1. Introduction
The liver plays a major role in maintaining normal blood glucose levels by regulating de novo glucose production and glycogen breakdown. Hepatic glucose production (gluconeogenesis) is essential for the supply of glucose as an energy source to other tissues. However, excessive hepatic gluconeogenesis causes hyperglycemia. Insulin resistance is defined as the disability of insulin to regulate glucose and lipid metabolism in peripheral tissues even at elevated insulin levels in the blood. Hepatic insulin resistance results in the elevation of hepatic glucose production and triglyceride (TG) accumulation by impairing insulin-mediated inhibition of gluconeogenesis and regulating insulin-mediated TG metabolism, respectively, which contributes to hyperglycemia and dyslipidemia [
1]. Therefore, the control of hepatic insulin resistance is an attractive therapeutic target for treating type 2 diabetes and hepatic steatosis.
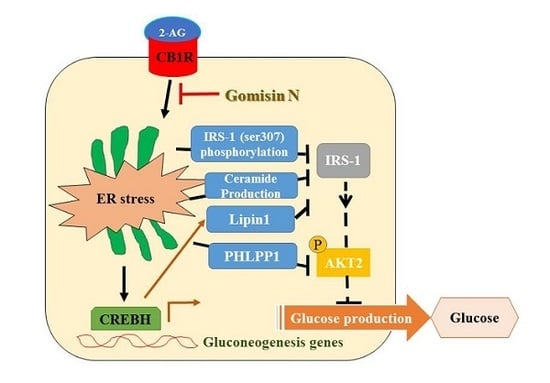
Endogenous cannabinoids, such as arachidonoyl ethanolamide (AEA) and 2-arachidonoylglycerol (2-AG), are bioactive lipid mediators that interact with the cannabinoid type 1 receptor (CB1R) and cannabinoid type 2 receptor (CB2R), and regulate numerous biochemical responses [
2]. CB1R is predominantly present in the brain and controls food behavior and energy expenditure [
2]. The activation of CB1R in the central nervous system facilitates food intake by modulating the release of orexigenic and anorexigenic neuropeptides in hypothalamic neurons. CB1R is also found in peripheral tissues and controls glucose and lipid metabolism [
2]. Activation of hepatic CB1R induces insulin resistance through several mechanisms in the endoplasmic reticulum (ER) stress-dependent manner [
3,
4,
5]. Activation of CB1R inhibits insulin signaling by elevating inhibitory serine-307 phosphorylation of insulin receptor substrate 1 (IRS1) and by stimulating the dephosphorylation of insulin-activated protein kinase B (PKB/AKT) through upregulation of the S/T phosphatase PH domain and leucine-rich repeats protein phosphatase 1 (
PHLPP1) [
3]. Furthermore, it stimulates the expression of
Lipin1, a phosphatidic acid phosphatase, via ER stress-inducible transcription factor, cAMP-responsive element-binding protein H (CREBH) [
4]. This subsequently leads to accumulation of diacylglycerol (DAG), resulting in the phosphorylation of protein kinase C with inhibition of insulin receptor signaling [
4]. In addition, CB1R-mediated ER stress increases the production of ceramide by upregulation of de novo ceramide synthesis and downregulation of ceramide degradation, which inhibits insulin signaling [
5]. Moreover, hepatic CB1R activation stimulates the lipogenesis transcription factor, sterol regulatory element-binding transcription factor 1c (SREBP1c), and results in increased TG accumulation by upregulating the expression of its downstream lipogenesis genes, including fatty acid synthase (
FAS), stearoyl-Coenzyme A desaturase 1 (
SCD1), and acetyl-CoA carboxylase (
ACC), which contributes to insulin resistance and steatosis [
6]. Furthermore, CB1R also increases the expression of gluconeogenesis genes via CREBH and results in the elevated glucose production [
7]. Thus, activation of hepatic CB1R plays a role in the development of insulin resistance, type 2 diabetes, and hepatic steatosis [
8]. In this regard, selective inhibition of hepatic CB1R signaling could be a potential molecular target for the treatment of type 2 diabetes and hepatic steatosis.
Schisandra chinensis has been used as a traditional herbal medicine in Asian countries, such as China, Korea, Japan, and Russia. It has diverse pharmacological activities, including anti-oxidant, anti-tumor, anti-obesity, anti-inflammatory, cardioprotective, and hepatoprotective effects [
9]. Recently, we reported that
S. chinensis has a protective effect against ER stress-induced hepatic steatosis [
10]. Gomisin N (GN), a lignan derived from
S. chinensis, possesses hepatoprotective, anti-cancer, and anti-inflammatory effects [
11]. Recently, we reported that GN exerts protective effects against obesity-induced hepatic steatosis and hyperglycemia through inhibition of ER stress and AMP-activated protein kinase (AMPK) activation, respectively [
12,
13]. As activation of hepatic CB1R signaling has been implicated in the development of insulin resistance, hyperglycemia, and hepatic steatosis, targeted inhibition of hepatic CB1R signaling might provide therapeutic approaches to restore insulin receptor signaling and improve hyperglycemia and hepatic steatosis.
Therefore, in the current study, we investigated the inhibitory effect of GN on hepatic CB1R and CB1R-mediated insulin resistance and gluconeogenesis in 2-arachidonoylglycerol (AG; an agonist of CB1R)-treated HepG2 cells and in high-fat diet (HFD)-induced obese mice.
3. Discussion
The activation of peripheral CB1Rs has been increasingly recognized as an important regulator of metabolic disorders due to deleterious effects on lipid and glucose metabolism [
2]. Among peripheral CB1Rs, activation of the hepatic CB1R induces ER stress-dependent hepatic insulin resistance and gluconeogenesis, resulting in hyperglycemia [
3,
7]. In addition, hepatic CB1R causes lipid accumulation by upregulation of lipogenesis genes, resulting in hepatic steatosis [
6]. Therefore, the inhibition of hepatic CB1R signaling is a promising target for treating type 2 diabetes and hepatic steatosis. GN is a phytochemical derived from
S. chinensis, a traditional medicinal herb [
11]. Previously, we demonstrated that GN exerts protective effects against HFD-induced hyperglycemia and hepatic steatosis in HFD obese mice [
12,
13,
14]. In the present study, we investigated whether GN-mediated improvement of hyperglycemia and hepatic steatosis is through inhibition of the hepatic CB1R signaling. We examined the inhibitory effect of GN on CB1R-induced insulin resistance and gluconeogenesis in vitro and in vivo.
Recent studies have demonstrated that obesity leads to activation of the hepatic CB1R signaling, which contributes to insulin resistance and gluconeogenesis in the ER stress dependent manner, resulting in hyperglycemia [
3,
4,
5,
6,
7]. Therefore, hepatic CB1R-mediated ER stress can be a potential target for HFD-induced hyperglycemia. Previously, we demonstrated that GN inhibited fatty acid-induced ER stress and prevented HFD-induced hepatic steatosis and hyperglycemia [
12,
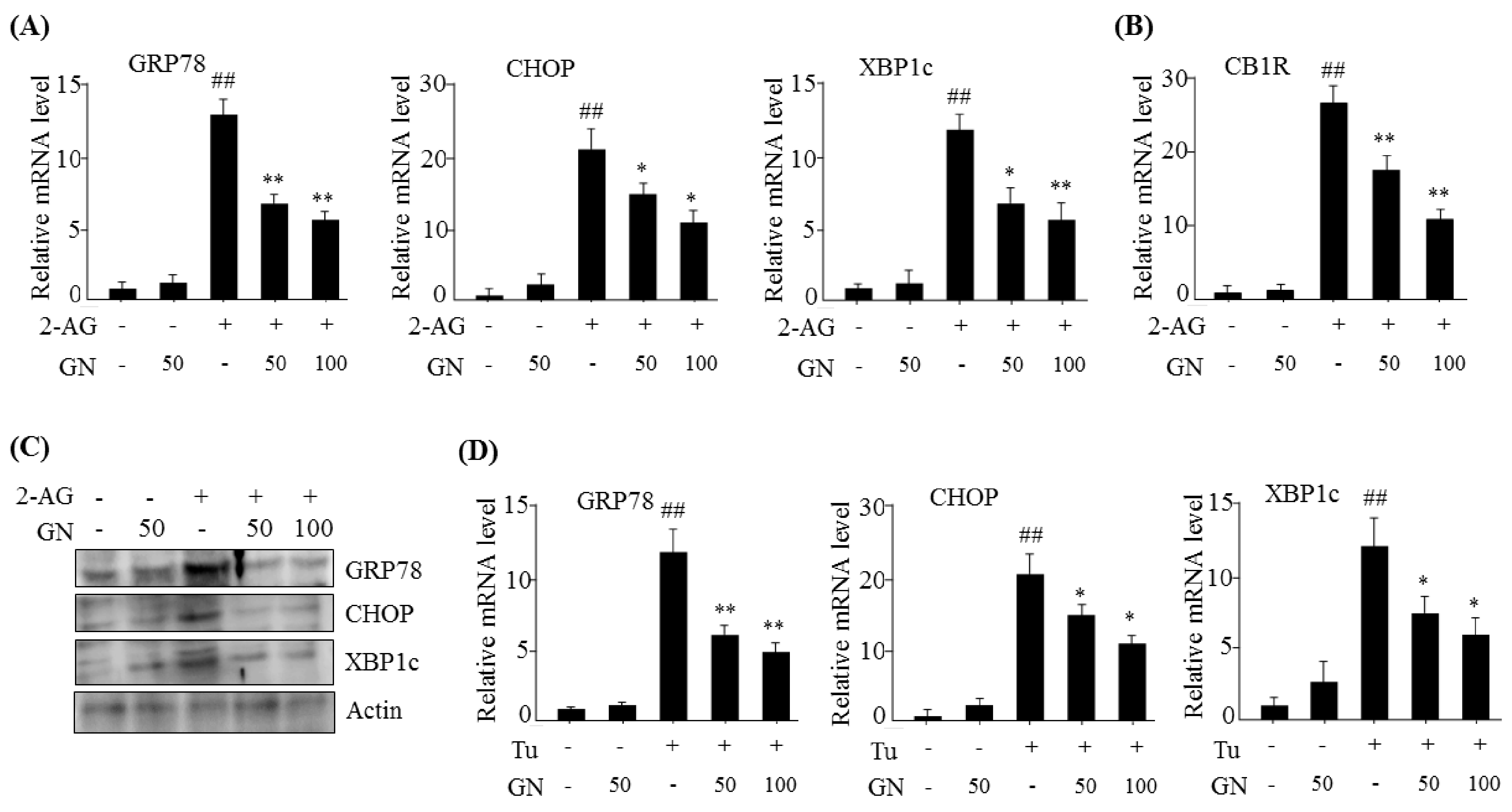
13]. In the current study, we investigated whether GN also inhibited CB1R-induced ER stress and subsequently improved CB1R-mediated insulin resistance and gluconeogenesis in HepG2 cells. Our data showed that treatment with 2-AG, a CB1R activator, promoted the expression of ER stress markers such as
GRP78,
CHOP, and
XBP1c in HepG2 cells, but GN significantly prevented 2-AG-induced expression of these genes, indicating that GN inhibits CB1R-induced ER stress in HepG2 cells.
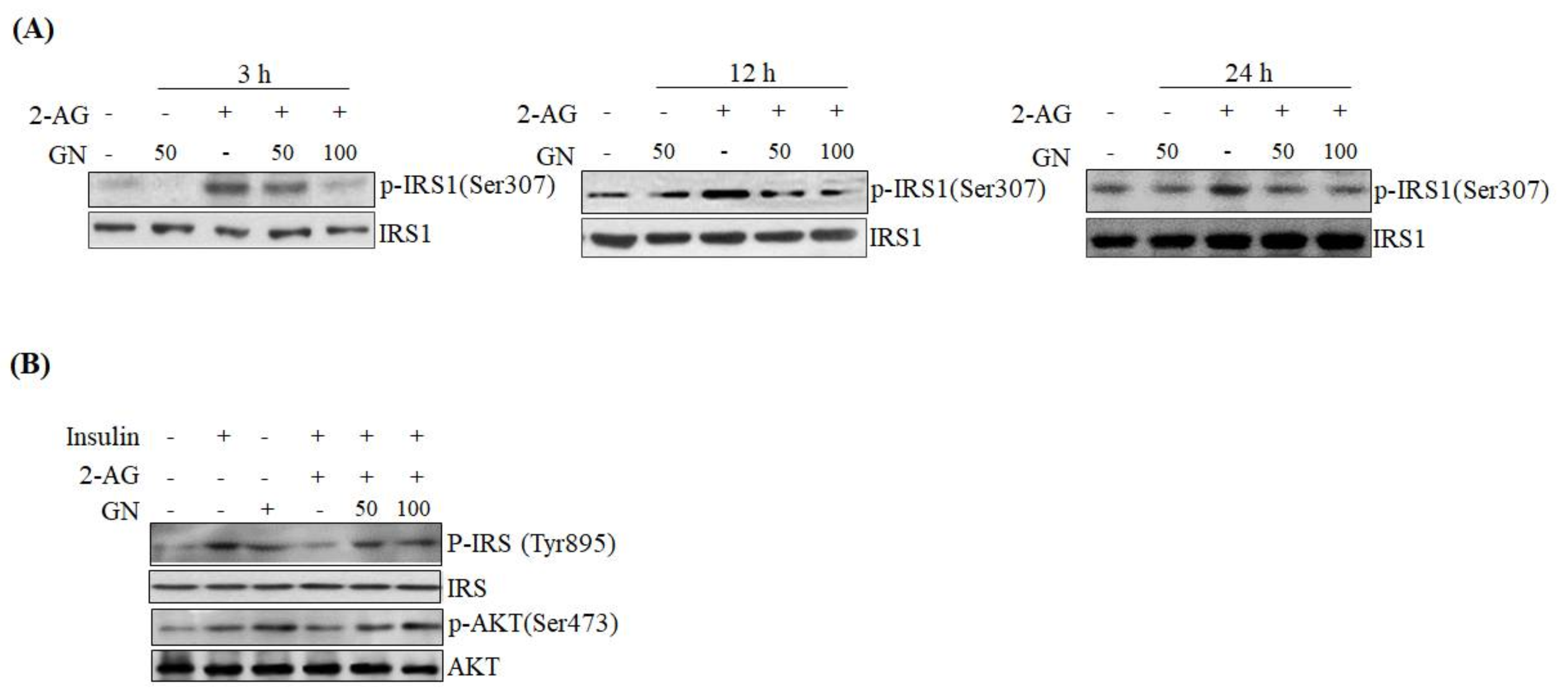
It has been reported that CB1R-induced ER stress causes insulin resistance via several mechanisms [
3,
4,
5,
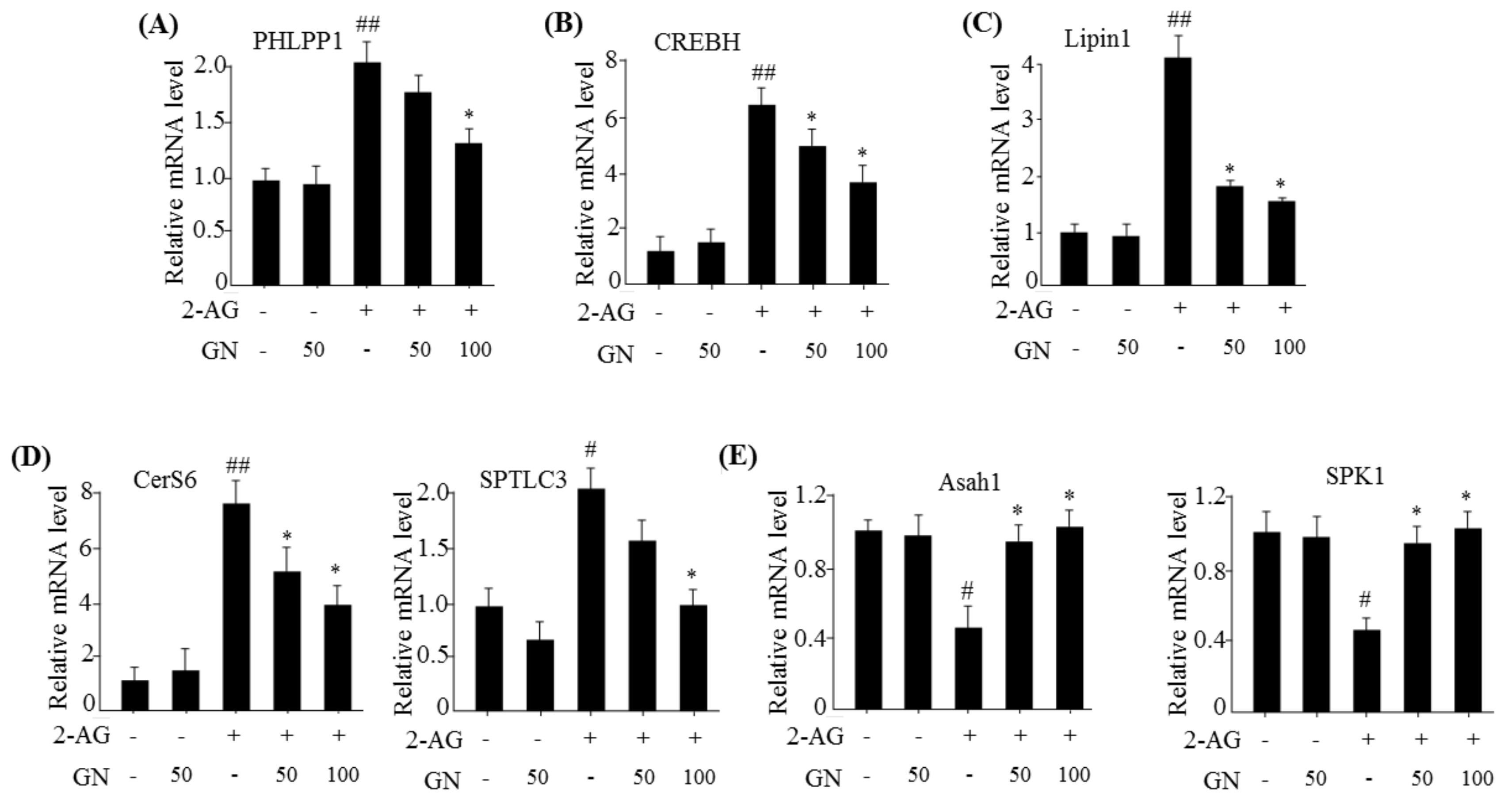
6]. As described previously, hepatic CB1R-induced ER stress leads to insulin resistance by upregulation of serine phosphatase PHLPP1, Lipin1, and ceramide production. PHLPP1 reverses insulin-activated AKT phosphorylation and inhibits insulin signaling. Lipin1 generates DAG, which subsequently suppresses insulin signaling via PKC activation. Therefore, we examined the inhibitory effects of GN on the expression of
PHLPP1 and
Lipin1 in 2-AG-treated HepG2 cells. Our data revealed that GN treatment significantly reversed 2-AG-induced mRNA levels of both
PHLPP1 and
Lipin1. CB1R-induced ceramide production, which is regulated by de novo synthesis and degradation of ceramide, also leads to inhibition of insulin signaling. To investigate whether GN affects CB1R-induced ceramide production, we examined the expression of de novo ceramide synthesis genes and ceramide degradation genes in 2-AG-treated HepG2 cells. GN reduced the 2-AG-induced mRNA expression of
CerS6 and
SPTLC3 involved in
de novo ceramide synthesis, but increased the expression of mRNA levels of
Asah1 and
SPK1 involved in ceramide degradation, which were decreased by 2-AG treatment, indicating that GN suppresses CB1R-induced ceramide production. Taken together, these results suggest that GN might contribute to improvement of hepatic insulin resistance by inhibition of CB1R-induced PHLPP1, Lipin1, and ceramide production. To confirm the ameliorative effect of GN on insulin resistance, we examined insulin signaling in 2-AG-treated HepG2 cells. Treatment with 2-AG increased serine-307 phosphorylation of IRS1, which inhibits AKT phosphorylation, and resulted in reduced insulin-stimulated phosphorylation of IRS1 (tyrosine-895) and AKT (serine-473); however, GN reduced the phosphorylation of IRS1 (serine-307), whereas increased the insulin-stimulated phosphorylation of IRS1 (tyrosine-895) and AKT (serine-473). These results demonstrated that GN reverses the hepatic CB1R-mediated inhibition of insulin signaling.
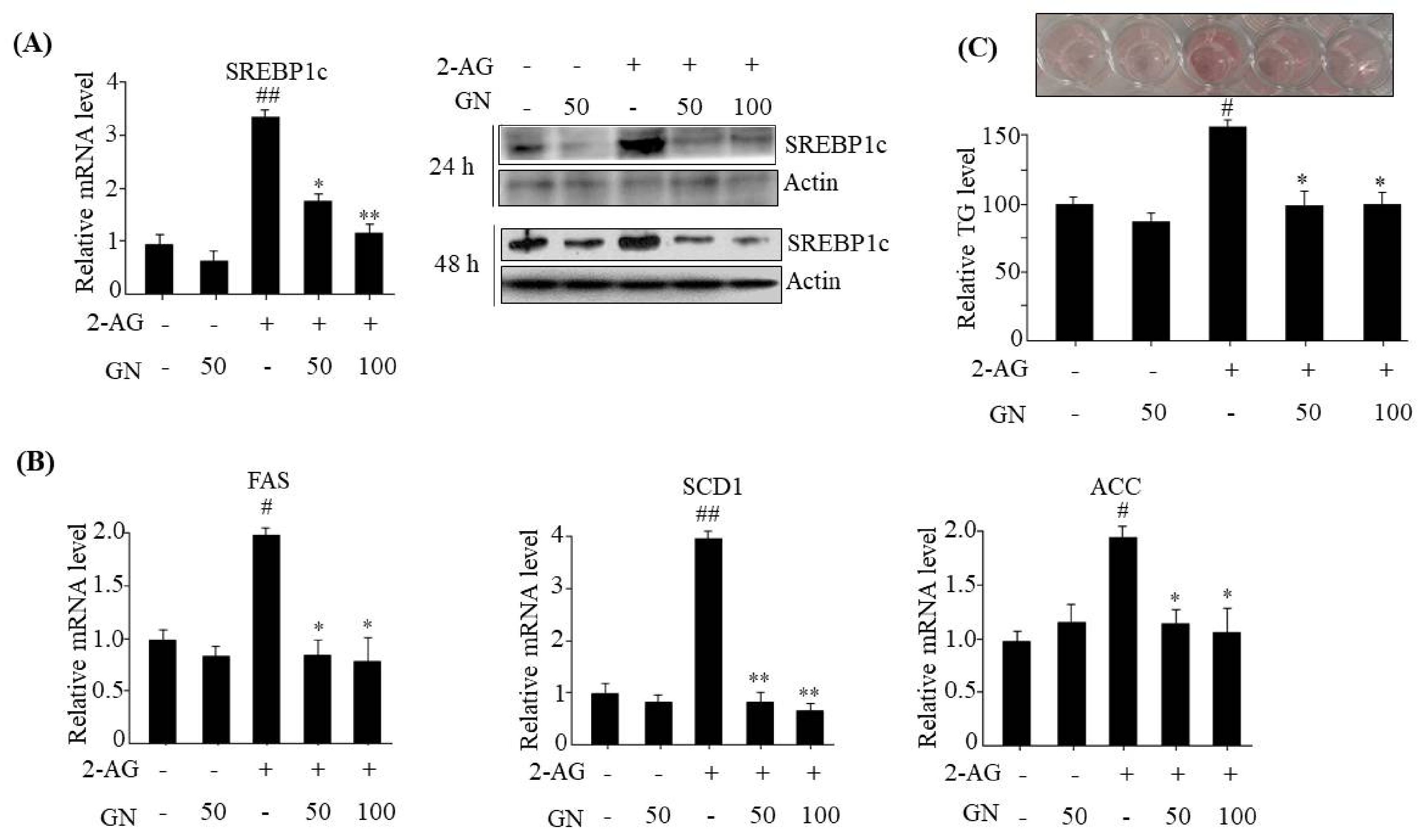
The hepatic CB1R has been reported to induce TG accumulation via upregulation of lipogenesis genes expression, which leads to dysregulation of insulin signaling [
6]. Thus, we tested whether GN inhibits CB1R-induced lipogenesis in 2-AG-treated HepG2 cells. We found that GN inhibited 2-AG-induced expression of lipogenesis genes including
SREBP1c,
FAS,
SCD1,
ACC, and subsequent intracellular TG accumulation in HepG2 cells. These results suggest that prevention of TG accumulation might also play an important role in GN-mediated improvement of insulin resistance and hepatic steatosis.
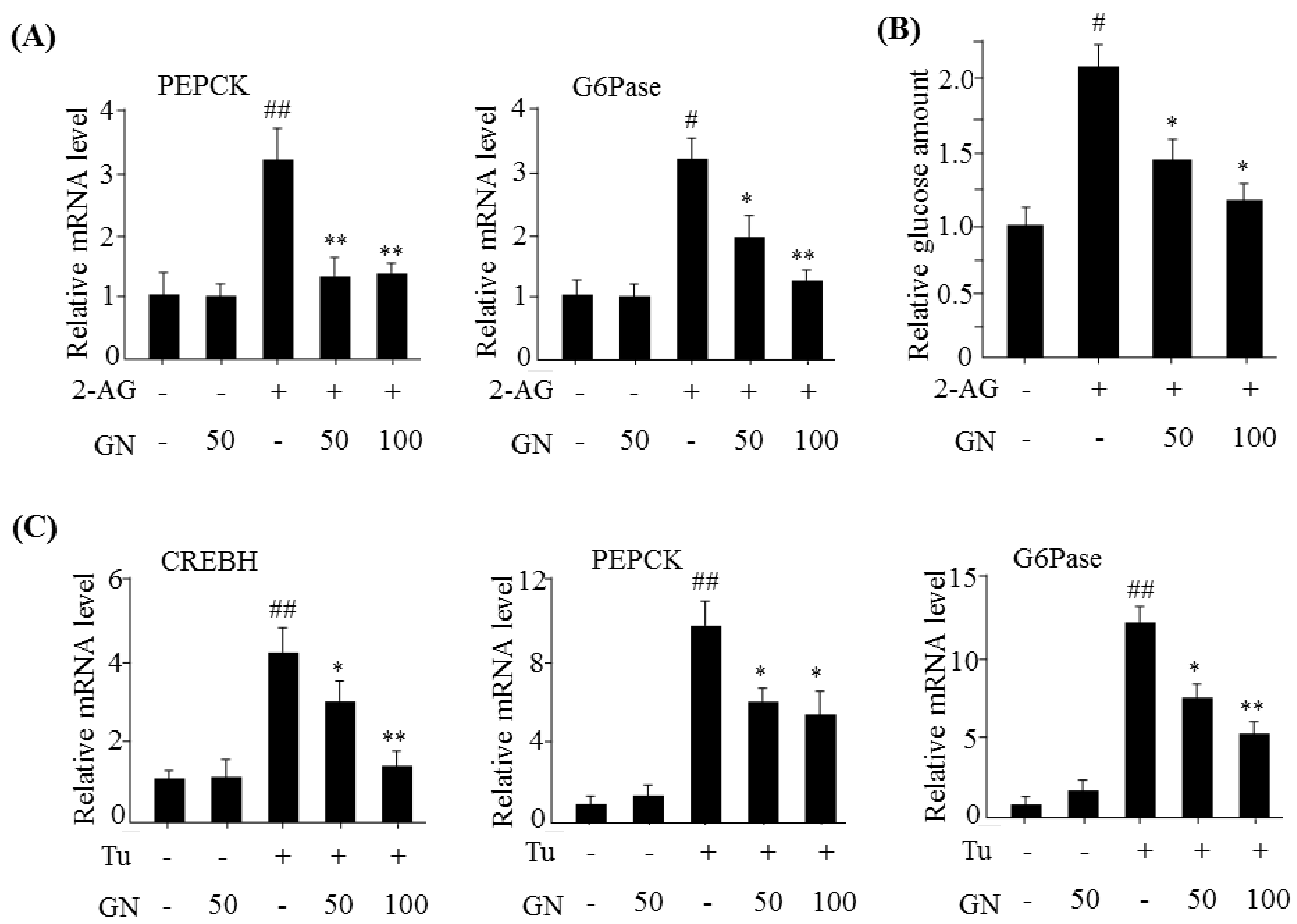
The hepatic CB1R stimulates gluconeogenesis via induction of ER stress, which plays a role in HFD-induced hyperglycemia [
7]. CB1R-induced ER stress stimulates the expression of gluconeogenesis genes via CREBH. Thus, we investigated the inhibitory effect of GN on gluconeogenesis in 2-AG-treated HepG2 cells. Treatment with 2-AG promoted the expression of
CREBH,
PEPCK, and
G6Pase, and resulted in increased glucose production. However, GN inhibited 2-AG-induced expression of gluconeogenesis genes and subsequent glucose production in HepG2 cells. To confirm whether GN-mediated inhibition of gluconeogenesis is through suppression of ER stress, we assessed the inhibitory effects of GN on the expression of gluconeogenesis genes in tunicamycin-treated HepG2 cells. Consistent with the results in 2-AG-treated HepG2 cells, GN treatment reversed tunicamycin-induced expression of gluconeogenesis genes including
CREBH,
PEPCK, and
G6Pase. Taken together, these results indicate that GN inhibits CB1R-induced gluconeogenesis via suppression of ER stress, which might contribute to improvement of hyperglycemia.
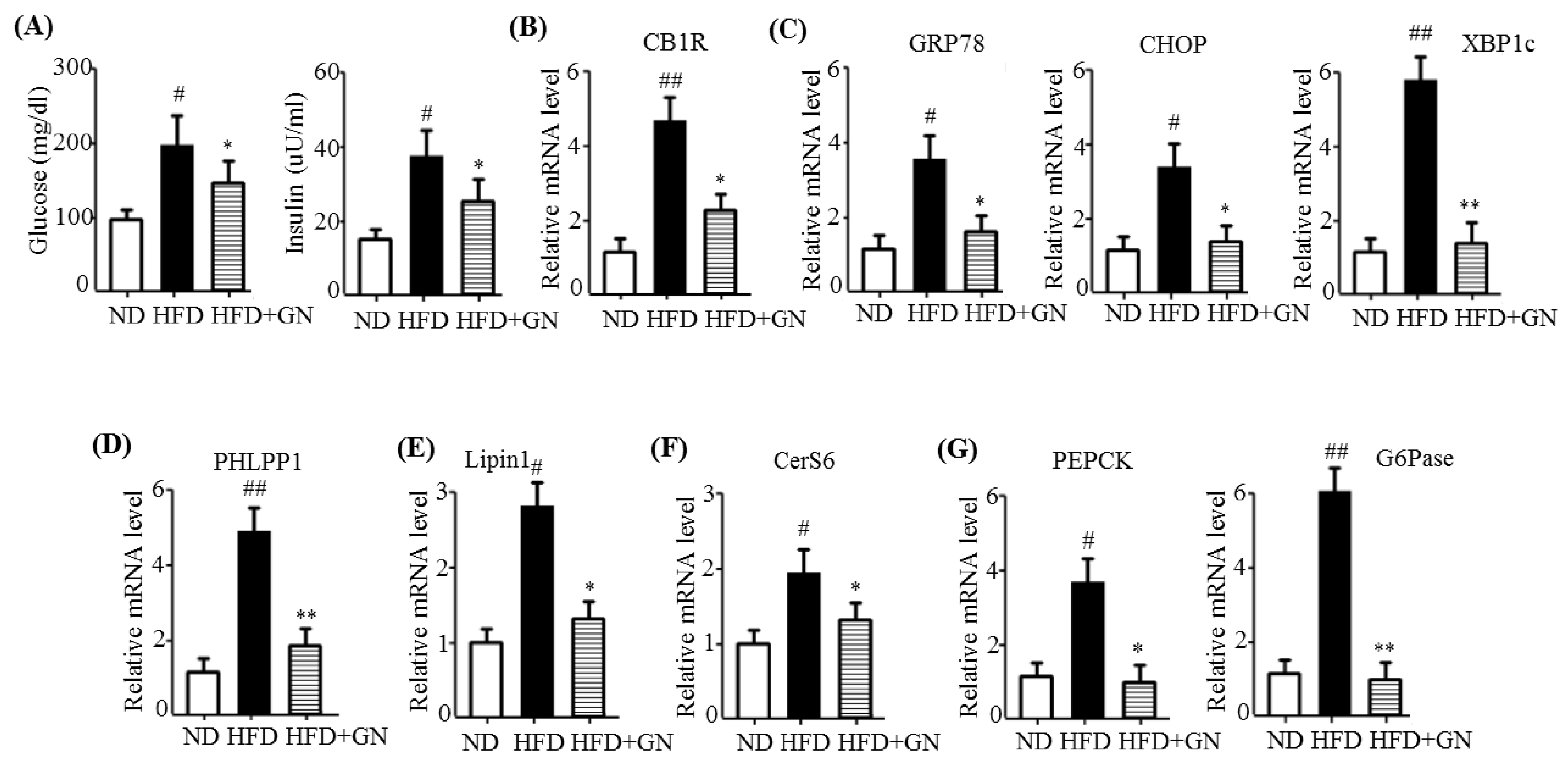
HFD activates hepatic CB1R signaling and results in insulin resistance and hyperglycemia [
3]. Our previous study revealed that GN administration to HFD obese mice efficiently reduced HFD-induced hyperglycemia, and improved glucose tolerance in mice [
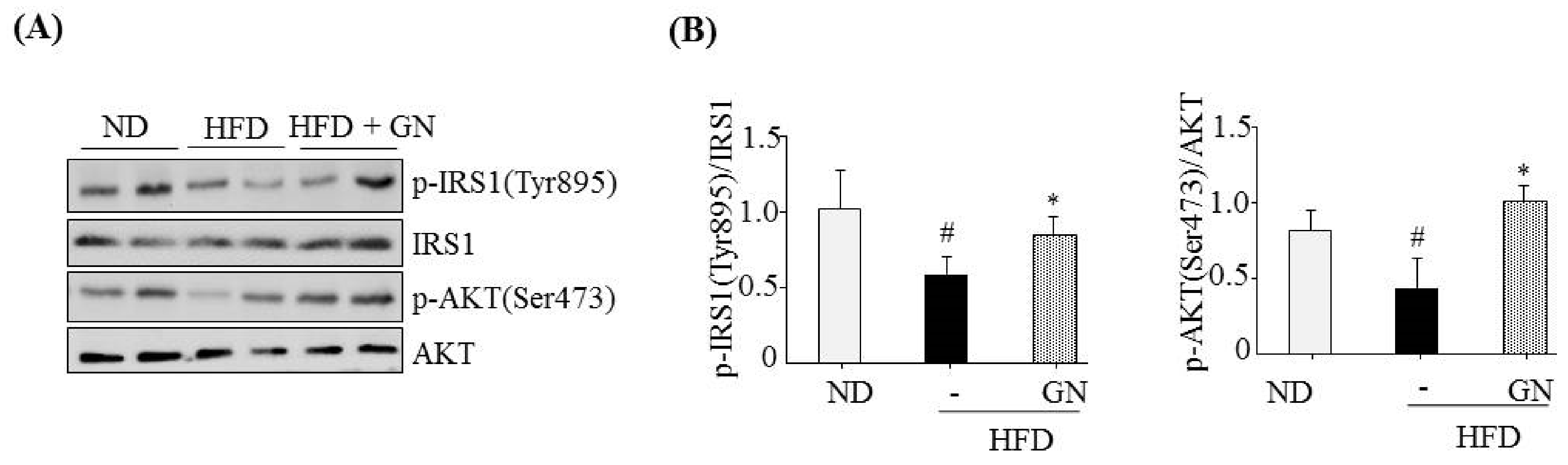
13]. Thus, in the current study, we investigated whether the inhibition of hepatic CB1R signaling plays a role in GN-mediated improvement of hyperglycemia and glucose tolerance. We assessed the inhibitory effects of GN on CB1R-mediated insulin resistance and gluconeogenesis in the liver of HFD obese mice. Results showed that GN administration to HFD obese mice reversed HFD-induced expression of
CB1R, ER stress markers,
PHLPP1, ceramide synthase
CerS6, and recovered reduced phosphorylation of IRS-1 at tyrosine 895 and AKT at serine 473 in the livers of HFD obese mice. Furthermore, GN efficiently reduced HFD-induced expression of gluconeogenesis genes,
PEPCK and
G6Pase. These results indicate that GN can ameliorate HFD-induced hyperglycemia through inhibition of CB1R-induced insulin resistance and gluconeogenesis.
Activation of CB1R signaling in the CNS increases food intake and induces obesity [
15]. Rimonabant, an inverse agonist of central CB1R, was used as an anti-obesity drug [
16]. However, it was withdrawn from the market because of its psychiatric side effects. Since the withdrawal of rimonabant, many investigators have studied peripheral CB1R inhibitors with lesser side effects. Natural products derived from medicinal herbs are usually considered less toxic with fewer side effects. GN is active component of
S. chinensis that has been used as a traditional herbal medicine and has diverse pharmacological activities [
9]. Our results suggest that GN is an attractive and potent compound that inhibits peripheral CB1R signaling and can be useful in the treatment of metabolic disorders including type 2 diabetes.
In conclusion, GN inhibits CB1R-induced ER stress and results in improvement of insulin resistance and gluconeogenesis, which might contribute to the amelioration of hyperglycemia.
4. Materials and Methods
4.1. Reagents
GN (≥98% purity) was obtained from ChemFaces (Wuhan, China). Tunicamycin and 2-AG were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM), penicillin–streptomycin, and fetal bovine serum (FBS) were obtained from Gibco BRL (Grand Island, NY, USA). Antibodies against GRP78, CHOP, and XBP1c were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against p-IRS1 (serine-307), p-IRS1 (tyrosine-895), p-AKT (serine-473), IRS1, and AKT were purchased from Cellular Signaling Technology (Danvers, MA, USA).
4.2. Cell Culture
The human hepatocellular carcinoma cell line HepG2 was obtained from the American Type Culture Collection (Manassas, VA, USA). HepG2 cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 20 U/mL penicillin, and 20 μg/mL streptomycin.
4.3. Quantitative Polymerase Chain Reaction (qPCR)
Total RNA was isolated from HepG2 cells and mouse livers using TRIzol
TM (Invitrogen, Darmstadt, Germany), as per manufacturer’s instructions. One microgram of the isolated RNA was reverse-transcribed by using TOPScript RT DryMix (Enzynomics, Daejeon, Korea). Quantitative real-time PCR was performed using a SYBR Green premixed Taq reaction mixture with gene-specific primers. The gene-specific primers used in this study are listed in
Supplementary Table S1.
4.4. Western Blots
Proteins (40 μg per well) were separated from HepG2 cells using SDS-PAGE on 8% gels and transferred to polyvinylidene fluoride membranes. The membranes were incubated with primary antibodies, followed by incubation with anti-rabbit or anti-mouse secondary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA) and protein bands were visualized using an enhanced chemiluminescence system (ECL Advance, GE Healthcare, Hatfield, UK).
4.5. Triglyceride (TG) Measurement
HepG2 cell suspensions were mixed with 750 μL of chloroform/methanol/H2O (8:4:3, v/v/v) to extract TG. The cell suspensions were incubated at room temperature for 1 h and centrifuged at 800× g for 10 min. The bottom layer (organic phage) obtained was dried overnight and then dissolved in ethanol, followed by measurement of TG concentrations using an AM 157S-K TG kit (Asan Pharmaceutical, Seoul, Korea), which was normalized to the protein concentration.
4.6. Oil Red O Staining
HepG2 were washed twice with phosphate-buffered saline (PBS) and fixed with 10% formalin for 60 min. And then, the cells were then stained with an Oil Red O (ORO) working solution (1.5 mg/mL ORO/60% isopropanol) for 60 min at room temperature. After staining, the cells were washed with distilled water and photographed under a light microscope.
4.7. Glucose Production
Glucose production from HepG2 cells was measured using a colorimetric glucose oxidase assay according to the manufacturer’s protocol (Sigma-Aldrich). Briefly, cells were washed three times with PBS and incubated in glucose production buffer (glucose-free DMEM (pH 7.4), 20 mM sodium lactate, 1 mM sodium pyruvate, and 15 mM HEPES) for 3 h at 37 °C in 5% CO2. Glucose concentration was normalized to cellular protein concentration.
4.8. Animal Study
C57BL/6 mice (male, 6-week-old) were purchased from Jung-Ang Lab Animal, Inc. (Seoul, Korea). The animals were housed in standard conditions of temperature (21–23 °C), humidity (40–60%), and a 12-h light/dark cycle, and were given free access to food and water. The mice were fed a normal diet (ND) or an HFD for 12 weeks. Subsequently, the HFD-fed mice were divided into the following two groups (n = 6 per group): HFD (distilled water-treated) group or HFD + GN (20 mg/kg of body weight) group. The experimental diets were TD.06414, a high-fat in which 60% calories are from fats, and the control diet in which 10% calories are from fat. GN was administered orally every day for 6 weeks. The animal protocol used in this study was reviewed and approved by the Pusan National University’s Institutional Animal Care and Use Committee in accordance with established ethical and scientific care procedures (approval number: PNU-2017–1456; 7 February 2017).
4.9. Biochemical Analysis
After starvation for 12 h, the mice were sacrificed. The blood samples were collected and centrifuged at 1000× g for 15 min at 4 °C to obtain serum, which was stored at −80°C until analysis. The concentrations of blood glucose and insulin were determined by using commercial analysis kits (Asan Pharmaceutical, Seoul, Korea).
4.10. Statistical Analysis
All data are presented as the mean ± standard error of the mean (SEM). The statistical differences between various groups were examined by one-way analysis of variance (ANOVA) followed by Tukey’s test. Values with p < 0.05 were considered statistically significant.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






