 40, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Hedhli, N.; Huang, Q.; Kalinowski, A.; Palmeri, M.; Hu, X.; Russell, R.R.; Russell, K.S. Endothelium-derived neuregulin protects the heart against ischemic injury. Circulation 2011, 123, 2254–2262. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.R.; Horrevoets, A.J.G.; van Royen, N. Cellular and Pharmacological Targets to Induce Coronary Arteriogenesis. Curr. Cardiol. Rev. 2014, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Gui, C.; Zhu, L.; Hu, M.; Lei, L.; Long, Q. Neuregulin-1/ErbB signaling is impaired in the rat model of diabetic cardiomyopathy. Cardiovasc. Pathol. 2012, 21, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Lin, M.; Xu, Y.; Shao, J.; Li, X.; Zhang, H.; Yang, S. Circulating neuregulin 4 levels are inversely associated with subclinical cardiovascular disease in obese adults. Sci. Rep. 2016, 6, 36710. [Google Scholar] [CrossRef] [PubMed]
- Dang, R.; Cai, H.; Zhang, L.; Liang, D.; Lv, C.; Guo, Y.; Yang, R.; Zhu, Y.; Jiang, P. Dysregulation of Neuregulin-1/ErbB signaling in the prefrontal cortex and hippocampus of rats exposed to chronic unpredictable mild stress. Physiol. Behav. 2016, 154, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Dang, R.; Guo, Y.; Zhang, L.; Chen, L.; Yang, R.; Jiang, P. Chronic stress and excessive glucocorticoid exposure both lead to altered Neuregulin-1/ErbB signaling in rat myocardium. Steroids 2016, 112, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Dang, R.; Guo, Y.; Zhu, Y.; Yang, R.; Cai, H.; Jiang, P. Chronic administration of calcitriol enhanced neuregulin-1/ErbB signaling in rat myocardium. Pharmazie 2016, 71, 192–195. [Google Scholar] [PubMed]
- Nawa, H.; Sotoyama, H.; Iwakura, Y.; Takei, N.; Namba, H. Neuropathologic implication of peripheral neuregulin-1 and EGF signals in dopaminergic dysfunction and behavioral deficits relevant to schizophrenia: Their target cells and time window. Biomed. Res. Int. 2014, 697935. [Google Scholar] [CrossRef] [PubMed]
- Paterson, C.; Law, A.J. Transient overexposure of neuregulin 3 during early postnatal development impactsselective behaviors in adulthood. PLoS ONE 2014, 9, e104172. [Google Scholar] [CrossRef] [PubMed]
- Mutafova-Yambolieva, V.N.; Durnin, L. The purinergic neurotransmitter revisited: A single substance or multiple players? Pharmacol. Ther. 2014, 144, 162–191. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic signalling. Br. J. Pharmacol. 2006, 147, S172–S181. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, D.; Shan, J.; Ayers-Ringler, A.; Oliveros, J.; Benitez, M.; Prieto, R.; McCullumsmith, D.; Choi, S. Purinergic Signaling and Energy Homeostasis in Psychiatric Disorders. Curr. Mol. Med. 2015, 15, 275–295. [Google Scholar] [CrossRef] [PubMed]
- Furuyashiki, T. Roles of dopamine and inflammation-related molecules in behavioral alterations caused by repeated stress. J. Pharmacol. Sci. 2012, 120, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.V.; Downey, J.M. Status of P2Y12 treatment must be considered in evaluation of myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2015, 106, 8. [Google Scholar] [CrossRef] [PubMed]
- Tomai, F.; Crea, F.; Chiariello, L.; Gioffrè, P.A. Ischemic preconditioning in humans: Models, mediators, and clinical relevance. Circulation 1999, 100, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Tomai, F.; Crea, F.; Gaspardone, A.; Versaci, F.; De Paulis, R.; Polisca, P.; Chiariello, L.; Gioffrè, P.A. Effects of A1 adenosine receptor blockade by bamiphylline on ischemic preconditioning during coronary angioplasty. Eur. Heart J. 1996, 17, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Claeys, M.J.; Vrints, C.J.; Bosmans, J.M.; Conraads, V.M.; Snoeck, J.P. Aminophylline inhibits adaptation to ischemia during angioplasty. Role of adenosine in ischemic preconditioning. Eur. Heart J. 1996, 17, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Leesar, M.A.; Stoddard, M.; Ahmed, M.; Broadbent, J.; Bolli, R. Preconditioning of human myocardium with adenosine during coronary angioplasty. Circulation 1997, 95, 2500–2507. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Cichoń, N.; Lach, D.; Dziedzic, A.; Bijak, M.; Saluk, J. The inflammatory processes in atherogenesis. Polski Merkuriusz Lekarski 2017, 42, 125–128. [Google Scholar] [PubMed]
- Salim, S.; Chugh, G.; Asghar, M. Inflammation in anxiety. Adv. Protein Chem. Struct. Biol. 2012, 88, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Guarner, V.; Rubio-Ruiz, M.E. Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip. Top. Gerontol. 2015, 40, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Inflammation in ischemic stroke subtypes. Curr. Pharm. Des. 2012, 18, 4289–4310. [Google Scholar] [CrossRef] [PubMed]
- Pitsavos, C.; Panagiotakos, D.B.; Papageorgiou, C.; Tsetsekou, E.; Soldatos, C.; Stefanadis, C. Anxiety in relation to inflammation and coagulation markers, among healthy adults: The ATTICA study. Atherosclerosis 2006, 185, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Thayer, J.F.; Friedman, B.H.; Borkovec, T.D. Autonomic characteristics of generalized anxiety disorder and worry. Biol. Psychiatry 1996, 39, 255–266. [Google Scholar] [CrossRef]
- Brosschot, J.; Gerin, W.; Thayer, J. The perseverative cognition hypothesis: A review of worry, prolonged stress-related physiological activation, and health. J. Psychosomat. Res. 2006, 60, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.T.; Doberenz, S.; Dietel, A.; Conrad, A.; Mueller, A.; Wollburg, E.; Meuret, A.E.; Barr Taylor, C.; Kim, S. Sympathetic activation in broadly defined generalized anxiety disorder. J. Psychiatr. Res. 2008, 42, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, S.V.; Petrov, S.V.; Volkov, A.I.; Chekhonin, V.P. The translocation of macromolecules via the hematoencephalic barrier. Vestnik Rossiiskoi Akademii Meditsinskikh Nauk 2007, 6, 37–49. [Google Scholar]
- Ito, K. Impact of post-translational modifications of proteins on the inflammatory process. Biochem. Soc. Trans. 2007, 35, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Horsburgh, S.; Robson-Ansley, P.; Adams, R.; Smith, C. Exercise and inflammation-related epigenetic modifications: Focus on DNA methylation. Exerc. Immunol. Rev. 2015, 21, 26–41. [Google Scholar] [PubMed]
- Bollaerts, I.; Van Houcke, J.; Andries, L.; De Groef, L.; Moons, L. Neuroinflammation as Fuel for Axonal Regeneration in the Injured Vertebrate Central Nervous System. Mediators Inflamm. 2017, 2017, 9478542. [Google Scholar] [CrossRef] [PubMed]
- El Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.C.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009, 58, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Balcerczyk, A.; Tothill, R.W.; Haviv, I.; Kaspi, A.; Lunke, S.; Ziemann, M.; Karagiannis, T.; Tonna, S.; Kowalczyk, A.; et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 2011, 21, 1601–1615. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, F.; Mizutani, S.; Ogawa, Y.; Sawada, N. Glucose-independent persistence of PAI-1 gene expression and H3K4 tri-methylation in type 1 diabetic mouse endothelium: Implication in metabolic memory. Biochem. Biophys. Res. Commun. 2013, 433, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Vecellio, M.; Spallotta, F.; Nanni, S.; Colussi, C.; Cencioni, C.; Derlet, A.; Bassetti, B.; Tilenni, M.; Carena, M.C.; Farsetti, A.; et al. The histone acetylase activator pentadecylidenemalonate 1b rescues proliferation and differentiation in human cardiac mesenchymal cells of type 2 diabetic patients. Diabetes 2014, 63, 2132–2147. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Mattson, M.P. Activity-dependent, stress-responsive BDNF signaling and the quest for optimal brain health and resilience throughout the lifespan. Neuroscience 2013, 239, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Henaomejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Bäckhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenment, L.T.; Knight, R.; et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef] [PubMed]
- De Palma, G.; Blennerhassett, P.; Lu, J.; Deng, Y.; Park, A.J.; Green, W.; Denou, E.; Silva, M.A.; Santacruz, A.; Sanz, Y.; et al. Microbiota and host determinants of behavioral phenotype in maternally separated mice. Nat. Commun. 2015, 6, 7735. [Google Scholar] [CrossRef] [PubMed]
- Hoban, A.E.; Stilling, R.M.; Moloney, G.; Shanahan, F.; Dinan, T.G.; Clarke, G.; Cryan, J.F. The microbiome regulates amygdala-dependent fear recall. Mol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]
- Tillisch, K.; Labus, J.; Kilpatrick, L.; Jiang, Z.; Stains, J.; Ebrat, B.; Guyonnet, D.; Legrain–Raspaud, S.; Trotin, B.; Naliboff, B.; et al. Consumption of Fermented Milk Product with Probiotic Modulates Brain Activity. Gastroenterology 2013, 144, 1394–1401. [Google Scholar] [CrossRef] [PubMed]
- Dinan, T.G.; Stanton, C.; Cryan, J.F. Psychobiotics: A novel class of psychotropic. Biol. Psychiatry 2013, 74, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Declerck, K.; Szarc vel Szic, K.; Palagani, A.; Heyninck, K.; Haegeman, G.; Morand, C.; Milenkovic, D.; Vanden Berghe, W. Epigenetic control of cardiovascular health by nutritional polyphenols involves multiple chromatin-modifying writer-reader-eraser proteins. Curr. Top. Med. Chem. 2016, 16, 788–806. [Google Scholar] [CrossRef] [PubMed]
- Peredo-Escárcega, A.E.; Guarner-Lans, V.; Pérez-Torres, I.; Ortega-Ocampo, S.; Carreón-Torres, E.; Castrejón-Tellez, V.; Díaz-Díaz, E.; Rubio-Ruiz, M.E. The combination of resveratrol and quercetin attenuates Metabolic Syndrome in rats by modifying the serum fatty acid composition and by up-regulating SIRT 1 and SIRT 2 expression in white adipose tissue. Evid. Based Complement. Altern. Med. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Singh-Taylor, A.; Molet, J.; Jiang, S.; Korosi, A.; Bolton, J.L.; Noam, Y.; Simeone, K.; Cope, J.; Chen, Y.; Mortazavi, A.; et al. NRSF-dependent epigenetic mechanisms contribute to programming of stress-sensitive neurons by neonatal experience, promoting resilience. Mol. Psychiatry 2018, 23, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Seth, K.A.; Majzoub, J.A. Repressor element silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) can act as an enhancer as well as a repressor of corticotropin-releasing hormone gene transcription. J. Biol. Chem. 2001, 276, 13917–13923. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- McGowan, P.O.; Meaney, M.J.; Szyf, M. Diet and the epigenetic re programming of phenotypic differences in behavior. Brain Res. 2008, 1237, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Takigawa-Imamura, H.; Sekine, T.; Murata, M.; Takayama, K.; Nakazawa, K.; Nakagawa, J. Stimulation of glucose uptake in muscle cells by prolonged treatment with scriptide, a histone deacetylase inhibitor. Biosci. Biotechnol. Biochem. 2003, 67, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Galmozzi, A.; Mitro, N.; Ferrari, A.; Gers, E.; Gilardi, F.; Godio, C.; Cermenati, G.; Gualerzi, A.; Donetti, E.; Rotili, D.; et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 2013, 62, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Crosson, C.E.; Mani, S.K.; Husain, S.; Alsarraf, O.; Menick, D.R. Inhibition of histone deacetylase protects the retina from ischemic injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3639–3645. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, O.; Flosseau, K.; Ma, F.X.; Blondeau, B.; Mai, A.; Bassel-Duby, R.; Ravassard, P.; Olson, E.N.; Haumaitre, C.; Scharfmann, R. Specific control of pancreatic endocrine beta- and delta-cell mass by class IIa histone deacetylases HDAC4, HDAC5, and HDAC9. Diabetes 2011, 60, 2861–2871. [Google Scholar] [CrossRef] [PubMed]
- Cencioni, C.; Spallotta, F.; Martelli, F.; Valente, S.; Mai, A.; Zeiher, A.M.; Gaetano, C. Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int. J. Mol. Sci. 2013, 14, 17643–17663. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.C.; Blaabjerg, L.; Storling, J.; Ronn, S.G.; Mascagni, P.; Dinarello, C.A.; Mandrup-Poulsen, T. The oral histone deacetylase inhibitor ITF2357 reduces cytokines and protects islet beta cells in vivo and in vitro. Mol. Med. 2011, 17, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Tedong, L.; Madiraju, P.; Martineau, L.C.; Vallerand, D.; Arnason, J.T.; Desire, D.D.; Lavoie, L.; Kamtchouing, P.; Haddad, P.S. Hydro-ethanolic extract of cashew tree (Anacardium occidentale) nut and its principal compound, anacardic acid, stimulate glucose uptake in C2C12 muscle cells. Mol. Nutr. Food Res. 2010, 54, 1753–1762. [Google Scholar] [CrossRef] [PubMed]
- Halili, M.A.; Andrews, M.R.; Labzin, L.I.; Schroder, K.; Matthias, G.; Cao, C.; Lovelace, E.; Reid, R.C.; Le, G.T.; Hume, D.A.; et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J. Leukoc. Biol. 2010, 87, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Luna Ortiz, P.; Guarner, V.; Farías, J.M.; Hernández-Pacheco, G.; Martínez, M. Importance of metabolic memory in the development of vascular complications in diabetic patients. J. Cardiothorac. Vasc. Anesth. 2016, 30, 1369–1378. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M.; McGowan, P.; Meaney, M.J. The social environment and the epigenome. Environ. Mol. Mutagen. 2008, 49, 46–60. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Neuro-and Cardio-Active Substance | Early Programming of Psychiatric Disorders | Early Programming of Cardiometabolic Diseases |
|---|---|---|
| Cortisol | Changes in concentration during early stages determine the risk to suffer stress or have an adequate level of resilience [37]. | High fasting levels during early development determine low birthweight which is associated with CMD [27,38]. |
| Oxytocin | Elevated estrogen levels during pregnancy determine oxytocin receptors in the limbic system which determine the risk of psychiatric disorders [39]. | Oxytocin levels during early life determine the risk of CMD in the adult [40]. |
| Vasopressin | Expression of vasopressin receptors is determined by stress in the early postnatal stages and induces a behavior similar to depression [41]. | Maternal water restriction induces risk of hypertension in the offspring when they reach adulthood, probably by alterations in vasopressin receptors [42]. |
| Natriuretic peptides | There are epigenetic modifications in the pattern of secretion of these peptides and of other proteins that determine the force of ventricular contraction throughout life [43]. | |
| Renin-angiotensin system | The improper activation of the renin-angiotensin-aldosterone system (RAAS) from early stages is implied in the development of hypertension linked to the fetal stage [44]. | |
| Neuregulins | Alterations of neuregulin 1 during neural development modify behavioral traits. This contributes to a hyperdopaminergic trait and to the pathogenesis of schizophrenia [45]. | Neuregulin 1 is essential for normal heart development, promoting survival of embryonary ventricular myocytes [46]. |
| Purinergic mediators | Adenine nucleotide signaling has an important role in progenitor cell proliferation, cell migration, interaction and differentiation of neurons and glia, and in the formation of synaptic nets during embryogenesis and may alter adult nervous system functioning in the adult [47]. | There is abnormal cardiac function in adult offspring of pregnant mice treated with adenosine antagonists [48,49]. |
| Inflammatory mediators | Premature infections cause alterations in the balance of neurotransmitters such as serotonin and may alter in a premature way the development of the fetal brain, having consequences in the adult [50,51]. | Cardiometabolic programming might be mediated by inflammatory mediators present during early development [50,51]. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapata-Martín del Campo, C.M.; Martínez-Rosas, M.; Guarner-Lans, V. Epigenetic Programming of Synthesis, Release, and/or Receptor Expression of Common Mediators Participating in the Risk/Resilience for Comorbid Stress-Related Disorders and Coronary Artery Disease. Int. J. Mol. Sci. 2018, 19, 1224. https://doi.org/10.3390/ijms19041224
Zapata-Martín del Campo CM, Martínez-Rosas M, Guarner-Lans V. Epigenetic Programming of Synthesis, Release, and/or Receptor Expression of Common Mediators Participating in the Risk/Resilience for Comorbid Stress-Related Disorders and Coronary Artery Disease. International Journal of Molecular Sciences. 2018; 19(4):1224. https://doi.org/10.3390/ijms19041224
Chicago/Turabian StyleZapata-Martín del Campo, Carlos Manuel, Martín Martínez-Rosas, and Verónica Guarner-Lans. 2018. "Epigenetic Programming of Synthesis, Release, and/or Receptor Expression of Common Mediators Participating in the Risk/Resilience for Comorbid Stress-Related Disorders and Coronary Artery Disease" International Journal of Molecular Sciences 19, no. 4: 1224. https://doi.org/10.3390/ijms19041224
APA StyleZapata-Martín del Campo, C. M., Martínez-Rosas, M., & Guarner-Lans, V. (2018). Epigenetic Programming of Synthesis, Release, and/or Receptor Expression of Common Mediators Participating in the Risk/Resilience for Comorbid Stress-Related Disorders and Coronary Artery Disease. International Journal of Molecular Sciences, 19(4), 1224. https://doi.org/10.3390/ijms19041224