Transcriptomic and GC-MS Metabolomic Analyses Reveal the Sink Strength Changes during Petunia Anther Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
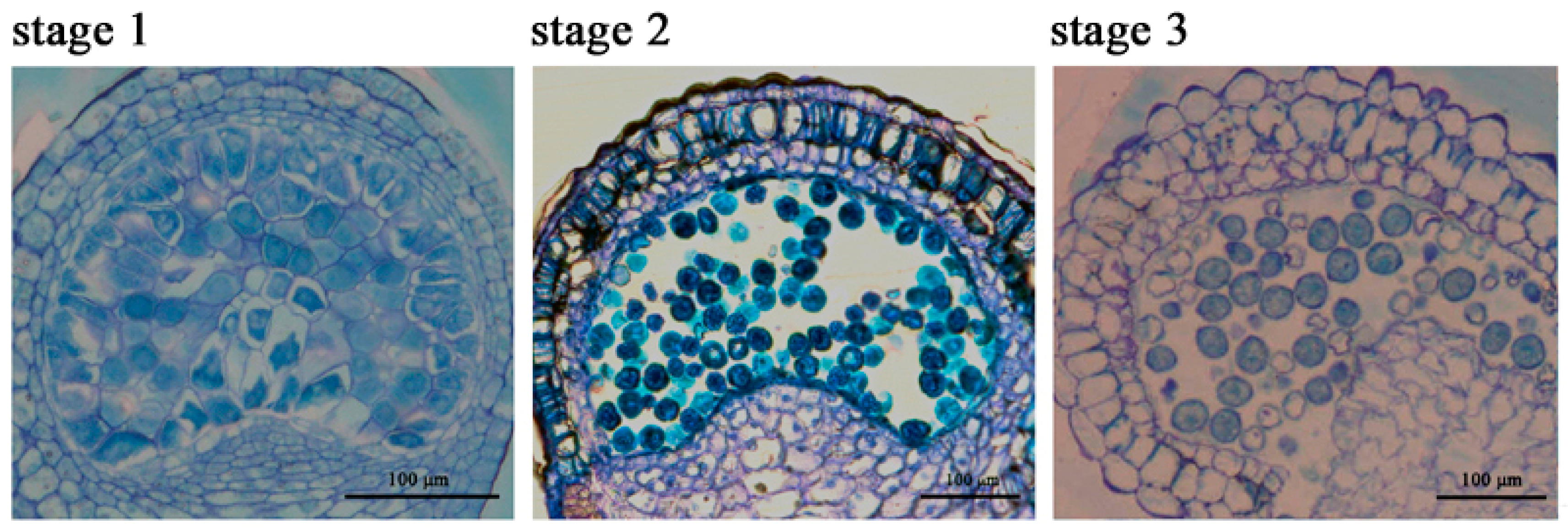
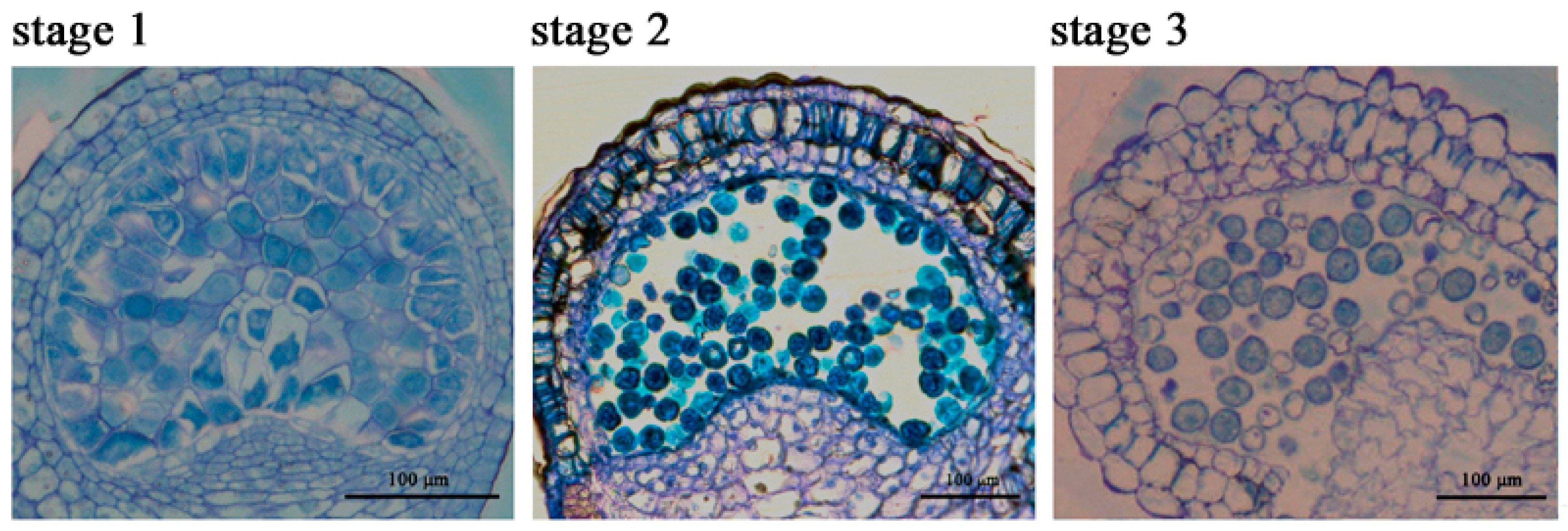
2.1. The Chlorophyll Content Is Reduced during the Anther Maturation Process
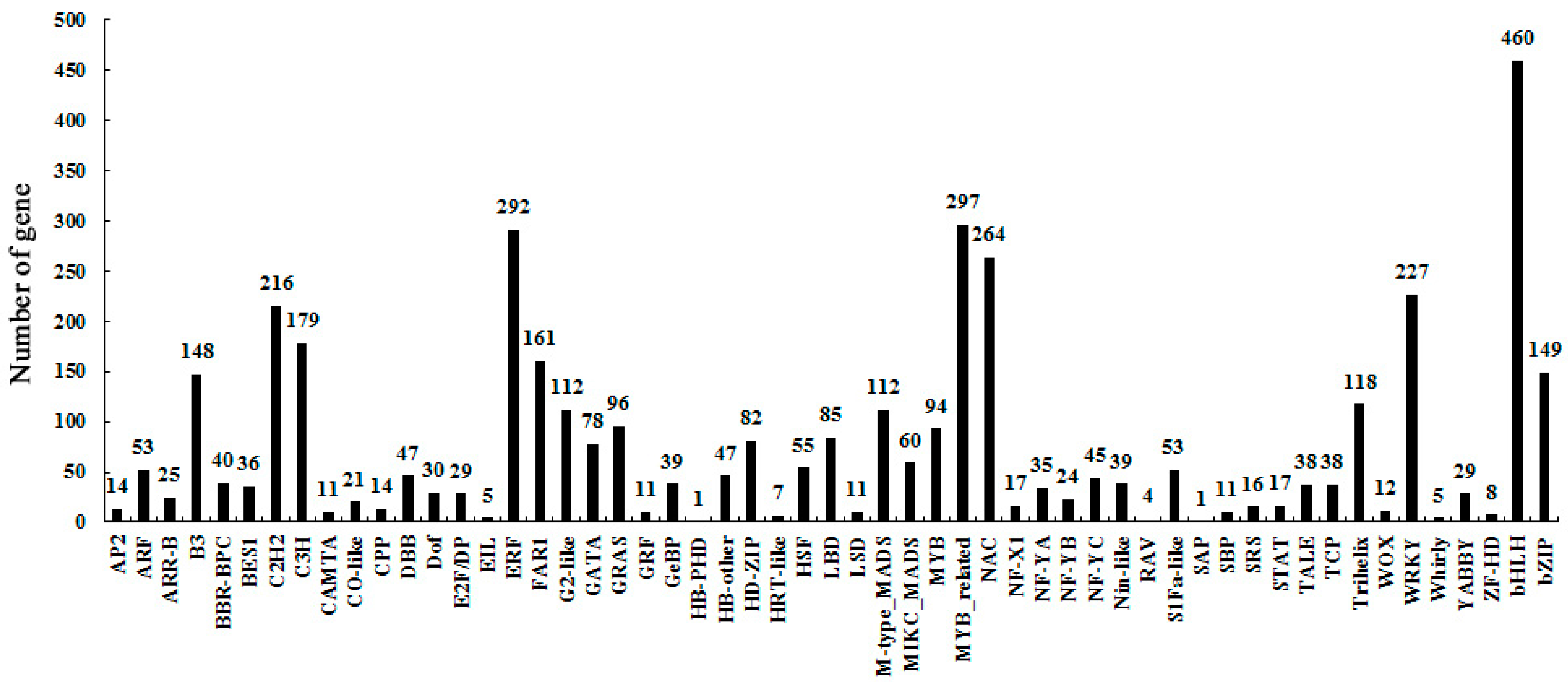
2.2. Transcriptome Sequencing
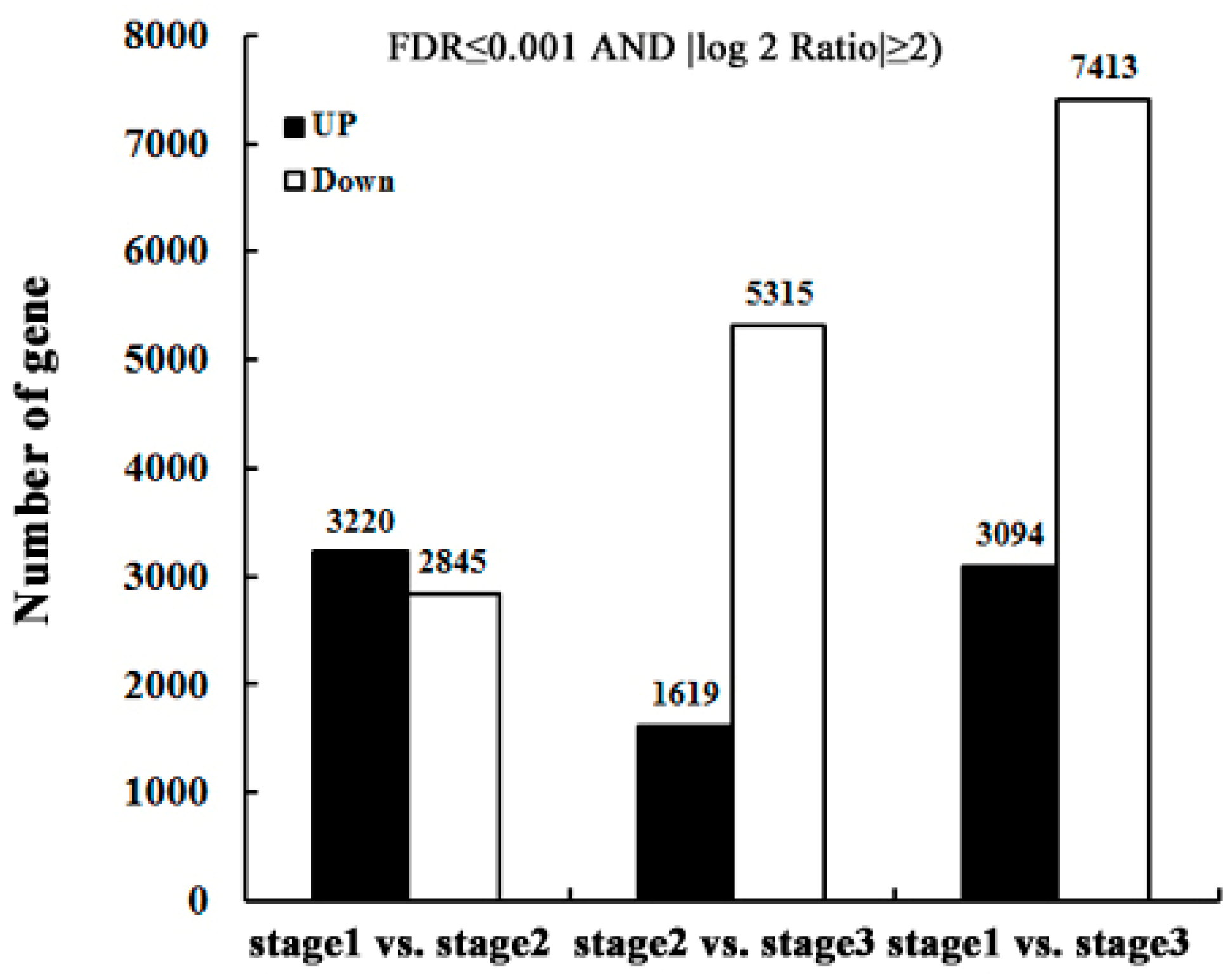
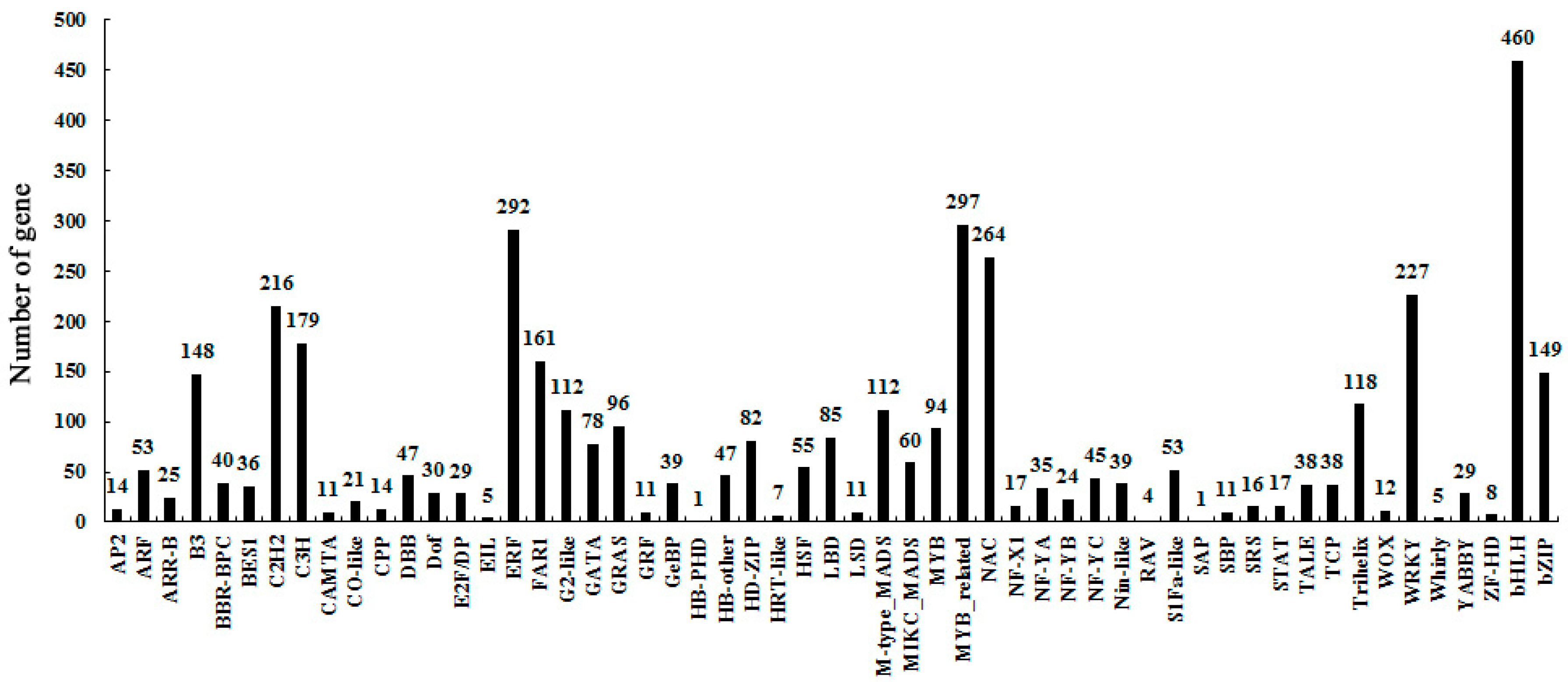
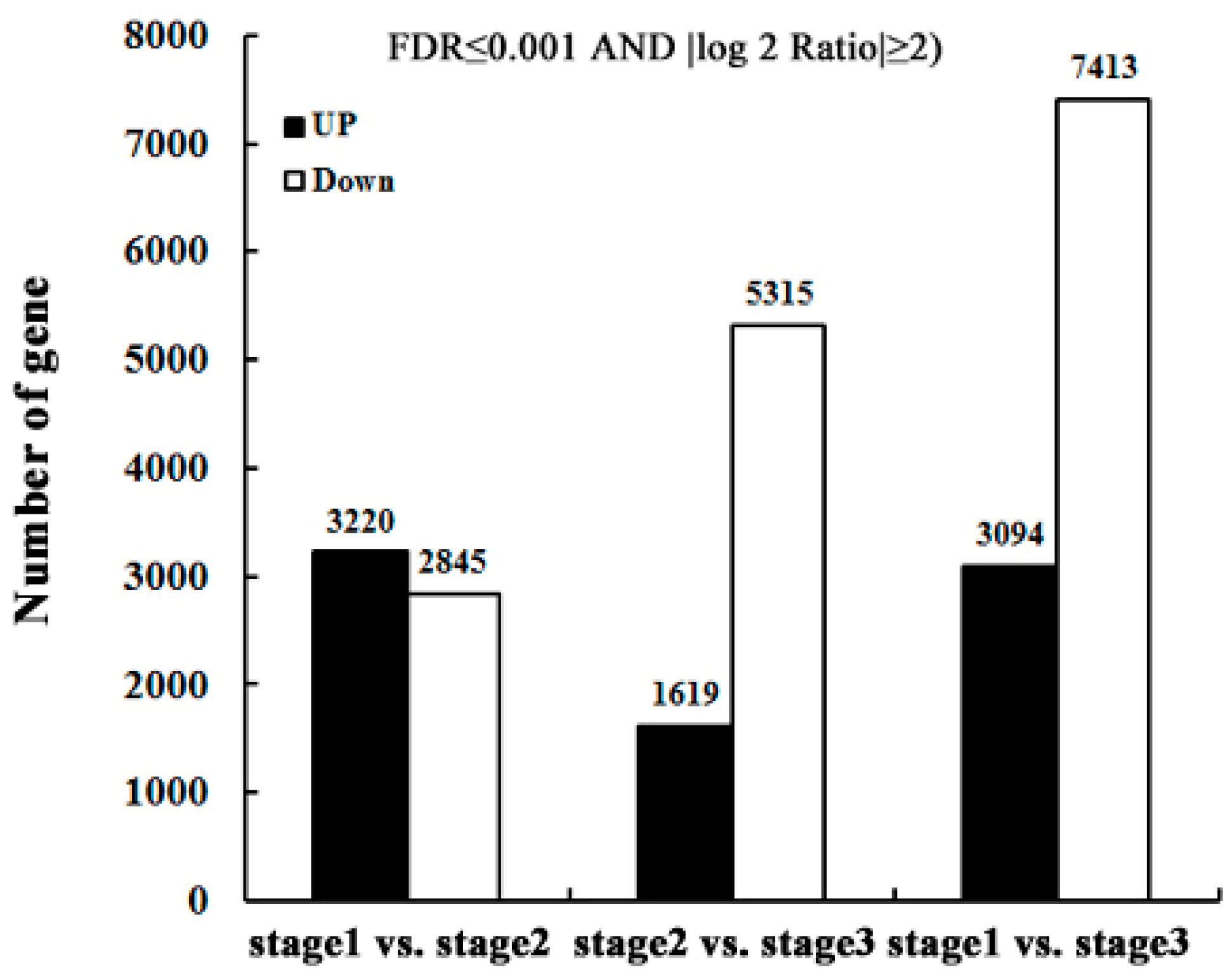
2.3. Identify Differentially-Expressed Unigenes between Anther Transcriptomes
2.4. KEGG Pathways Enrichment
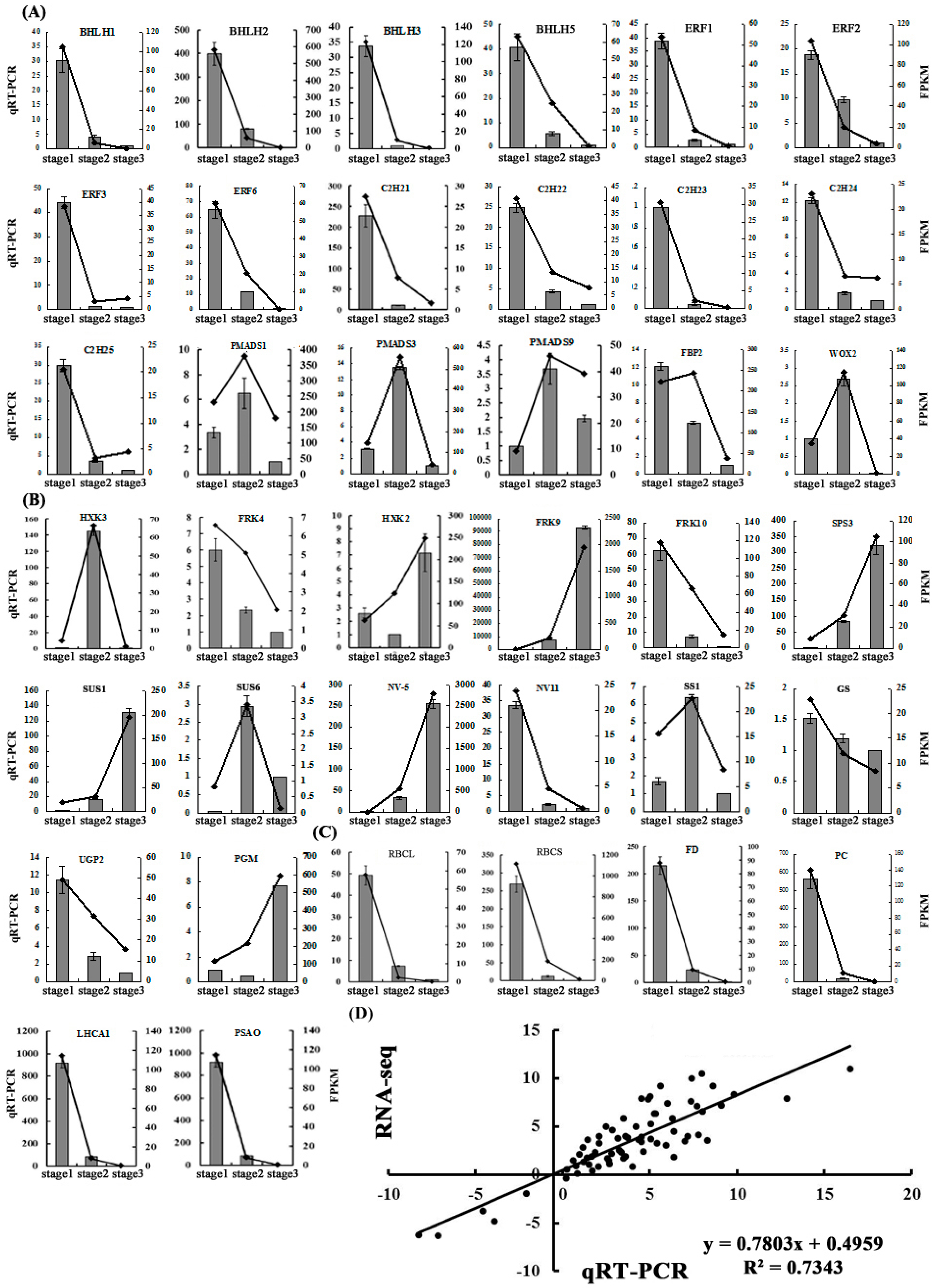
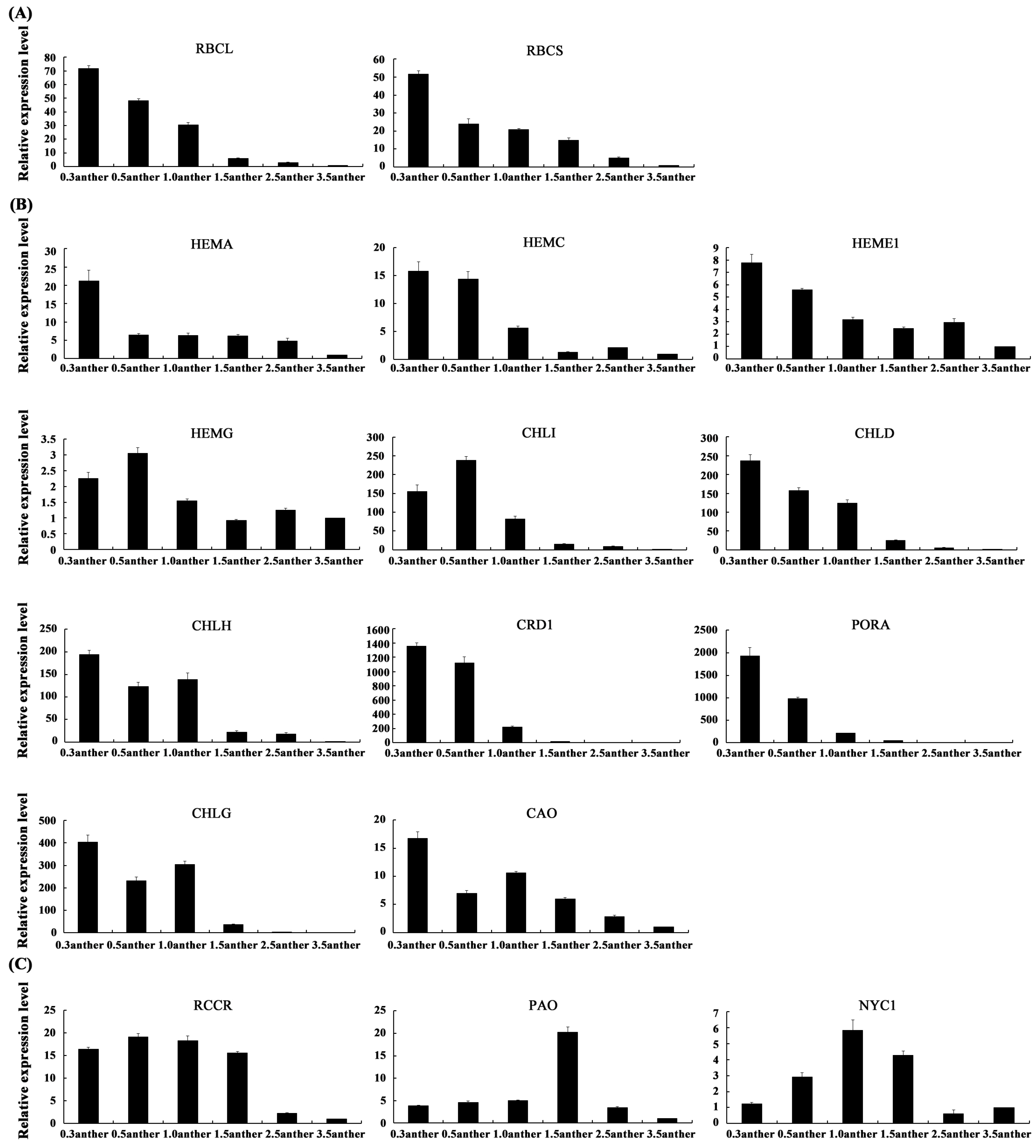
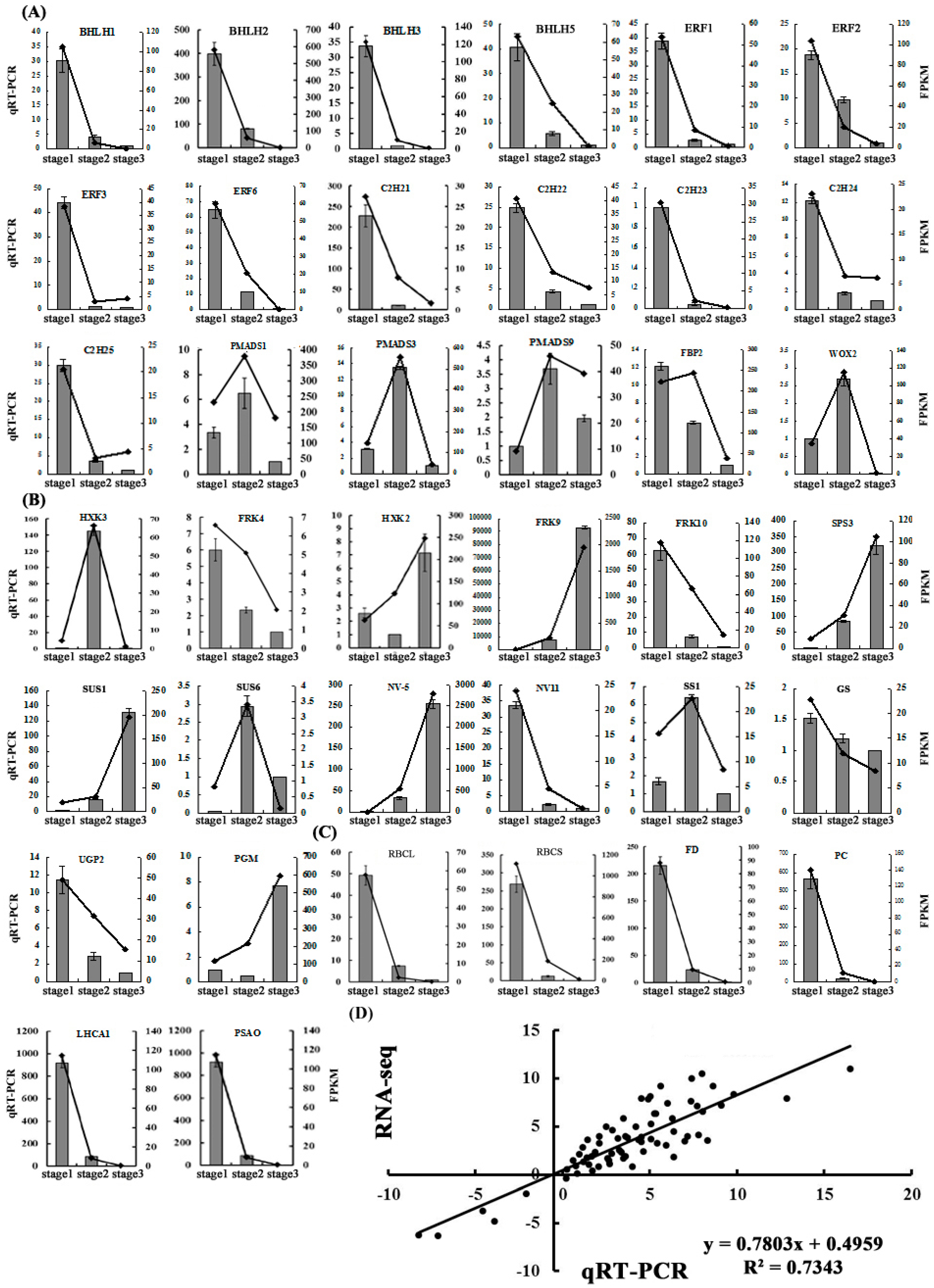
2.5. Validation of the Gene Expression Profiles by qRT-PCR
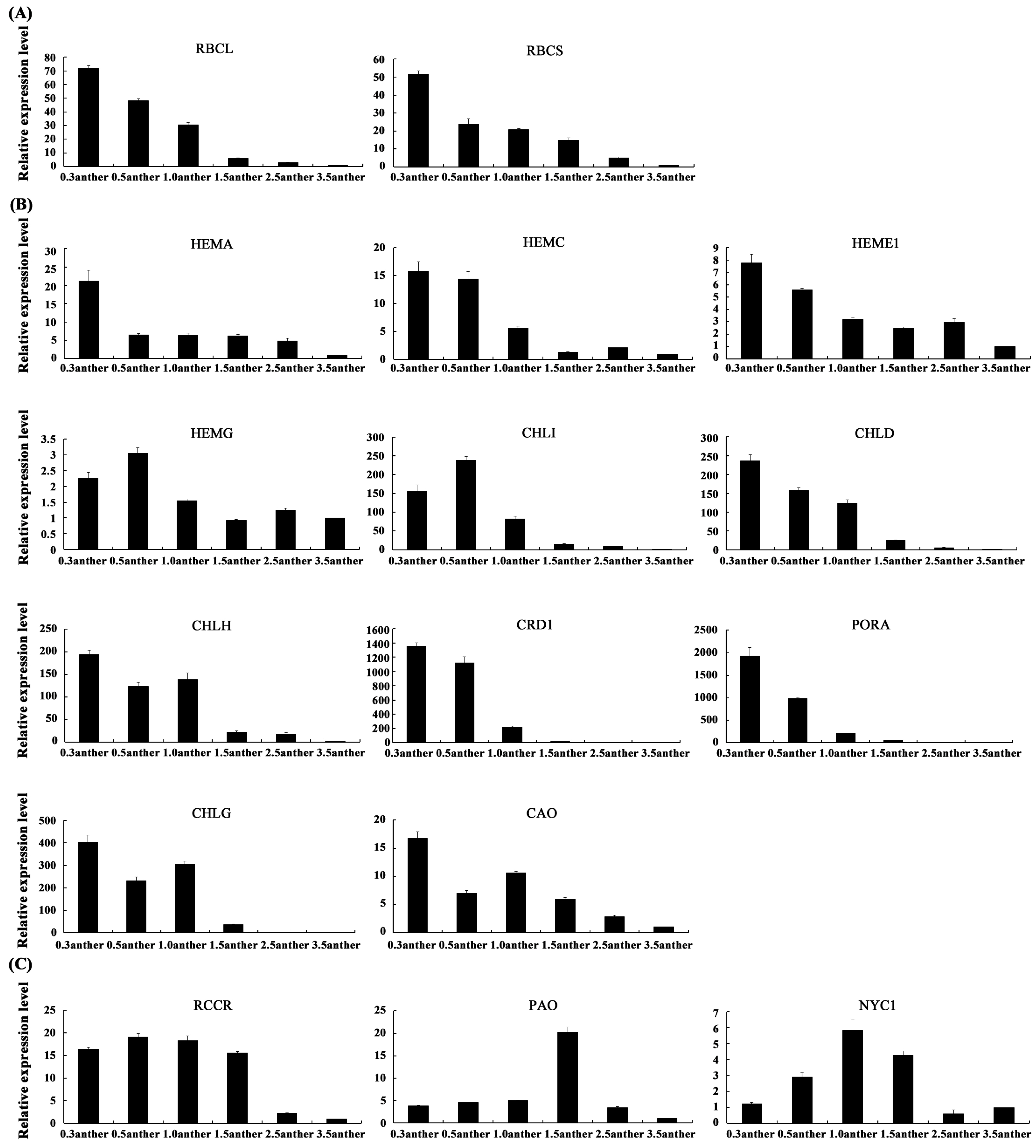
2.6. The Expression Patterns of Photosynthesis-Related Genes
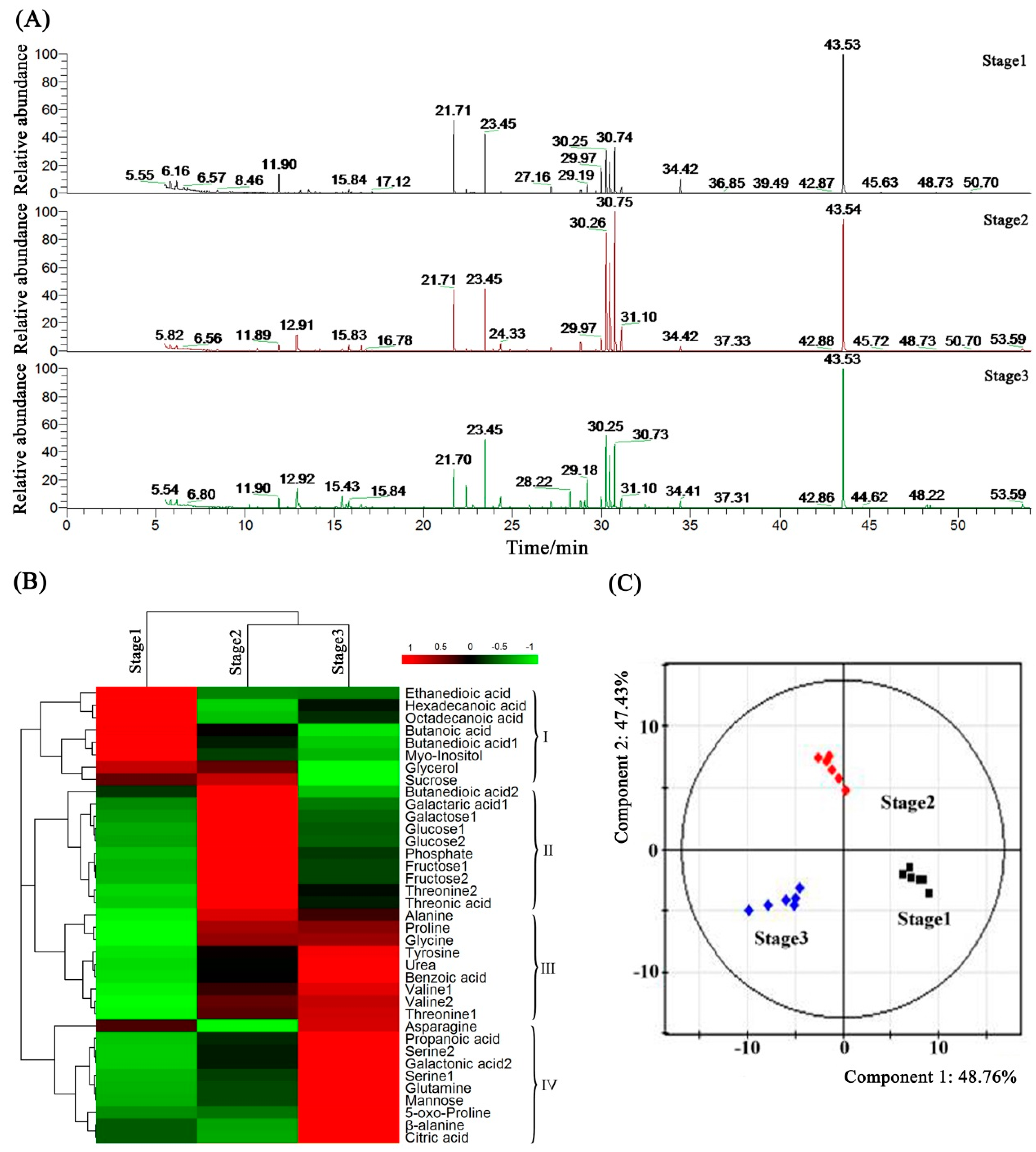
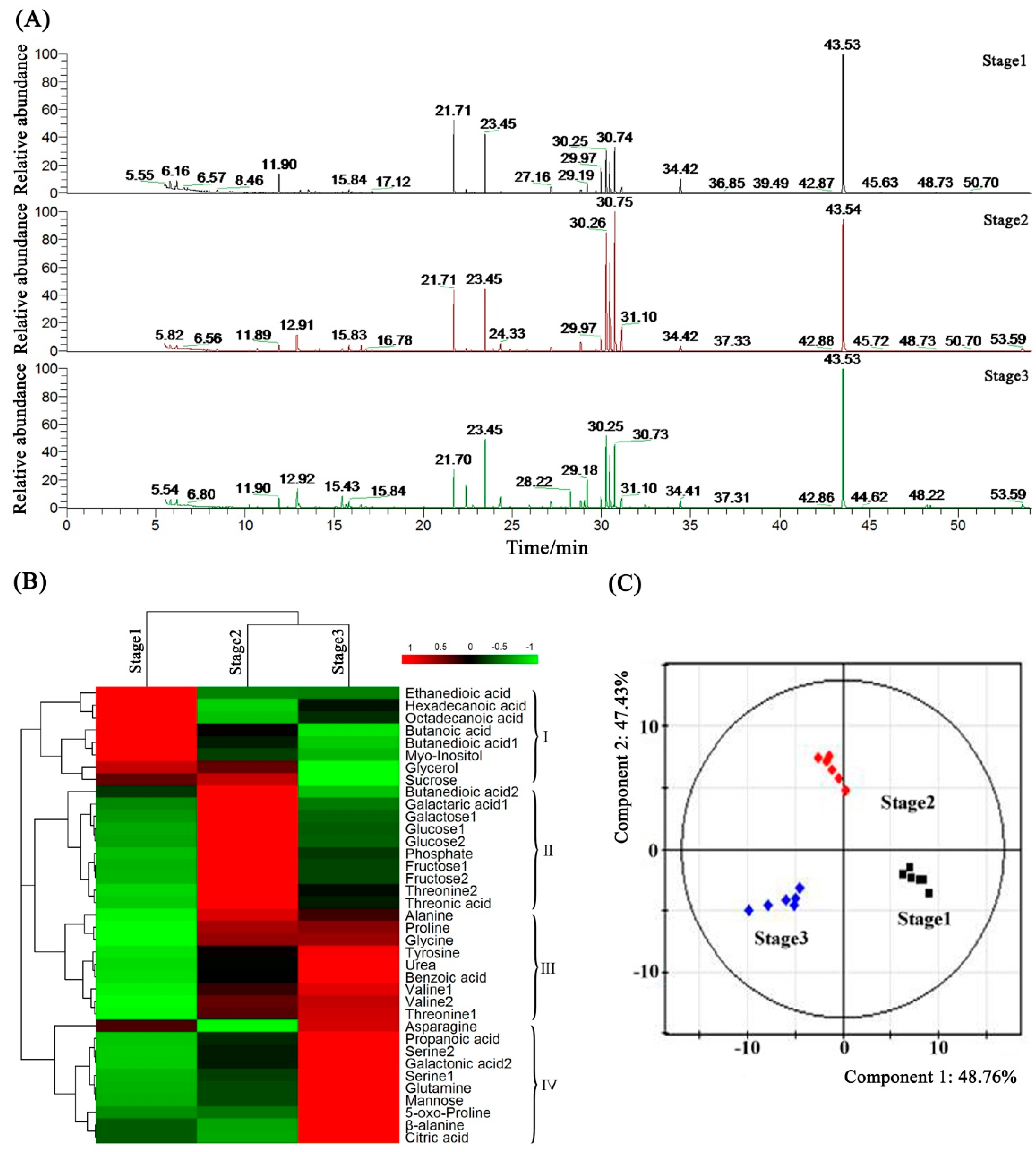
2.7. Metabolome Analysis of Petunia Anther Development by GC-MS
2.8. Metabolic Profiling and Core Differential Metabolites’ Identification
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Chlorophylls and Carotenoids Content Assay
4.3. RNA Extraction
4.4. RNA-Seq and Reads Mapping
4.5. Gene Expression Difference Analysis
4.6. qRT-PCR Analysis
4.7. Metabolite Extraction and Derivatization
4.8. GC-MS Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McCormick, S. Molecular analysis of male gametogenesis in plants. Trends Genet. 1991, 7, 298–303. [Google Scholar] [CrossRef]
- Gerats, T.; Vandenbussche, M. A model system for comparative research: Petunia. Trends Plant Sci. 2005, 10, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Zenoni, S.; D’Agostino, N.; Tornielli, G.B.; Quattrocchio, F.; Chiusano, M.L.; Koes, R.; Zethof, J.; Guzzo, F.; Delledonne, M.; Frusciante, L.; et al. Revealing impaired pathways in the an11 mutant by high-throughput characterization of Petunia axillaris and Petunia inflata transcriptomes. Plant J. 2011, 68, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Vandenbussche, M.; Chambrier, P.; Rodrigues Bento, S.; Morel, P. Petunia, Your Next Supermodel? Front. Plant Sci. 2016, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Twell, D.; Kuang, Y.F.; Liao, J.P.; Zhou, X.Q. Transcriptome analysis of Hamelia patens (Rubiaceae) anthers reveals candidate genes for tapetum and pollen wall development. Front. Plant Sci. 2016, 7, 1991. [Google Scholar] [CrossRef] [PubMed]
- Deveshwar, P.; Bovill, W.D.; Sharma, R.; Able, J.A.; Kapoor, S. Analysis of anther transcriptomes to identify genes contributing to meiosis and male gametophyte development in rice. BMC Plant Biol. 2011, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Hobo, T.; Suwabe, K.; Aya, K.; Suzuki, G.; Yano, K.; Ishimizu, T.; Fujita, M.; Kikuchi, S.; Hamada, K.; Miyano, M.; et al. Various spatiotemporal expression profiles of anther-expressed genes in rice. Plant Cell Physiol. 2008, 49, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Golkari, S.; Gilbert, J.; Prashar, S.; Procunier, J.D. Microarray analysis of Fusarium graminearum-induced wheat genes: Identification of organ-specific and differentially expressed genes. Plant Biotechnol. J. 2007, 5, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Duncan, D.; Morrow, D.J.; Fernandes, J.; Walbot, V. Transcriptome profiling of maize anthers using genetic ablation to analyze pre-meiotic and tapetal cell types. Plant J. 2007, 50, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.F.; Tzeng, J.D.; Liu, M.C.; Yei, F.L.; Chung, M.C.; Wang, C.S. Identification of anther-specific/predominant genes regulated by gibberellin during development of lily anthers. J. Plant Physiol. 2008, 165, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Bokvaj, P.; Hafidh, S.; Honys, D. Transcriptome profiling of male gametophyte development in Nicotiana tabacum. Genomics Data 2015, 3, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Cnudde, F.; Moretti, C.; Porceddu, A.; Pezzotti, M.; Gerats, T. Transcript profiling on developing Petunia hybrida floral organs. Sex. Plant Reprod. 2003, 16, 77–85. [Google Scholar] [CrossRef]
- Cnudde, F.L.; Hedatale, V.; de Jong, H.; Pierson, E.S.; Rainey, D.Y.; Zabeau, M.; Weterings, K.; Gerats, T.; Peters, J.L. Changes in gene expression during male meiosis in Petunia hybrida. Chromosome Res. 2006, 14, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, K.; Ishimizu, T.; Nishimura, K.; Matsubara, K.; Kodama, H.; Watanabe, H.; Hase, S.; Ando, T. Analysis of expressed sequence tags from Petunia flowers. Plant Sci. 2007, 173, 495–500. [Google Scholar] [CrossRef]
- Roessner, U.; Wagner, C.; Kopka, J.; Trethewey, R.N.; Willmitzer, L. Simultaneous analysis of metabolites in potato tuber by gas chromatography–mass spectrometry. Plant J. 2000, 23, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.D.; Whiting, M.D.; Rudell, D.R. Metabolic profiling of ethephon-treated sweet cherry (Prunus avium L.). Metabolomics 2011, 7, 126–133. [Google Scholar] [CrossRef]
- Jänkänpää, H.J.; Mishra, Y.; Schröder, W.P.; Jansson, S. Metabolic profiling reveals metabolic shifts in Arabidopsis plants grown under different light conditions. Plant Cell Environ. 2012, 35, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Gai, Y.P.; Han, X.J.; Li, Y.Q.; Yuan, C.Z.; Mo, Y.Y.; Guo, F.Y.; Liu, Q.X.; Ji, X.L. Metabolomic analysis reveals the potential metabolites and pathogenesis involved in mulberry yellow dwarf disease. Plant Cell Environ. 2014, 37, 1474–1490. [Google Scholar] [CrossRef] [PubMed]
- Cevalloscevallos, J.M.; Futch, D.B.; Shilts, T.; Folimonova, S.Y.; Reyesdecorcuera, J.I. GC-MS metabolomic differentiation of selected citrus varieties with different sensitivity to citrus huanglongbing. Plant Physiol. Biochem. 2012, 53, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, J.C.; Ch, R.D.V.; Verhoeven, H.A.; Haring, M.A.; van Tunen, A.J.; Schuurink, R.C. Regulation of floral scent production in petunia revealed by targeted metabolomics. Phytochemistry 2003, 62, 997–1008. [Google Scholar] [CrossRef]
- Oyama-Okubo, N.; Sakai, T.; Ando, T.; Nakayama, M.; Soga, T. Metabolome profiling of floral scent production in Petunia axillaris. Phytochemistry 2013, 90, 37. [Google Scholar] [CrossRef] [PubMed]
- Yun, Z.; Qu, H.X.; Wang, H.; Zhu, F.; Zhang, Z.K.; Duan, X.W.; Yang, B.; Cheng, Y.J.; Jiang, Y.M. Comparative transcriptome and metabolome provides new insights into the regulatory mechanisms of accelerated senescence in litchi fruit after cold storage. Sci. Rep. 2016, 6, 19356. [Google Scholar] [CrossRef] [PubMed]
- Matus, J.T. Transcriptomic and metabolomic networks in the grape berry illustrate that it takes more than flavonoids to fight against ultraviolet radiation. Front. Plant Sci. 2016, 7, 1337. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, A.M.; Martinelli, F.; Reagan, R.L.; Uratsu, S.L.; Vo, A.; Tinoco, M.A.; Phu, M.L.; Chen, Y.; Rocke, D.M.; Dandekar, A.M. Transcriptome and metabolome analysis of citrus fruit to elucidate puffing disorder. Plant Sci. 2014, 218, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Ritter, A.; Dittami, S.M.; Goulitquer, S.; Correa, J.A.; Boyen, C.; Potin, P.; Tonon, T. Transcriptomic and metabolomic analysis of copper stress acclimation in Ectocarpus siliculosus highlights signaling and tolerance mechanisms in brown algae. BMC Plant Biol. 2014, 14, 116. [Google Scholar] [CrossRef] [PubMed]
- Bielecka, M.; Watanabe, M.; Morcuende, R.; Scheible, W.R.; Hawkesford, M.J.; Hesse, H.; Hoefgen, R. Transcriptome and metabolome analysis of plant sulfate starvation and resupply provides novel information on transcriptional regulation of metabolism associated with sulfur, nitrogen and phosphorus nutritional responses in Arabidopsis. Front. Plant Sci. 2015, 5, 805. [Google Scholar] [CrossRef] [PubMed]
- Bombarely, A.; Moser, M.; Amrad, A.; Bao, M.Z.; Bapaume, L.; Barry, C.S.; Bliek, M.; Boersma, M.R.; Borghi, L.; Bruggmann, R.; et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants 2016, 2, 16074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Y.Z.; Yin, C.Q.; Guo, R.; Peng, H.; Yang, Z.N.; Liu, G.F.; Bao, M.Z.; Hu, H.R. An anther-specific gene PhGRP is regulated by PhMYC2 and causes male sterility when overexpressed in petunia anthers. Plant Cell Rep. 2017, 36, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.Z.; Yang, D.; Sun, J.; Peng, H.; Yin, C.Q.; Guo, R.; Ning, G.G.; Hu, H.R. A novel PhLRR gene promoter is sufficient for engineering male sterility in petunia. Plant Mol. Biol. Rep. 2016, 34, 970–977. [Google Scholar] [CrossRef]
- Porceddu, A.; Reale, L.; Lanfaloni, L.; Moretti, C.; Sorbolini, S.; Tedeschini, E.; Ferranti, F.; Pezzotti, M. Cloning and expression analysis of a Petunia hybrida flower specific mitotic-like cyclin. FEBS Lett. 1999, 462, 211–215. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Y.; Li, D.; Wang, G.; Li, X.; Xia, Y.P. Transcriptomic analysis of the underground renewal buds during dormancy transition and release in “Hangbaishao” peony (Paeonia lactiflora). PLoS ONE 2015, 10, e0119118. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, Z.Y.; Zhang, W.J.; Zhao, X.M.; Li, X.; Zhang, D.; Liu, Q.Q.; Tang, W.H. Global gene profiling of laser-captured pollen mother cells indicates molecular pathways and gene subfamilies involved in rice meiosis. Plant Physiol. 2010, 154, 1855–1870. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Huang, W.J.; He, J.; Zhang, D.; Tang, W.H. Stage-specific gene profiling of germinal cells helps delineate the mitosis/meiosis transition. Plant Physiol. 2018, 176, 1610–1626. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, U.; Grimm, B.; Hortensteiner, S. Recent advances in chlorophyll biosynthesis and breakdown in higher plants. Plant Mol. Biol. 2004, 56, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schelbert, S.; Aubry, S.; Burla, B.; Agne, B.; Kessler, F.; Krupinska, K.; Hörtensteiner, S. Pheophytin pheophorbide hydrolase (Pheophytinase) is involved in chlorophyll breakdown during leaf senescence in Arabidopsis. Plant Cell 2009, 21, 767–785. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Guo, Y.J.; Kuai, B.K. Isolation and characterization of a chlorophyll degradation regulatory gene from tall fescue. Plant Cell Rep. 2011, 30, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Miyake, C.; Iwano, M.; Sekine, M.; Shinmyo, A.; Kato, K. Ribulose 1,5-bisphosphate carboxylase/oxygenase large subunit translation is regulated in a small subunit-independent manner in the expanded leaves of tobacco. Plant Cell Physiol. 2008, 49, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Klotz, K.L.; Finger, F.L.; Anderson, M.D. Wounding increases glycolytic but not soluble sucrolytic activities in stored sugarbeet root. Postharvest Biol. Technol. 2006, 41, 48–55. [Google Scholar] [CrossRef]
- Min, L.; Li, Y.Y.; Hu, Q.; Zhu, L.F.; Gao, W.H.; Wu, Y.L.; Ding, Y.H.; Liu, S.M.; Yang, X.Y.; Zhang, X.L. Sugar and auxin signaling pathways respond to high-temperature stress during anther development as revealed by transcript profiling analysis in Cotton. Plant Physiol. 2014, 164, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.Z.; Ma, F.F.; Huang, X.; Bao, M.Z.; Liu, G.F.; Hu, H.R. Transcriptional profile of differentially expressed genes related to abortive flower buds under short light period stress in petunia. Sci. Hortic. 2013, 164, 323–332. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhu, Q.G.; Meng, Q.W.; Wu, C.A. Transcript profiling during salt stress of young cotton (Gossypium hirsutum) seedlings via Solexa sequencing. Acta Physiol. Plant. 2012, 34, 107–115. [Google Scholar] [CrossRef]
- Mosca, F.; Tritto, E.; Muzzi, A.; Monaci, E.; Bagnoli, F.; Iavarone, C.; O’Hagan, D.; Rappuoli, R.; De Gregorio, E. Molecular and cellular signatures of human vaccine adjuvants. Proc. Natl. Acad. Sci. USA 2008, 105, 10501–10506. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Song, Y.; Zhang, G.; Ju, L.; Zhang, J.; Yu, Y.G.; Niu, N.; Wang, J.W.; Ma, S.C. De novo assembly and transcriptome analysis of wheat with male sterility induced by the chemical hybridizing agent SQ-1. PLoS ONE 2015, 10, e0123556. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, Y.; Tian, S.; Wang, Y.; Ma, H.; Liu, S.; Wang, Y.; Hu, H. Transcriptomic and GC-MS Metabolomic Analyses Reveal the Sink Strength Changes during Petunia Anther Development. Int. J. Mol. Sci. 2018, 19, 955. https://doi.org/10.3390/ijms19040955
Yue Y, Tian S, Wang Y, Ma H, Liu S, Wang Y, Hu H. Transcriptomic and GC-MS Metabolomic Analyses Reveal the Sink Strength Changes during Petunia Anther Development. International Journal of Molecular Sciences. 2018; 19(4):955. https://doi.org/10.3390/ijms19040955
Chicago/Turabian StyleYue, Yuanzheng, Shaoze Tian, Yu Wang, Hui Ma, Siyu Liu, Yuqiao Wang, and Huirong Hu. 2018. "Transcriptomic and GC-MS Metabolomic Analyses Reveal the Sink Strength Changes during Petunia Anther Development" International Journal of Molecular Sciences 19, no. 4: 955. https://doi.org/10.3390/ijms19040955
APA StyleYue, Y., Tian, S., Wang, Y., Ma, H., Liu, S., Wang, Y., & Hu, H. (2018). Transcriptomic and GC-MS Metabolomic Analyses Reveal the Sink Strength Changes during Petunia Anther Development. International Journal of Molecular Sciences, 19(4), 955. https://doi.org/10.3390/ijms19040955