CDG Therapies: From Bench to Bedside

 , ,
, ,  ,
,  ,
,
Abstract
:
1. Introduction
2. Disease Models
3. Biomarkers
4. Dietary Supplementation Therapies
4.1. Defects Located in the Cytosol
4.1.1. Defects in Protein N-Glycosylation
MPI-CDG
PMM2-CDG
4.1.2. Defects in Monosacharide/Nucleotide Synthesis
CAD-CDG
GNE-CDG
NANS-CDG
PGM1-CDG
PGM3-CDG
4.2. Defects Located in the Endoplasmic Reticulum (ER)
4.2.1. Defects in Protein N-Glycosylation
ALG1-CDG
ALG13-CDG
MAGT1-CDG
4.2.2. Defects in Lipid Glycosylation and GPI Synthesis
PIGA-CDG
PIGM-CDG
PIGO-CDG
4.3. Defects Located in the Golgi Apparatus
4.3.1. Defects in Nucleotide-Sugar Transporters
SLC35A1-CDG
SLC35A2-CDG
SLC35C1-CDG
4.3.2. Other Defects
TMEM165-CDG
4.4. Defects Located in the ER-Golgi Intermediate Compartment( ERGIC)
4.4.1. Defects in Multiple and Other Glycosylation Pathways
CCDC115-CDG
TMEM199-CDG
4.5. Defects Located at the Plasma Membrane
4.5.1. Defects in Multiple and Other Glycosylation Pathways
SLC39A8-CDG
4.6. Defects Located at the Sarcolemma Membrane
Defects in O-Mannosylglycan Synthesis
ISPD-CDG
5. Other Therapeutic Strategies
5.1. Pharmacological Chaperones
5.2. Antisense Therapy
5.3. Gene Therapy
5.4. Transplantation Options
5.4.1. Liver Transplantation
5.4.2. Heart Transplantation
5.4.3. Cell Transplantation
6. Observational and Interventional Clinical Trials
6.1. Natural History Studies
6.2. Interventional Clinical Trials
6.2.1. GNE-CDG
6.2.2. PGM1-CDG
7. Discussion
8. Methods
- (a)
- Only English-written manuscripts were included;
- (b)
- Articles reporting biomarkers, in vitro and/or in vivo models, compassionate use or clinical trials of therapies in CDG were included;
- (c)
- Only articles reporting CDG with therapies under development (compassionate use, clinical research) or already approved were included;
- (d)
- Reviews were excluded, although we have included some examples for contextualization purposes;
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| α–DG | α-Dystroglycan |
| AAV | Adeno-associated virus |
| Ac4ManNAc | Peracetylated N-acetylmannosamine |
| ACE | Angiotensin-converting enzyme |
| AGA | Aspartylglucosaminidase |
| AMO | Antisense morpholino oligonucleotides |
| ApoC-III | Apolipoprotein C-III |
| ATase | Amidophosphoribosyltransferase |
| ATCase | Aspartate transcarbamoylase |
| BBB | Blood brain barrier |
| CCFDN | Congenital cataracts-facial dysmorphism-neuropathy syndrome |
| CDG | Congenital disorder(s) of glycosylation |
| CDP | Cytidine diphosphate |
| CHO | Chinese hamster ovary |
| CMP | Cytidine monophosphate |
| CNS | Central nervous system |
| CPSase | Carbamoyl phosphate synthetase |
| CPY | Carboxypeptidase Y |
| CTP | Cytosine triphosphate |
| DC | Dilated cardiomyopathy |
| DFCs | Dorsal forerunner cells |
| DHOase | Dihydroorotase |
| DIGE | 2D-Differential gel electrophoresis |
| ER | Endoplasmic reticulum |
| FKTN | Fukutin |
| FKRP | Fukutin related protein |
| Fuc | Fucose |
| GABA | γ-Amino butyric acid |
| Gal | Galactose |
| GalNAc | N-Acetylgalactosamine |
| GALE | UDP-galactose-4-epimerase |
| GALK | Galactokinase |
| GALT | Galactose-1-phosphate uridyl transferase |
| GDP | Guanosine diphosphate |
| Glc | Glucose |
| GlcNAc | N-Acetylglucosamine |
| GPI | Glycosylphosphatidylinositol |
| GSL | Glycosphingolipid |
| HDAC | Histone deacetylase inhibitors |
| HIBM | Hereditary inclusion body myopathy |
| HIFs | Hypoxia inducible factors |
| HIF1α | Hypoxia inducible factor 1-alpha |
| IBM | Inclusion body myopathy |
| ICAM-1 | Intercellular adhesion molecule 1 |
| IEM | Inborn errors of metabolism |
| IM | Intramuscular |
| IMD | Inherited metabolic disease |
| iPSCs | Induced pluripotent stem cells |
| IV | Intravenous |
| KV | Kupffer’s vesicle |
| LLO | Lipid-linked oligosaccharides |
| Magts | Mannoside acetylglucosaminyltransferases |
| Man | Mannose |
| Man-1-P | Mannose-1-phosphate |
| Man-6-P | Mannose-6-phosphate |
| ManN | d-mannosamine |
| ManNAc | N-acetylmannosamine |
| MCAHS2 | Multiple congenital anomalies-hypotonia-seizures syndrome-2 |
| MPG1 | Mannose-1-phosphate guanylyltransferase |
| MPI | Phosphomannose isomerase |
| MPP | Matrix metalloproteinase |
| MS | Mass spectrometry |
| NCAM | Neural cell adhesion molecule |
| NGT | UDP-N-acetylglucosamine transporter |
| PI | Phosphatidylinositol |
| PC | Pharmacological chaperone |
| PMM2 | Phosphomannomutase 2 |
| PUFA | Polyunsaturated fatty acids |
| Rbo | Ribitol |
| SA | Sialic acid |
| TF4 | Thyroxine |
| TS | trans-Splicing |
| TSH | Thyroid stimulating hormone |
| UDP | Uridine diphosphate |
| UGT | UDP-galactose transporter |
| UMPS | Uridine monophosphate synthetase |
| uPAR | Urokinase plasminogen activator receptor |
| UPR | Unfolded protein response |
| UTP | Uridine triphosphate |
| VLC-PUFA | Very long chain polyunsaturated fatty acids |
| Vma3p | 17-kDa Proteolipid subunit of vacuolar ATPase |
| Vma11p | V-Type proton ATPase 16 kDa proteolipid subunit 2 |
| Vph1p | 100 kDa Subunit a of vacuolar-ATPase V0 domain |
| XMEN | X-linked immunodeficiency with magnesium defect, EBV infections and neoplasia |
References
- Péanne, R.; de Lonlay, P.; Foulquier, F.; Kornak, U.; Lefeber, D.J.; Morava, E.; Pérez, B.; Seta, N.; Thiel, C.; Van Schaftingen, E.; et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur. J. Med. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hennet, T. Diseases of glycosylation beyond classical congenital disorders of glycosylation. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 1306–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques-da-Silva, D.; dos Reis Ferreira, V.; Monticelli, M.; Janeiro, P.; Videira, P.A.; Witters, P.; Jaeken, J.; Cassiman, D. Liver involvement in congenital disorders of glycosylation (CDG). A systematic review of the literature. J. Inherit. Metab. Dis. 2017, 40, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Van Scherpenzeel, M.; Willems, E.; Lefeber, D.J. Clinical diagnostics and therapy monitoring in the congenital disorders of glycosylation. Glycoconj. J. 2016, 33, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Péanne, R. What is new in CDG? J. Inherit. Metab. Dis. 2017, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J. Congenital disorders of glycosylation (CDG): It’s (nearly) all in it! J. Inherit. Metab. Dis. 2011, 34, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cerdá, C.; Girós, M.L.; Serrano, M.; Ecay, M.J.; Gort, L.; Pérez Dueñas, B.; Medrano, C.; García-Alix, A.; Artuch, R.; Briones, P.; et al. A Population-Based Study on Congenital Disorders of Protein N- and Combined with O-glycosylation Experience in Clinical and Genetic Diagnosis. J. Pediatr. 2017, 183, 170–177.e1. [Google Scholar] [CrossRef] [PubMed]
- Bakar, N.A.; Lefeber, D.J.; Scherpenzeel, M. Van Clinical glycomics for the diagnosis of congenital disorders of glycosylation. J. Inherit. Metab. Dis. 2018. [Google Scholar] [CrossRef]
- Marques-da-Silva, D.; Francisco, R.; Webster, D.; dos Reis Ferreira, V.; Jaeken, J.; Pulinilkunnil, T. Cardiac complications of congenital disorders of glycosylation (CDG): A systematic review of the literature. J. Inherit. Metab. Dis. 2017, 40, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Francisco, R.; Pascoal, C.; Marques-da-Silva, D.; Morava, E.; Gole, G.A.; Coman, D.; Jaeken, J.; Dos Reis Ferreira, V. Keeping an eye on congenital disorders of O-glycosylation: A systematic literature review. J. Inherit. Metab. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Timal, S.; Hoischen, A.; Lehle, L.; Adamowicz, M.; Huijben, K.; Sykut-cegielska, J.; Paprocka, J.; Jamroz, E.; Van spronsen, F.J.; Körner, C.; et al. Gene identification in the congenital disorders of glycosylation type i by whole-exome sequencing. Hum. Mol. Genet. 2012, 21, 4151–4161. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Cuéllar, G.; Gauthier, J.; Désilets, V.; Lachance, C.; Lemire-Girard, M.; Rypens, F.; Le Deist, F.; Decaluwe, H.; Duval, M.; Bouron-Dal Soglio, D.; et al. A Novel PGM3 Mutation Is Associated With a Severe Phenotype of Bone Marrow Failure, Severe Combined Immunodeficiency, Skeletal Dysplasia, and Congenital Malformations. J. Bone Miner. Res. 2017, 32, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Hennet, T.; Matthijs, G.; Freeze, H.H. CDG nomenclature: Time for a change! Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 825–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, C.; Körner, C. Therapies and therapeutic approaches in Congenital Disorders of Glycosylation. Glycoconj. J. 2013, 30, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Witters, P.; Cassiman, D.; Morava, E. Nutritional therapies in congenital disorders of glycosylation (CDG). Nutrients 2017, 9, 1222. [Google Scholar] [CrossRef] [PubMed]
- Thiel, C.; Körner, C. Mouse models for congenital disorders of glycosylation. J. Inherit. Metab. Dis. 2011, 34, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Deutscher, S.L.; Nuwayhid, N.; Stanley, P.; Briles, E.I.B.; Hirschberg, C.B. Translocation across golgi vesicle membranes: A CHO glycosylation mutant deficient in CMP-sialic acid transport. Cell 1984, 39, 295–299. [Google Scholar] [CrossRef]
- Fernandez, F.; Shridas, P.; Jiang, S.; Aebi, M.; Waechter, C.J. Expression and characterization of a human cDNA that complements the temperature-sensitive defect in dolichol kinase activity in the yeast sec59-1 mutant: The enzymatic phosphorylation of dolichol and diacylglycerol are catalyzed by separate CTP-mediated. Glycobiology 2002, 12, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Stanley, P. Lec3 Chinese Hamster Ovary Mutants Lack UDP-N-acetylglucosamine 2-Epimerase Activity because of Mutations in the Epimerase Domain of the Gne Gene. J. Biol. Chem. 2003, 278, 53045–53054. [Google Scholar] [CrossRef] [PubMed]
- Maszczak-Seneczko, D.; Sosicka, P.; Majkowski, M.; Olczak, T.; Olczak, M. UDP-N-acetylglucosamine transporter and UDP-galactose transporter form heterologous complexes in the Golgi membrane. FEBS Lett. 2012, 586, 4082–4087. [Google Scholar] [CrossRef] [PubMed]
- Maszczak-Seneczko, D.; Sosicka, P.; Kaczmarek, B.; Majkowski, M.; Luzarowski, M.; Olczak, T.; Olczak, M. UDP-galactose (SLC35A2) and UDP-N-acetylglucosamine (SLC35A3) transporters form glycosylation-related complexes with mannoside acetylglucosaminyltransferases (Mgats). J. Biol. Chem. 2015, 290, 15475–15486. [Google Scholar] [CrossRef] [PubMed]
- Lübke, T.; Marquardt, T.; Etzioni, A.; Hartmann, E.; Von Figura, K.; Körner, C. Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nat. Genet. 2001, 28, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, A. Insights into leukocyte adhesion deficiency type 2 from a novel mutation in the GDP-fucose transporter gene. Blood 2003, 101, 1705–1712. [Google Scholar] [CrossRef] [PubMed]
- Grubenmann, C.E. Deficiency of the first mannosylation step in the N-glycosylation pathway causes congenital disorder of glycosylation type Ik. Hum. Mol. Genet. 2004, 13, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Duncker, I.; Dupré, T.; Piller, V.; Piller, F.; Candelier, J.J.; Trichet, C.; Tchernia, G.; Oriol, R.; Mollicone, R. Genetic complementation reveals a novel human congenital disorder of glycosylation of type II, due to inactivation of the Golgi CMP-sialic acid transporter. Blood 2005, 105, 2671–2676. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-J.; Kang, J.Y.; Kim, Y.U.; Yoon, J.S.; Choy, H.E.; Maeda, Y.; Kinoshita, T.; Hong, Y. Isolation of new CHO cell mutants defective in CMP-sialic acid biosynthesis and transport. Mol. Cells 2006, 22, 343–352. [Google Scholar] [PubMed]
- Helmus, Y.; Denecke, J.; Yakubenia, S.; Robinson, P.; Lühn, K.; Watson, D.L.; McGrogan, P.J.; Vestweber, D.; Marquardt, T.; Wild, M.K. Leukocyte adhesion deficiency II patients with a dual defect of the GDP-fucose transporter. Blood 2006, 107, 3959–3966. [Google Scholar] [CrossRef] [PubMed]
- Jay, C.; Nemunaitis, G.; Nemunaitis, J.; Senzer, N.; Hinderlich, S.; Darvish, D.; Ogden, J.; Eager, J.; Tong, A.; Maples, P.B. Preclinical assessment of wt GNE gene plasmid for management of hereditary inclusion body myopathy 2 (HIBM2). Gene Regul. Syst. Biol. 2008, 2008, 243–252. [Google Scholar] [CrossRef]
- Lefeber, D.J.; de Brouwer, A.P.M.; Morava, E.; Riemersma, M.; Schuurs-Hoeijmakers, J.H.M.; Absmanner, B.; Verrijp, K.; van den Akker, W.M.R.; Huijben, K.; Steenbergen, G.; et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet. 2011, 7, e1002427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosnoblet, C.; Legrand, D.; Demaegd, D.; Hacine-Gherbi, H.; De Bettignies, G.; Bammens, R.; Borrego, C.; Duvet, S.; Morsomme, P.; Matthijs, G.; et al. Impact of disease-causing mutations on TMEM165 subcellular localization, a recently identified protein involved in CDG-II. Hum. Mol. Genet. 2013, 22, 2914–2928. [Google Scholar] [CrossRef] [PubMed]
- Rohlfing, A.K.; Rust, S.; Reunert, J.; Tirre, M.; Du Chesne, I.; Wemhoff, S.; Meinhardt, F.; Hartmann, H.; Das, A.M.; Marquardt, T. ALG1-CDG: A new case with early fatal outcome. Gene 2014, 534, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Riemersma, M.; Froese, D.S.; Van Tol, W.; Engelke, U.F.; Kopec, J.; Van Scherpenzeel, M.; Ashikov, A.; Krojer, T.; Von Delft, F.; Tessari, M.; et al. Human ISPD Is a Cytidyltransferase Required for Dystroglycan O-Mannosylation. Chem. Biol. 2015, 22, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Dörre, K.; Olczak, M.; Wada, Y.; Sosicka, P.; Grüneberg, M.; Reunert, J.; Kurlemann, G.; Fiedler, B.; Biskup, S.; Hörtnagel, K.; et al. A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): Molecular basis, clinical phenotype, and therapeutic approach. J. Inherit. Metab. Dis. 2015, 38, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.G.; Shiryaev, S.A.; Rymen, D.; Eklund, E.A.; Raymond, K.; Kircher, M.; Abdenur, J.E.; Alehan, F.; Midro, A.T.; Bamshad, M.J.; et al. ALG1-CDG: Clinical and Molecular Characterization of 39 Unreported Patients. Hum. Mutat. 2016, 37, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, J.; Mimatsu, H.; Mizuno, S.; Okamoto, N.; Fukushi, D.; Tominaga, K.; Kidokoro, H.; Muramatsu, Y.; Nishi, E.; Nakamura, S.; et al. Phenotype–genotype correlations of PIGO deficiency with variable phenotypes from infantile lethality to mild learning difficulties. Hum. Mutat. 2017, 38, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Potelle, S.; Dulary, E.; Climer, L.; Duvet, S.; Morelle, W.; Vicogne, D.; Lebredonchel, E.; Houdou, M.; Spriet, C.; Krzewinski-Recchi, M.-A.; et al. Manganese-induced turnover of TMEM165. Biochem. J. 2017, 474, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Krawitz, P.M.; Murakami, Y.; Hecht, J.; Krüger, U.; Holder, S.E.; Mortier, G.R.; Delle Chiaie, B.; De Baere, E.; Thompson, M.D.; Roscioli, T.; et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 2012, 91, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.; Guillard, M.; Schmidt, S.; Gerardy-schahn, R.; Deen, P.M.T.; Wevers, R.A.; Lefeber, D.J.; Morava, E. Intellectual disability and bleeding diathesis due to deficient CMP—Sialic acid transport. Neurology 2013, 81, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.; Arya, R. Role of UDP-N-acetylglucosamine2-Epimerase/N-acetylmannosamine Kinase (GNE) in β1-Integrin-Mediated Cell Adhesion. Mol. Neurobiol. 2014, 50, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Hinderlich, S.; Salama, I.; Eisenberg, I.; Potikha, T.; Mantey, L.R.; Yarema, K.J.; Horstkorte, R.; Argov, Z.; Sadeh, M.; Reutter, W.; et al. The homozygous M712T mutation of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase results in reduced enzyme activities but not in altered overall cellular sialylation in hereditary inclusion body myopathy. FEBS Lett. 2004, 566, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Janik, A.; Sosnowska, M.; Kruszewska, J.; Krotkiewski, H.; Lehle, L.; Palamarczyk, G. Overexpression of GDP-mannose pyrophosphorylase in Saccharomyces cerevisiae corrects defects in dolichol-linked saccharide formation and protein glycosylation. Biochim. Biophys. Acta Gen. Subj. 2003, 1621, 22–30. [Google Scholar] [CrossRef]
- Stray-Pedersen, A.; Backe, P.H.; Sorte, H.S.; Mørkrid, L.; Chokshi, N.Y.; Erichsen, H.C.; Gambin, T.; Elgstøen, K.B.P.; Bjørås, M.; Wlodarski, M.W.; et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am. J. Hum. Genet. 2014, 95, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Lundin, K.E.; Hamasy, A.; Backe, P.H.; Moens, L.N.; Falk-Sörqvist, E.; Elgstøen, K.B.; Mørkrid, L.; Bjørås, M.; Granert, C.; Norlin, A.C.; et al. Susceptibility to infections, without concomitant hyper-IgE, reported in 1976, is caused by hypomorphic mutation in the phosphoglucomutase 3 (PGM3) gene. Clin. Immunol. 2015, 161, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-T.; Wang, N.; Xu, S.; Yin, J.; Nakanishi, H.; Dean, N.; Gao, X.-D. Quantitative study of yeast Alg1 β-1,4 mannosyltransferase activity, a key enzyme involved in protein N-glycosylation. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2934–2941. [Google Scholar] [CrossRef] [PubMed]
- Seppala, R.; Tietze, F.; Krasnewich, D.; Weiss, P.; Ashwell, G.; Barsh, G.; Thomas, G.H.; Packman, S.; Gahl, W.A. Sialic acid metabolism in sialuria fibroblasts. J. Biol. Chem. 1991, 266, 7456–7461. [Google Scholar] [PubMed]
- Panneerselvam, K.; Freeze, H.H. Mannose corrects altered N-glycosylation in carbohydrate-deficient glycoprotein syndrome fibroblasts. J. Clin. Investig. 1996, 97, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, K.; Etchison, J.R.; Skovby, F.; Freeze, H.H. Abnormal metabolism of mannose in families with carbohydrate-deficient glycoprotein syndrome Type 1. Biochem. Mol. Med. 1997, 61, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, T.; Fukushima, K.; Kurisaki, A.; Sagami, H.; Ogura, K.; Ohno, K.; Hara-Kuge, S.; Yamashita, K. A partial deficiency of dehydrodolichol reduction is a cause of carbohydrate-deficient glycoprotein syndrome type I. J. Biol. Chem. 1997, 272, 6868–6875. [Google Scholar] [CrossRef] [PubMed]
- Niehues, R.; Hasilik, M.; Alton, G.; Körner, C.; Schiebe-Sukumar, M.; Koch, H.G.; Zimmer, K.P.; Wu, R.; Harms, E.; Reiter, K.; et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J. Clin. Investig. 1998, 101, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Karsan, A.; Cornejo, C.J.; Winn, R.K.; Schwartz, B.R.; Way, W.; Lannir, N.; Gershoni-Baruch, R.; Etzioni, A.; Ochs, H.D.; Harlan, J.M. Leukocyte adhesion deficiency type II is a generalized defect of de novo GDP-fucose biosynthesis: Endothelial cell fucosylation is not required for neutrophil rolling on human nonlymphoid endothelium. J. Clin. Investig. 1998, 101, 2438–2445. [Google Scholar] [CrossRef] [PubMed]
- Lübke, T.; Marquardt, T.; Von Figura, K.; Körner, C. A new type of carbohydrate-deficient glycoprotein syndrome due to a decreased import of GDP-fucose into the Golgi. J. Biol. Chem. 1999, 274, 25986–25989. [Google Scholar] [CrossRef] [PubMed]
- Rush, J.S.; Panneerselvam, K.; Waechter, C.J.; Freeze, H.H. Mannose supplementation corrects GDP-mannose deficiency in cultured fibroblasts from some patients with Congenital Disorders of Glycosylation (CDG). Glycobiology 2000, 10, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, T.; Lühn, K.; Srikrishna, G.; Freeze, H.H.; Harms, E.; Vestweber, D. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood 1999, 94, 3976–3985. [Google Scholar] [PubMed]
- Lühn, K.; Wild, M.K.; Eckhardt, M.; Gerardy-Schahn, R.; Vestweber, D. The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat. Genet. 2001, 28, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Sturla, L.; Puglielli, L.; Tonetti, M.; Berninsone, P.; Hirschberg, C.B.; de Flora, A.; Etzioni, A. Impairment of the Golgi GDP-L-fucose transport and unresponsiveness to fucose replacement therapy in LAD II patients. Pediatr. Res. 2001, 49, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Sparks, S.E.; Ciccone, C.; Lalor, M.; Orvisky, E.; Klootwijk, R.; Savelkoul, P.J.; Dalakas, M.C.; Krasnewich, D.M.; Gahl, W.A.; Huizing, M. Use of a cell-free system to determine UDP-N-acetylglucosamine 2-epimerase and N-acetylmannosamine kinase activities in human hereditary inclusion body myopathy. Glycobiology 2005, 15, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.M.; Murakami, Y.; Baker, A.; Maeda, Y.; Roberts, I.A.G.; Kinoshita, T.; Layton, D.M.; Karadimitris, A. Targeted Therapy for Inherited GPI Deficiency. N. Engl. J. Med. 2007, 356, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hou, X.; Shi, S.; Körner, C.; Stanley, P. Slc35c2 promotes Notch1 fucosylation and is required for optimal Notch signaling in mammalian cells. J. Biol. Chem. 2010, 285, 36245–36254. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Ichikawa, M.; He, P.; Bravo, Y.; Dahl, R.; Ng, B.G.; Cosford, N.D.P.; Freeze, H.H. Phosphomannose isomerase inhibitors improve N-glycosylation in selected phosphomannomutase-deficient fibroblasts. J. Biol. Chem. 2011, 286, 39431–39438. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-Y.; Lenardo, M.J.; Chaigne-Delalande, B. Loss of MAGT1 abrogates the Mg2+ flux required for T cell signaling and leads to a novel human primary immunodeficiency. Magnes. Res. 2011, 24, S109–S114. [Google Scholar] [CrossRef] [PubMed]
- Mitrani-Rosenbaum, S.; Yakovlev, L.; Becker Cohen, M.; Telem, M.; Elbaz, M.; Yanay, N.; Yotvat, H.; Ben Shlomo, U.; Harazi, A.; Fellig, Y.; et al. Sustained expression and safety of human GNE in normal mice after gene transfer based on AAV8 systemic delivery. Neuromuscul. Disord. 2012, 22, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, Q.; Liu, F.; Zhang, X.; Li, W.; Liu, S.; Zhao, Y.; Gong, Y.; Yan, C. Unfolded Protein Response and Activated Degradative Pathways Regulation in GNE Myopathy. PLoS ONE 2013, 8, e58116. [Google Scholar] [CrossRef] [PubMed]
- Chaigne-Delalande, B.; Li, F.-Y.; O’Connor, G.M.; Lukacs, M.J.; Jiang, P.; Zheng, L.; Shatzer, A.; Biancalana, M.; Pittaluga, S.; Matthews, H.F.; et al. Mg2+ Regulates Cytotoxic Functions of NK and CD8 T Cells in Chronic EBV Infection Through NKG2D. Science 2013, 341, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.R.; Caputo, V.S.; Makarona, K.; Layton, D.M.; Roberts, I.A.G.; Almeida, A.M.; Karadimitris, A. Cell-type-specific transcriptional regulation of PIGM underpins the divergent hematologic phenotype in inherited GPl deficiency. Blood 2014, 124, 3151–3154. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, L.C.; Rust, S.; van Scherpenzeel, M.; Ng, B.G.; Losfeld, M.-E.; Timal, S.; Raymond, K.; He, P.; Ichikawa, M.; Veltman, J.; et al. Multiple Phenotypes in Phosphoglucomutase 1 Deficiency. N. Engl. J. Med. 2014, 370, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patzel, K.A.; Yardeni, T.; Le Poëc-Celic, E.; Leoyklang, P.; Dorward, H.; Alonzi, D.S.; Kukushkin, N.V.; Xu, B.; Zhang, Y.; Sollogoub, M.; et al. Non-specific accumulation of glycosphingolipids in GNE myopathy. J. Inherit. Metab. Dis. 2014, 37, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, X.; Ichikawa, M.; Lyons, J.J.; Datta, S.; Lamborn, I.T.; Jing, H.; Kim, E.S.; Biancalana, M.; Wolfe, L.A.; et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J. Allergy Clin. Immunol. 2014, 133, 1400–1409.e5. [Google Scholar] [CrossRef] [PubMed]
- Gerin, I.; Ury, B.; Breloy, I.; Bouchet-Seraphin, C.; Bolsée, J.; Halbout, M.; Graff, J.; Vertommen, D.; Muccioli, G.G.; Seta, N.; et al. ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto α-dystroglycan. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Mayr, J.A.; Alhaddad, B.; Rauscher, C.; Bierau, J.; Kovacs-Nagy, R.; Coene, K.L.M.; Bader, I.; Holzhacker, M.; Prokisch, H.; et al. CAD mutations and uridine-responsive epileptic encephalopathy. Brain 2017, 140, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Morelle, W.; Potelle, S.; Witters, P.; Wong, S.; Climer, L.; Lupashin, V.; Matthijs, G.; Gadomski, T.; Jaeken, J.; Cassiman, D.; et al. Galactose supplementation in patients with TMEM165-CDG rescues the glycosylation defects. J. Clin. Endocrinol. Metab. 2017, 102, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.G.; Asteggiano, C.G.; Kircher, M.; Buckingham, K.J.; Raymond, K.; Nickerson, D.A.; Shendure, J.; Bamshad, M.J.; Ensslen, M.; Freeze, H.H. Encephalopathy caused by novel mutations in the CMP-sialic acid transporter, SLC35A1. Am. J. Med. Genet. Part A 2017, 173, 2906–2911. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Nayak, J.; DeRossi, C.; Charbono, A.; Ichikawa, M.; Ng, B.G.; Grajales-Esquivel, E.; Srivastava, A.; Wang, L.; He, P.; et al. Mannose supplements induce embryonic lethality and blindness in phosphomannose isomerase hypomorphic mice. FASEB J. 2014, 28, 1854–1869. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, G.; Amiji, M. Use of CRISPR/ Cas9 gene-editing tools for developing models in drug discovery. Drug Discov. Today 2018. [Google Scholar] [CrossRef] [PubMed]
- Schwarzkopf, M.; Knobeloch, K.-P.; Rohde, E.; Hinderlich, S.; Wiechens, N.; Lucka, L.; Horak, I.; Reutter, W.; Horstkorte, R. Sialylation is essential for early development in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 5267–5270. [Google Scholar] [CrossRef] [PubMed]
- Thiel, C.; Lubke, T.; Matthijs, G.; von Figura, K.; Korner, C. Targeted Disruption of the Mouse Phosphomannomutase 2 Gene Causes Early Embryonic Lethality. Mol. Cell. Biol. 2006, 26, 5615–5620. [Google Scholar] [CrossRef] [PubMed]
- Sela, I.; Yakovlev, L.; Becker Cohen, M.; Elbaz, M.; Yanay, N.; Ben Shlomo, U.; Yotvat, H.; Fellig, Y.; Argov, Z.; Mitrani-Rosenbaum, S. Variable phenotypes of knockin mice carrying the M712T Gne mutation. NeuroMol. Med. 2013, 15, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Thiel, C.; Rindermann, J.; Derossi, C.; Popovici, D.; Hoffmann, G.F.; Gröne, H.J.; Körner, C. Successful prenatal mannose treatment for congenital disorder of glycosylation-Ia in mice. Nat. Med. 2012, 18, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Niethamer, T.K.; Yardeni, T.; Leoyklang, P.; Ciccone, C.; Astiz-Martinez, A.; Jacobs, K.; Dorward, H.M.; Zerfas, P.M.; Gahl, W.A.; Huizing, M. Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Mol. Genet. Metab. 2012, 107, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Sugihara, K.; Asaka, T.; Toyama, T.; Yoshihara, T.; Furuichi, K.; Wada, T.; Asano, M. Glycoprotein hyposialylation gives rise to a nephrotic-like syndrome that is prevented by sialic acid administration in GNE V572L point-mutant mice. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Yardeni, T.; Jacobs, K.; Niethamer, T.K.; Ciccone, C.; Anikster, Y.; Kurochkina, N.; Gahl, W.A.; Huizing, M. Murine isoforms of UDP-GlcNAc 2-epimerase/ManNAc kinase: Secondary structures, expression profiles, and response to ManNAc therapy. Glycoconj. J. 2013, 30, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Galeano, B.; Klootwijk, R.; Manoli, I.; Sun, M.; Ciccone, C.; Darvish, D.; Starost, M.F.; Zerfas, P.M.; Hoffmann, V.J.; Hoogstraten-Miller, S.; et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J. Clin. Investig. 2007, 117, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Malicdan, M.C.V.; Noguchi, S.; Hayashi, Y.K.; Nonaka, I.; Nishino, I. Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat. Med. 2009, 15, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Malicdan, M.C.V.; Noguchi, S.; Tokutomi, T.; Goto, Y.I.; Nonaka, I.; Hayashi, Y.K.; Nishino, I. Peracetylated N-acetylmannosamine, a synthetic sugar molecule, efficiently rescues muscle phenotype and biochemical defects in mouse model of sialic acid-deficient myopathy. J. Biol. Chem. 2012, 287, 2689–2705. [Google Scholar] [CrossRef] [PubMed]
- Yonekawa, T.; Malicdan, M.C.V.; Cho, A.; Hayashi, Y.K.; Nonaka, I.; Mine, T.; Yamamoto, T.; Nishino, I.; Noguchi, S. Sialyllactose ameliorates myopathic phenotypes in symptomatic GNE myopathy model mice. Brain 2014, 137, 2670–2679. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef] [PubMed]
- The Jackson Laboratory. Available online: https://www.jax.org/ (accessed on 18 January 2018).
- Runge, K.W.; Huffaker, T.C.; Robbins, P.W. Two yeast mutations in glucosylation steps of the asparagine glycosylation pathway. J. Biol. Chem. 1984, 259, 412–417. [Google Scholar] [PubMed]
- Uchimura, S.; Sugiyama, M.; Nikawa, J. Effects of N-glycosylation and Inositol on the ER Stress Response in Yeast Saccharomyces cerevisiae. Biosci. Biotechnol. Biochem. 2005, 69, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Shrimal, S.; Gilmore, R. Reduced expression of the oligosaccharyltransferase exacerbates protein hypoglycosylation in cells lacking the fully assembled oligosaccharide donor. Glycobiology 2015, 25, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Clapham, D.E. Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 15750–15755. [Google Scholar] [CrossRef] [PubMed]
- DeRossi, C.; Bode, L.; Eklund, E.A.; Zhang, F.; Davis, J.A.; Westphal, V.; Wang, L.; Borowsky, A.D.; Freeze, H.H. Ablation of mouse phosphomannose isomerase (Mpi) causes mannose 6-phosphate accumulation, toxicity, and embryonic lethality. J. Biol. Chem. 2006, 281, 5916–5927. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Mir, A.; Gao, N.; Rosa, S.; Monson, C.; Sharma, V.; Steet, R.; Freeze, H.H.; Lehrman, M.A.; Sadler, K.C. A zebrafish model of congenital disorders of glycosylation with phosphomannose isomerase deficiency reveals an early opportunity for corrective mannose supplementation. Dis. Model. Mech. 2013, 6, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Thiesler, C.T.; Cajic, S.; Hoffmann, D.; Thiel, C.; van Diepen, L.; Hennig, R.; Sgodda, M.; Weiβmann, R.; Reichl, U.; Steinemann, D.; et al. Glycomic Characterization of Induced Pluripotent Stem Cells Derived from a Patient Suffering from Phosphomannomutase 2 Congenital Disorder of Glycosylation (PMM2-CDG). Mol. Cell. Proteom. 2016, 15, 1435–1452. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.; Clasquin, M.; Smolen, G.A.; Histen, G.; Powe, J.; Chen, Y.; Lin, Z.; Lu, C.; Liu, Y.; Cang, Y.; et al. A mouse model of a human congenital disorder of glycosylation caused by loss of PMM2. Hum. Mol. Genet. 2016, 25, 2182–2193. [Google Scholar] [CrossRef] [PubMed]
- Cline, A.; Gao, N.; Flanagan-Steet, H.; Sharma, V.; Rosa, S.; Sonon, R.; Azadi, P.; Sadler, K.C.; Freeze, H.H.; Lehrman, M.A.; et al. A zebrafish model of PMM2-CDG reveals altered neurogenesis and a substrate-accumulation mechanism for N-linked glycosylation deficiency. Mol. Biol. Cell 2012, 23, 4175–4187. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, W.M.; Dookwah, M.; Dear, M.L.; Gatto, C.L.; Aoki, K.; Tiemeyer, M.; Broadie, K. Synaptic roles for phosphomannomutase type 2 in a new Drosophila congenital disorder of glycosylation disease model. Dis. Model. Mech. 2016, 9, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Jansen, E.J.R.; Timal, S.; Ryan, M.; Ashikov, A.; Van Scherpenzeel, M.; Graham, L.A.; Mandel, H.; Hoischen, A.; Iancu, T.C.; Raymond, K.; et al. ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokey, J.J.; Dasgupta, A.; Amack, J.D. The V-ATPase accessory protein Atp6ap1b mediates dorsal forerunner cell proliferation and left–right asymmetry in zebrafish. Dev. Biol. 2015, 407, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Schoonderwoert, V.T.G.; Martens, G.J.M. Targeted disruption of the mouse gene encoding the V-ATPase accessory subunit Ac45. Mol. Membr. Biol. 2002, 19, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.G.; Wolfe, L.A.; Ichikawa, M.; Markello, T.; He, M.; Tifft, C.J.; Gahl, W.A.; Freeze, H.H. Biallelic mutations in CAD, impair de novo pyrimidine biosynthesis and decrease glycosylation precursors. Hum. Mol. Genet. 2015, 24, 3050–3057. [Google Scholar] [CrossRef] [PubMed]
- Franks, D.M.; Izumikawa, T.; Kitagawa, H.; Sugahara, K.; Okkema, P.G.C. elegans pharyngeal morphogenesis requires both de novo synthesis of pyrimidines and synthesis of heparan sulfate proteoglycans. Dev. Biol. 2006, 296, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Norby, S. A specific nutritional requirement for pyrimidines in rudimentary mutants of Drosophila melanogaster. Hereditas 1970, 66, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Willer, G.B.; Lee, V.M.; Gregg, R.G.; Link, B.A. Analysis of the zebrafish perplexed mutation reveals tissue-specific roles for de novo pyrimidine synthesis during development. Genetics 2005, 170, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.A.; LaMora, A.; Johnson, S.L.; Voigt, M.M. Novel role for carbamoyl phosphate synthetase 2 in cranial sensory circuit formation. Int. J. Dev. Neurosci. 2014, 33, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.J.; Stevens, T.H. Vma22p is a novel endoplasmic reticulum-associated protein required for assembly o fthe yeast vacuolar H(+)-ATPase complex. J. Biol. Chem. 1995, 270, 22329–22336. [Google Scholar] [CrossRef] [PubMed]
- Miles, A.L.; Burr, S.P.; Grice, G.L.; Nathan, J.A. The vacuolar-ATPase complex and assembly factors, TMEM199 and CCDC115, control HIF1α prolyl hydroxylation by regulating cellular Iron levels. eLife 2017, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Bork, K.; Reutter, W.; Weidemann, W.; Horstkorte, R. Enhanced sialylation of EPO by overexpression of UDP-GlcNAc 2-epimerase/ManAc kinase containing a sialuria mutation in CHO cells. FEBS Lett. 2007, 581, 4195–4198. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.D.; Jeong, Y.T.; Park, S.Y.; Kim, J.H. Enhanced sialylation of recombinant human erythropoietin in Chinese hamster ovary cells by combinatorial engineering of selected genes. Glycobiology 2011, 21, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Möller, H.; Böhrsch, V.; Lucka, L.; Hackenberger, C.P.R.; Hinderlich, S. Efficient metabolic oligosaccharide engineering of glycoproteins by UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) knock-down. Mol. Biosyst. 2011, 7, 2245–2251. [Google Scholar] [CrossRef] [PubMed]
- Keppler, O.T.; Hinderlich, S.; Langner, J.; Schwartz-Albiez, R.; Reutter, W.; Pawlita, M. UDP-GlcNAc 2-Epimerase: A regulator of cell surface sialylation. Science 1999, 284, 1372–1376. [Google Scholar] [CrossRef] [PubMed]
- Mantey, L.R.; Keppler, O.T.; Pawlita, M.; Reutter, W.; Hinderlich, S. Efficient biochemical engineering of cellular sialic acids using an unphysiological sialic acid precursor in cells lacking UDP-N-acetylglucosamine 2-epimerase. FEBS Lett. 2001, 503, 80–84. [Google Scholar] [CrossRef]
- Krause, S.; Hinderlich, S.; Amsili, S.; Horstkorte, R.; Wiendl, H.; Argov, Z.; Mitrani-Rosenbaum, S.; Lochmüller, H. Localization of UDP-GlcNAc 2-epimerase/ManAc kinase (GNE) in the Golgi complex and the nucleus of mammalian cells. Exp. Cell Res. 2005, 304, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, W.; Klukas, C.; Klein, A.; Simm, A.; Schreiber, F.; Horstkorte, R. Lessons from GNE-deficient embryonic stem cells: Sialic acid biosynthesis is involved in proliferation and gene expression. Glycobiology 2010, 20, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Milman Krentsis, I.; Sela, I.; Eiges, R.; Blanchard, V.; Berger, M.; Becker Cohen, M.; Mitrani-Rosenbaum, S. GNE is involved in the early development of skeletal and cardiac muscle. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, W.; Hering, J.; Bennmann, D.; Thate, A.; Horstkorte, R. The key enzyme of the sialic acid metabolism is involved in embryoid body formation and expression of marker genes of germ layer formation. Int. J. Mol. Sci. 2013, 14, 20555–20563. [Google Scholar] [CrossRef] [PubMed]
- Gagiannis, D.; Orthmann, A.; Danßmann, I.; Schwarzkopf, M.; Weidemann, W.; Horstkorte, R. Reduced sialylation status in UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase (GNE)-deficient mice. Glycoconj. J. 2007, 24, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Kakani, S.; Yardeni, T.; Poling, J.; Ciccone, C.; Niethamer, T.; Klootwijk, E.D.; Manoli, I.; Darvish, D.; Hoogstraten-Miller, S.; Zerfas, P.; et al. The Gne M712T mouse as a model for human glomerulopathy. Am. J. Pathol. 2012, 180, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Malicdan, M.C.V.; Noguchi, S.; Nonaka, I.; Hayashi, Y.K.; Nishino, I. A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum. Mol. Genet. 2007, 16, 2669–2682. [Google Scholar] [CrossRef] [PubMed]
- Anada, R.P.; Wong, K.T.; Malicdan, M.C.; Goh, K.J.; Hayashi, Y.; Nishino, I.; Noguchi, S. Absence of β-amyloid deposition in the central nervous system of a transgenic mouse model of distal myopathy with rimmed vacuoles. Amyloid 2014, 21, 138–139. [Google Scholar] [CrossRef] [PubMed]
- Kreuzmann, D.; Horstkorte, R.; Kohla, G.; Kannicht, C.; Bennmann, D.; Thate, A.; Bork, K. Increased Polysialylation of the Neural Cell Adhesion Molecule in a Transgenic Mouse Model of Sialuria. ChemBioChem 2017, 18, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Daya, A.; Vatine, G.D.; Becker-Cohen, M.; Tal-Goldberg, T.; Friedmann, A.; Gothilf, Y.; Du, S.J.; Mitrani-Rosenbaum, S. Gne depletion during zebrafish development impairs skeletal muscle structure and function. Hum. Mol. Genet. 2014, 23, 3350–3361. [Google Scholar] [CrossRef] [PubMed]
- Van Karnebeek, C.D.M.; Bonafé, L.; Wen, X.Y.; Tarailo-Graovac, M.; Balzano, S.; Royer-Bertrand, B.; Ashikov, A.; Garavelli, L.; Mammi, I.; Turolla, L.; et al. NANS-mediated synthesis of sialic acid is required for brain and skeletal development. Nat. Genet. 2016, 48, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Inagaki, N.; Hayashi, T.; Shichi, D.; Sato, A.; Hinohara, K.; Vatta, M.; Towbin, J.A.; Chikamori, T.; Yamashina, A.; et al. Impaired binding of ZASP/Cypher with phosphoglucomutase 1 is associated with dilated cardiomyopathy. Cardiovasc. Res. 2009, 83, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Greig, K.T.; Antonchuk, J.; Metcalf, D.; Morgan, P.O.; Krebs, D.L.; Zhang, J.-G.; Hacking, D.F.; Bode, L.; Robb, L.; Kranz, C.; et al. Agm1/Pgm3-Mediated Sugar Nucleotide Synthesis Is Essential for Hematopoiesis and Development. Mol. Cell. Biol. 2007, 27, 5849–5859. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, S.K.; Stanley, P. Mouse large can modify complex N- and mucin O-glycans on α-dystroglycan to induce laminin binding. J. Biol. Chem. 2005, 280, 20851–20859. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.F.; Lee, M.M.; Zhang, P.; Song, Z. The golgi CMP-sialic acid transporter: A new CHO mutant provides functional insights. Glycobiology 2008, 18, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Cipollo, J.F.; Awad, A.M.; Costello, C.E.; Hirschberg, C.B. srf-3, a mutant of Caenorhabditis elegans, resistant to bacterial infection and to biofilm binding, is deficient in glycoconjugates. J. Biol. Chem. 2004, 279, 52893–52903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Haryadi, R.; Chan, K.F.; Teo, G.; Goh, J.; Pereira, N.A.; Feng, H.; Song, Z. Identification of functional elements of the GDP-fucose transporter SLC35C1 using a novel Chinese hamster ovary mutant. Glycobiology 2012, 22, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Haryadi, R.; Zhang, P.; Chan, K.F.; Song, Z. CHO-gmt5, a novel CHO glycosylation mutant for producing afucosylated and asialylated recombinant antibodies. Bioengineered 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.F.; Shahreel, W.; Wan, C.; Teo, G.; Hayati, N.; Tay, S.J.; Tong, W.H.; Yang, Y.; Rudd, P.M.; Zhang, P.; et al. Inactivation of GDP-fucose transporter gene (Slc35c1) in CHO cells by ZFNs, TALENs and CRISPR-Cas9 for production of fucose-free antibodies. Biotechnol. J. 2016, 11, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Stadlmann, J.; Taubenschmid, J.; Wenzel, D.; Gattinger, A.; Dürnberger, G.; Dusberger, F.; Elling, U.; Mach, L.; Mechtler, K.; Penninger, J.M. Comparative glycoproteomics of stem cells identifies new players in ricin toxicity. Nature 2017, 549, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Hellbusch, C.C.; Sperandio, M.; Frommhold, D.; Yakubenia, S.; Wild, M.K.; Popovici, D.; Vestweber, D.; Gröne, H.J.; Von Figura, K.; Lübke, T.; et al. Golgi GDP-fucose transporter-deficient mice mimic congenital disorder of glycosylation IIc/leukocyte adhesion deficiency II. J. Biol. Chem. 2007, 282, 10762–10772. [Google Scholar] [CrossRef] [PubMed]
- Yakubenia, S.; Frommhold, D.; Schölch, D.; Hellbusch, C.C.; Körner, C.; Petri, B.; Jones, C.; Ipe, U.; Bixel, M.G.; Krempien, R.; et al. Leukocyte trafficking in a mouse model for leukocyte adhesion deficiency II/congenital disorder of glycosylation IIc. Blood 2008, 112, 1472–1481. [Google Scholar] [CrossRef] [PubMed]
- Rabionet, M.; Van Der Spoel, A.C.; Chuang, C.C.; Von Tümpling-Radosta, B.; Litjens, M.; Bouwmeester, D.; Hellbusch, C.C.; Körner, C.; Wiegandt, H.; Gorgas, K.; et al. Male germ cells require polyenoic sphingolipids with complex glycosylation for completion of meiosis: A link to ceramide synthase-3. J. Biol. Chem. 2008, 283, 13357–13369. [Google Scholar] [CrossRef] [PubMed]
- Panzer, J.A.; Gibbs, S.M.; Dosch, R.; Wagner, D.; Mullins, M.C.; Granato, M.; Balice-Gordon, R.J. Neuromuscular synaptogenesis in wild-type and mutant zebrafish. Dev. Biol. 2005, 285, 340–357. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Willer, J.R.; Scherer, P.C.; Panzer, J.A.; Kugath, A.; Skordalakes, E.; Gregg, R.G.; Willer, G.B.; Balice-Gordon, R.J. Neural and synaptic defects in slytherin, a zebrafish model for human congenital disorders of glycosylation. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Jiang, H.; Wu, P.; Marlow, F.L. Negative feedback regulation of Wnt signaling via N-linked fucosylation in zebrafish. Dev. Biol. 2014, 395, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; He, L.; Dong, H.; Dalton, T.P.; Nebert, D.W. Generation of a Slc39a8 Hypomorph Mouse: Markedly Decreased ZIP8 Zn2+/(HCO3−)2 Transporter Expression. Biochem. Biophys. Res. Commun. 2011, 410, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Gálvez-Peralta, M.; He, L.; Jorge-Nebert, L.F.; Wang, B.; Miller, M.L.; Eppert, B.L.; Afton, S.; Nebert, D.W. Zip8 zinc transporter: Indispensable role for both multiple-organ organogenesis and hematopoiesis in utero. PLoS ONE 2012, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cantagrel, V.; Lefeber, D.J.; Ng, B.G.; Guan, Z.; Silhavy, J.L.; Bielas, S.L.; Lehle, L.; Hombauer, H.; Adamowicz, M.; Swiezewska, E.; et al. SRD5A3 Is Required for Converting Polyprenol to Dolichol and Is Mutated in a Congenital Glycosylation Disorder. Cell 2010, 142, 203–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colinet, A.S.; Sengottaiyan, P.; Deschamps, A.; Colsoul, M.L.; Thines, L.; Demaegd, D.; Duchêne, M.C.; Foulquier, F.; Hols, P.; Morsomme, P. Yeast Gdt1 is a Golgi-localized calcium transporter required for stress-induced calcium signaling and protein glycosylation. Sci. Rep. 2016, 6, 24282. [Google Scholar] [CrossRef] [PubMed]
- Potelle, S.; Morelle, W.; Dulary, E.; Duvet, S.; Vicogne, D.; Spriet, C.; Krzewinski-Recchi, M.A.; Morsomme, P.; Jaeken, J.; Matthijs, G.; et al. Glycosylation abnormalities in Gdt1p/TMEM165 deficient cells result from a defect in Golgi manganese homeostasis. Hum. Mol. Genet. 2016, 25, 1489–1500. [Google Scholar] [CrossRef] [PubMed]
- Bammens, R.; Mehta, N.; Race, V.; Foulquier, F.; Jaeken, J.; Tiemeyer, M.; Steet, R.; Matthijs, G.; Flanagan-Steet, H. Abnormal cartilage development and altered N-glycosylation in Tmem165-deficient zebrafish mirrors the phenotypes associated with TMEM165-CDG. Glycobiology 2014, 25, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.D.; Stevens, T.H. VMA12 encodes a yeast endoplasmic reticulum protein required for vacuolar H+-ATPase assembly. J. Biol. Chem. 1997, 272, 25928–25934. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Li, Z.; Baines, A.C.; Gavriilaki, E.; Ye, Z.; Wen, Z.; Braunstein, E.M.; Biesecker, L.G.; Cheng, L.; Dong, X.; et al. A hypomorphic PIGA gene mutation causes severe defects in neuron development and susceptibility to complement-mediated toxicity in a human iPSC model. PLoS ONE 2017, 12, e0174074. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, K.; Kitamura, D.; Okabe, M.; Taniuchi, I.; Ikawa, M.; Watanabe, T.; Kinoshita, T.; Takeda, J. Glycosylphosphatidylinositol-anchor-deficient mice: Implications for clonal dominance of mutant cells in paroxysmal nocturnal hemoglobinuria. Blood 1996, 87, 3600–3606. [Google Scholar] [PubMed]
- Tremml, G.; Dominguez, C.; Rosti, V.; Zhang, Z.; Pandolfi, P.P.; Keller, P.; Bessler, M. Increased sensitivity to complement and a decreased red blood cell life span in mice mosaic for a nonfunctional Piga gene. Blood 1999, 94, 2945–2954. [Google Scholar] [PubMed]
- Kim, Y.U.; Hong, Y. Functional analysis of the first mannosyltransferase (PIG-M) involved in glycosylphosphatidylinositol synthesis in Plasmodium falciparum. Mol. Cells 2007, 24, 294–300. [Google Scholar] [PubMed]
- Kim, Y.U.; Ashida, H.; Mori, K.; Maeda, Y.; Hong, Y.; Kinoshita, T. Both mammalian PIG-M and PIG-X are required for growth of GPI14-disrupted yeast. J. Biochem. 2007, 142, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Kanagawa, M.; Kobayashi, K.; Tajiri, M.; Manya, H.; Kuga, A.; Yamaguchi, Y.; Akasaka-Manya, K.; Furukawa, J.I.; Mizuno, M.; Kawakami, H.; Shinohara, Y.; et al. Identification of a Post-translational Modification with Ribitol-Phosphate and Its Defect in Muscular Dystrophy. Cell Rep. 2016, 14, 2209–2223. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.M.; Lyon, K.A.; Leung, H.; Leahy, D.J.; Ma, L.; Ginty, D.D. Dystroglycan Organizes Axon Guidance Cue Localization and Axonal Pathfinding. Neuron 2012, 76, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Lloyd, K.C.K. Conditional targeting of Ispd using paired Cas9 nickase and a single DNA template in mice. FEBS Open Bio 2014, 4, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Roscioli, T.; Kamsteeg, E.; Buysse, K.; Maystadt, I.; van Reeuwijk, J.; van den Elzen, C.; van Beusekom, E.; Riemersma, M.; Pfundt, R.; Vissers, L.E.L.M.; et al. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat. Genet. 2012, 44, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Zühlsdorf, A.; Park, J.H.; Wada, Y.; Rust, S.; Reunert, J.; DuChesne, I.; Grüneberg, M.; Marquardt, T. Transferrin variants: Pitfalls in the diagnostics of Congenital disorders of glycosylation. Clin. Biochem. 2015, 48, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Heywood, W.E.; Bliss, E.; Mills, P.; Yuzugulen, J.; Carreno, G.; Clayton, P.T.; Muntoni, F.; Worthington, V.C.; Torelli, S.; Sebire, N.J.; et al. Global serum glycoform profiling for the investigation of dystroglycanopathies & Congenital Disorders of Glycosylation. Mol. Genet. Metab. Rep. 2016, 7, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cerdá, C.; Quelhas, D.; Vega, A.I.; Ecay, J.; Vilarinho, L.; Ugarte, M. Screening using serum percentage of carbohydrate-deficient transferrin for congenital disorders of glycosylation in children with suspected metabolic disease. Clin. Chem. 2008, 54, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Wopereis, S.; Grünewald, S.; Morava, É.; Penzien, J.M.; Briones, P.; García-Silva, M.T.; Demacker, P.N.M.; Huijben, K.M.L.C.; Wevers, R.A. Apolipoprotein C-III Isofocusing in the Diagnosis of Genetic Defects in O-Glycan Biosynthesis. Clin. Chem. 2003, 49, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Daci, A.; Bozalija, A.; Jashari, F.; Krasniqi, S. Individualizing Treatment Approaches for Epileptic Patients with Glucose Transporter Type1 (GLUT-1) Deficiency. Int. J. Mol. Sci. 2018, 19, 122. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.I.; Rubio-Gozalbo, M.E.; Vicente, J.B.; Rivera, I. Sweet and sour: An update on classic galactosemia. J. Inherit. Metab. Dis. 2017, 40, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Babovic-Vuksanovic, D.; Patterson, M.C.; Schwenk, W.F.; O’Brien, J.F.; Vockley, J.; Freeze, H.H.; Mehta, D.P.; Michels, V.V. Severe hypoglycemia as a presenting symptom of carbohydrate-deficient glycoprotein syndrome. J. Pediatr. 1999, 135, 775–781. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; McClean, P.; Henderson, M.J.; Keir, D.G.; Worthington, V.C.; Imtiaz, F.; Schollen, E.; Matthijs, B.G.; Winchester, G. Successful treatment of carbohydrate deficient glycoprotein syndrome type 1b with oral mannose. Arch. Dis. Child. 2001, 85, 339–340. [Google Scholar] [CrossRef] [PubMed]
- Westphal, V.; Kjaergaard, S.; Davis, J.A.; Peterson, S.M.; Skovby, F.; Freeze, H.H. Genetic and metabolic analysis of the first adult with congenital disorder of glycosylation type Ib: Long-term outcome and effects of mannose supplementation. Mol. Genet. Metab. 2001, 73, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Harms, H.K.; Zimmer, K.P.; Kurnik, K.; Bertele-Harms, R.M.; Weidinger, S.; Reiter, K. Oral mannose therapy persistently corrects the severe clinical symptoms and biochemical abnormalities of phosphomannose isomerase deficiency. Acta Paediatr. Int. J. Paediatr. 2002, 91, 1065–1072. [Google Scholar] [CrossRef]
- Penel-Capelle, D.; Dobbelaere, D.; Jaeken, J.; Klein, A.; Cartigny, M.; Weill, J. Congenital disorder of glycosylation Ib (CDG-Ib) without gastrointestinal symptoms. J. Inherit. Metab. Dis. 2003, 26, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Damen, G.; de Klerk, H.; Huijmans, J.; den Hollander, J.; Sinaasappel, M. Gastrointestinal and other clinical manifestations in 17 children with congenital disorders of glycosylation type Ia, Ib, and Ic. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Clayton, P.; Grunewald, S.; Keir, G.; Mills, K.; Mills, P.; Winchester, B.; Worthington, V.; Young, E. Elevation of plasma aspartylglucosaminidase is a useful marker for the congenital disorders of glycosylation type I (CDG I). J. Inherit. Metab. Dis. 2005, 28, 1197–1198. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.; Mills, P.; Jackson, M.; Worthington, V.; Beesley, C.; Mann, A.; Clayton, P.; Grunewald, S.; Keir, G.; Young, L.; et al. Diagnosis of congenital disorders of glycosylation type-I using protein chip technology. Proteomics 2006, 6, 2295–2304. [Google Scholar] [CrossRef] [PubMed]
- Mention, K.; Lacaille, F.; Valayannopoulos, V.; Romano, S.; Kuster, A.; Cretz, M.; Zaidan, H.; Galmiche, L.; Jaubert, F.; de Keyzer, Y.; et al. Development of liver disease despite mannose treatment in two patients with CDG-Ib. Mol. Genet. Metab. 2008, 93, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.S.; Khosravi, M.J.; Patterson, M.C.; Conover, C.A. IGF system in children with congenital disorders of glycosylation. Clin. Endocrinol. (Oxf). 2009, 70, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Liem, Y.S.; Bode, L.; Freeze, H.H.; Leebeek, F.W.G.; Zandbergen, A.A.M.; Paul Wilson, J.H. Using heparin therapy to reverse protein-losing enteropathy in a patient with CDG-Ib. Nat. Clin. Pract. Gastroenterol. Hepatol. 2008, 5, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.C.H.; de Kleine, R.H.; van den Berg, A.P.; Heijdra, Y.; van Scherpenzeel, M.; Lefeber, D.J.; Morava, E. Successful Liver Transplantation and Long-Term Follow-up in a Patient With MPI-CDG. Pediatrics 2014, 134, e279–e283. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Matthijs, G.; Saudubray, J.-M.; Dionisi-Vici, C.; Bertini, E.; de Lonlay, P.; Henri, H.; Carchon, H.; Schollen, E.; Van Schaftingen, E. Phosphomannose Isomerase Deficiency: A Carbohydrate-Deficient Glycoprotein Syndrome with Hepatic-Intestinal Presentation. Am. J. Hum. Genet. 1998, 62, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.S.; Kappler, M.; Bonfert, M.; Borggraefe, I.; Schoen, C.; Reiter, K. Seizures and stupor during intravenous mannose therapy in a patient with CDG syndrome type 1b (MPI-CDG). J. Inherit. Metab. Dis. 2010, 33. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Ng, B.G.; Losfeld, M.E.; Zhu, W.; Freeze, H.H. Identification of intercellular cell adhesion molecule 1 (ICAM-1) as a hypoglycosylation marker in congenital disorders of glycosylation cells. J. Biol. Chem. 2012, 287, 18210–18217. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Srikrishna, G.; Freeze, H.H. N-glycosylation deficiency reduces ICAM-1 induction and impairs inflammatory response. Glycobiology 2014, 24, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Bengtson, P.; Ng, B.G.; Jaeken, J.; Matthijs, G.; Freeze, H.H.; Eklund, E.A. Serum transferrin carrying the xeno-tetrasaccharide NeuAc-Gal-GlcNAc2 is a biomarker of ALG1-CDG. J. Inherit. Metab. Dis. 2016, 39, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Burda, P.; Borsig, L.; de Rijk-Van Andel, J.; Wevers, R.; Jaeken, J.; Carchon, H.; Berger, E.G.; Aebi, M. A novel carbohydrate-deficient glycoprotein syndrome characterized by a deficiency in glucosylation of the dolichol-linked oligosaccharide. J. Clin. Investig. 1998, 102, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; James, P.M.; Ng, B.G.; Li, X.; Xia, B.; Rong, J.; Asif, G.; Raymond, K.; Jones, M.A.; Hegde, M.; et al. A Novel N-Tetrasaccharide in Patients with Congenital Disorders of Glycosylation, Including Asparagine-Linked Glycosylation Protein 1, Phosphomannomutase 2, and Mannose Phosphate Isomerase Deficiencies. Clin. Chem. 2016, 62, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Losfeld, M.E.; Soncin, F.; Ng, B.G.; Singec, I.; Freeze, H.H. A sensitive green fluorescent protein biomarker of N-glycosylation site occupancy. FASEB J. 2012, 26, 4210–4217. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, S.; Huyben, K.; de Jong, J.G.; Smeitink, J.A.; Rubio, E.; Boers, G.H.; Conradt, H.S.; Wendel, U.; Wevers, R. A β-Trace protein in human cerebrospinal fluid: A diagnostic marker for N-glycosylation defects in brain. Biochim. Biophys. Acta 1999, 1455, 54–60. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, Z.; Li, A.V.; Yarema, K.J. Roles for UDP-GlcNAc 2-epimerase/ManNAc 6-kinase outside of sialic acid biosynthesis: Modulation of sialyltransferase and BiP expression, GM3 and GD3 biosynthesis, proliferation, and apoptosis, and ERK1/2 phosphorylation. J. Biol. Chem. 2006, 281, 27016–27028. [Google Scholar] [CrossRef] [PubMed]
- Paccalet, T.; Coulombe, Z.; Tremblay, J.P. Ganglioside GM3 levels are altered in a mouse model of hibm: GM3 as a cellular marker of the disease. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Valles-Ayoub, Y.; Esfandiarifard, S.; Sinai, P.; Carbajo, R.; Khokher, Z.; No, D.; Pietruszka, M.; Darvish, B.; Kakkis, E.; Darvish, D. Serum Neural Cell Adhesion Molecule Is Hyposialylated in Hereditary Inclusion Body Myopathy. Genet. Test. Mol. Biomark. 2012, 16, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Leoyklang, P.; Malicdan, M.C.; Yardeni, T.; Celeste, F.; Ciccone, C.; Li, X.; Jiang, R.; Gahl, W.A.; Carrillo-Carrasco, N.; He, M.; et al. Sialylation of Thomsen–Friedenreich antigen is a noninvasive blood-based biomarker for GNE myopathy. Biomark. Med. 2014, 8, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Zdebska, E.; Musielak, M.; Jaeken, J.; Kościelak, J. Band 3 glycoprotein and glycophorin A from erythrocytes of children with congenital disorder of glycosylation type-Ia are underglycosylated. Proteomics 2001, 1, 269–274. [Google Scholar] [CrossRef]
- Sala, G.; Dupré, T.; Seta, N.; Codogno, P.; Ghidoni, R. Increased biosynthesis of glycosphingolipids in congenital disorder of glycosylation Ia (CDG-Ia) fibroblasts. Pediatr. Res. 2002, 52, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Harrison, H.H.; Miller, K.L.; Harbison, M.D.; Slonim, A.E. Multiple serum protein abnormalities in carbohydrate-deficient glycoprotein syndrome: Pathognomonic finding of two-dimensional electrophoresis? Clin. Chem. 1992, 38, 1390–1392. [Google Scholar] [PubMed]
- Mohamed, M.; Theodore, M.; van der Grinten, H.C.; Van Herwaarden, A.E.; Huijben, K.; van Dongen, L.; Kouwenberg, D.; Lefeber, D.J.; Wevers, R.A.; Morava, E. Thyroid function in PMM2-CDG: Diagnostic approach and proposed management. Mol. Genet. Metab. 2012, 105, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Pohl, S.; Hoffmann, A.; Rüdiger, A.; Nimtz, M.; Jaeken, J.; Conradt, H.S. Hypoglycosylation of a brain glycoprotein (β-trace protein) in CDG syndromes due to phosphomannomutase deficiency and N-acetylglucosaminyl-transferase II deficiency. Glycobiology 1997, 7, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Gadomski, T.E.; Bolton, M.; Alfadhel, M.; Dvorak, C.; Ogunsakin, O.A.; Nelson, S.L.; Morava, E. ALG13-CDG in a male with seizures, normal cognitive development, and normal transferrin isoelectric focusing. Am. J. Med. Genet. Part A 2017, 173, 2772–2775. [Google Scholar] [CrossRef] [PubMed]
- Cromphout, K.; Vleugels, W.; Heykants, L.; Schollen, E.; Keldermans, L.; Sciot, R.; D’Hooge, R.; de Deyn, P.P.; von Figura, K.; Hartmann, D.; et al. The Normal Phenotype of Pmm1-Deficient Mice Suggests that Pmm1 Is Not Essential for Normal Mouse Development. Mol. Cell. Biol. 2006, 26, 5621–5635. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Da-Cunha, M.; Vleugels, W.; Maliekal, P.; Matthijs, G.; Van Schaftingen, E. Mammalian phosphomannomutase PMM1 is the brain IMP-sensitive glucose-1,6-bisphosphatase. J. Biol. Chem. 2008, 283, 33988–33993. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Cimmaruta, C.; Liguori, L.; Viscido, G.; Cubellis, M.V.; Andreotti, G. A mutant of phosphomannomutase1 retains full enzymatic activity, but is not activated by IMP: Possible implications for the disease PMM2-CDG. PLoS ONE 2017, 12, e0189629. [Google Scholar] [CrossRef] [PubMed]
- Körner, C.; Lehle, L.; Von Figura, K. Carbohydrate-deficient glycoprotein syndrome type 1: Correction of the glycosylation defect by deprivation of glucose or supplementation of mannose. Glycoconj. J. 1998, 15, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Alton, G.; Kjaergaard, S.; Etchison, J.R.; Skovby, F.; Freeze, H.H. Oral ingestion of mannose elevates blood mannose levels: A first step toward a potential therapy for carbohydrate-deficient glycoprotein syndrome type I. Biochem. Mol. Med. 1997, 60, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Mayatepek, E.; Kohlmuller, D. Mannose supplementation in carbohydrate-deficient glycoprotein syndrome type I and phosphomannomutase deficiency. Eur. J. Pediatr. 1998, 157, 605–606. [Google Scholar] [CrossRef] [PubMed]
- Mayatepek, E.; Schröder, M.; Kohlmüller, D.; Bieger, W.P.; Nützenadel, W. Continuous mannose infusion in carbohydrate-deficient glycoprotein syndrome type I. Acta Paediatr. 1997, 86, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, S.; Kristiansson, B.; Stibler, H.; Freeze, H.H.; Schwartz, M.; Martinsson, T.; Skovby, F. Failure of short-term mannose therapy of patients with carbohydrate-deficient glycoprotein syndrome type 1A. Acta Paediatr. 1998, 87, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, T.; Denecke, J. Congenital disorders of glycosylation: Review of their molecular bases, clinical presentations and specific therapies. Eur. J. Pediatr. 2003, 162, 359–379. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Lehrman, M.A. Metformin-stimulated Mannose Transport in Dermal Fibroblasts. J. Biol. Chem. 2004, 279, 9703–9712. [Google Scholar] [CrossRef] [PubMed]
- Rutschow, S.; Thiem, J.; Kranz, C.; Marquardt, T. Membrane-permeant derivatives of mannose-1-phosphate. Bioorg. Med. Chem. 2002, 10, 4043–4049. [Google Scholar] [CrossRef]
- Eklund, E.A.; Merbouh, N.; Ichikawa, M.; Nishikawa, A.; Clima, J.M.; Dorman, J.A.; Norberg, T.; Freeze, H.H. Hydrophobic Man-1-P derivatives correct abnormal glycosylation in Type I congenital disorder of glycosylation fibroblasts. Glycobiology 2005, 15, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Hardré, R.; Khaled, A.; Willemetz, A.; Dupré, T.; Moore, S.; Gravier-Pelletier, C.; Merrer, Y. le Mono, di and tri-mannopyranosyl phosphates as mannose-1-phosphate prodrugs for potential CDG-Ia therapy. Bioorg. Med. Chem. Lett. 2007, 17, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Glycomine. Available online: http://glycomine.com/ (accessed on 19 January 2018).
- Balasubramaniam, S.; Duley, J.A.; Christodoulou, J. Inborn errors of pyrimidine metabolism: Clinical update and therapy. J. Inherit. Metab. Dis. 2014, 37, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Hinderlich, S.; Berger, M.; Keppler, O.T.; Pawlita, M.; Reutter, W. Biosynthesis of N-acetylneuraminic acid in cells lacking UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. Biol. Chem. 2001, 382, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.D.; Pang, P.C.; Krishnamurthy, S.; Wands, A.M.; Grassi, P.; Dell, A.; Haslam, S.M.; Kohler, J.J. Effects of altered sialic acid biosynthesis on N-linked glycan branching and cell surface interactions. J. Biol. Chem. 2017, 292, 9637–9651. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Y.W.; Gadomski, T.; Van Scherpenzeel, M.; Honzik, T.; Hansikova, H.; Holmefjord, K.S.B.; Mork, M.; Bowling, F.; Sykut-Cegielska, J.; Koch, D.; et al. Oral d-galactose supplementation in PGM1-CDG. Genet. Med. 2017, 19, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Schrapers, E.; Tegtmeyer, L.C.; Simic-Schleicher, G.; Debus, V.; Reunert, J.; Balbach, S.; Klingel, K.; Du Chesne, I.; Seelhöfer, A.; Fobker, M.; et al. News on Clinical Details and Treatment in PGM1-CDG. JIMD Rep. 2016, 26, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Nolting, K.; Park, J.H.; Tegtmeyer, L.C.; Zühlsdorf, A.; Grüneberg, M.; Rust, S.; Reunert, J.; Du Chesne, I.; Debus, V.; Schulze-Bahr, E.; et al. Limitations of galactose therapy in phosphoglucomutase 1 deficiency. Mol. Genet. Metab. Rep. 2017, 13, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Preisler, N.; Laforêt, P.; Echaniz-Laguna, A.; Ørngreen, M.C.; Lonsdorfer-Wolf, E.; Doutreleau, S.; Geny, B.; Stojkovic, T.; Piraud, M.; Petit, F.M.; et al. Fat and Carbohydrate Metabolism During Exercise in Phosphoglucomutase Type 1 Deficiency. J. Clin. Endocrinol. Metab. 2013, 98, E1235–E1240. [Google Scholar] [CrossRef] [PubMed]
- Voermans, N.C.; Preisler, N.; Madsen, K.L.; Janssen, M.C.H.; Kusters, B.; Abu Bakar, N.; Conte, F.; Lamberti, V.M.L.; Nusman, F.; van Engelen, B.G.; et al. PGM1 deficiency: Substrate use during exercise and effect of treatment with galactose. Neuromuscul. Disord. 2017, 27, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Koda, Y.; Soejima, M.; Kimura, H. Identification of human phosphoglucomutase 3 (PGM3) as N-acetylglucosamine-phosphate mutase (AGM1). Ann. Hum. Genet. 2002, 66, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, N.A.; Gilmore, R. Mammalian cells lacking either the cotranslational or posttranslocational oligosaccharyltransferase complex display substrate-dependent defects in asparagine linked glycosylation. Sci. Rep. 2016, 6, 20946. [Google Scholar] [CrossRef] [PubMed]
- Patiroglu, T.; Haluk Akar, H.; Gilmour, K.; Unal, E.; Akif Ozdemir, M.; Bibi, S.; Burns, S.; Chiang, S.C.; Schlums, H.; Bryceson, Y.T.; et al. A case of XMEN syndrome presented with severe auto-immune disorders mimicking autoimmune lymphoproliferative disease. Clin. Immunol. 2015, 159, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Joshi, C.; Kolbe, D.L.; Mansilla, M.A.; Mason, S.; Smith, R.J.H.; Campbell, C.A. Ketogenic diet—A novel treatment for early epileptic encephalopathy due to PIGA deficiency. Brain Dev. 2016, 38, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The ketogenic diet for the treatment of childhood epilepsy: A randomised controlled trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef]
- Taha, A.Y.; Burnham, W.M.; Auvin, S. Polyunsaturated fatty acids and epilepsy. Epilepsia 2010, 51, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Layton, M.; Karadimitris, A. Inherited glycosylphosphatidyl inositol deficiency: A treatable CDG. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Kuki, I.; Takahashi, Y.; Okazaki, S.; Kawawaki, H.; Ehara, E.; Inoue, N.; Kinoshita, T.; Murakami, Y. Vitamin B6-responsive epilepsy due to inherited GPI deficiency. Neurology 2013, 81, 1467–1469. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.G.; Buckingham, K.J.; Raymond, K.; Kircher, M.; Turner, E.H.; He, M.; Smith, J.D.; Eroshkin, A.; Szybowska, M.; Losfeld, M.E.; et al. Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. Am. J. Hum. Genet. 2013, 92, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Etzioni, A.; Sturla, L.; Antonellis, A.; Green, E.D.; Gershoni-Baruch, R.; Berninsone, P.M.; Hirschberg, C.B.; Tonetti, M. Leukocyte adhesion deficiency (LAD) type II/carbohydrate deficient glycoprotein (CDG) IIc founder effect and genotype/phenotype correlation. Am. J. Med. Genet. 2002, 110, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Etzioni, A.; Tonetti, M. Fucose supplementation in leukocyte adhesion deficiency type II. Blood 2000, 95, 3641–3643. [Google Scholar] [PubMed]
- Jansen, J.C.; Cirak, S.; van Scherpenzeel, M.; Timal, S.; Reunert, J.; Rust, S.; Pérez, B.; Vicogne, D.; Krawitz, P.; Wada, Y.; et al. CCDC115 Deficiency Causes a Disorder of Golgi Homeostasis with Abnormal Protein Glycosylation. Am. J. Hum. Genet. 2016, 98, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.C.; Timal, S.; Van Scherpenzeel, M.; Michelakakis, H.; Vicogne, D.; Ashikov, A.; Moraitou, M.; Hoischen, A.; Huijben, K.; Steenbergen, G.; et al. TMEM199 Deficiency is a Disorder of Golgi Homeostasis Characterized by Elevated Aminotransferases, Alkaline Phosphatase, and Cholesterol and Abnormal Glycosylation. Am. J. Hum. Genet. 2016, 98, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Roscioli, T.; Thorburn, D.R.; Prelog, K.; Bahlo, M.; Sue, C.M.; Balasubramaniam, S.; Christodoulou, J. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J. Inherit. Metab. Dis. 2017, 40, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Hogrebe, M.; Grüneberg, M.; Duchesne, I.; von der Heiden, A.L.; Reunert, J.; Schlingmann, K.P.; Boycott, K.M.; Beaulieu, C.L.; Mhanni, A.A.; et al. SLC39A8 Deficiency: A Disorder of Manganese Transport and Glycosylation. Am. J. Hum. Genet. 2015, 97, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Hogrebe, M.; Fobker, M.; Brackmann, R.; Fiedler, B.; Reunert, J.; Rust, S.; Tsiakas, K.; Santer, R.; Grüneberg, M.; et al. SLC39A8 deficiency: Biochemical correction and major clinical improvement by manganese therapy. Genet. Med. 2017, 20, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Gámez, A.; Yuste-Checa, P.; Brasil, S.; Briso-Montiano, Á.; Desviat, L.R.; Ugarte, M.; Pérez-Cerdá, C.; Pérez, B. Protein misfolding diseases: Prospects of pharmacological treatment. Clin. Genet. 2017, 93, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Brasil, S.; Briso-Montiano, A.; Gámez, A.; Underhaug, J.; Flydal, M.I.; Desviat, L.; Merinero, B.; Ugarte, M.; Martinez, A.; Pérez, B. New perspectives for pharmacological chaperoning treatment in methylmalonic aciduria cblB type. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Yuste-Checa, P.; Brasil, S.; Gámez, A.; Underhaug, J.; Desviat, L.R.; Ugarte, M.; Pérez-Cerdá, C.; Martinez, A.; Pérez, B. Pharmacological Chaperoning: A Potential Treatment for PMM2-CDG. Hum. Mutat. 2017, 38, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; de Vaca, I.C.; Poziello, A.; Monti, M.C.; Guallar, V.; Cubellis, M.V. Conformational response to ligand binding in Phosphomannomutase2: Insights into inborn glycosylation disorder. J. Biol. Chem. 2014, 289, 34900–34910. [Google Scholar] [CrossRef] [PubMed]
- Vega, A.I.; Pérez-Cerdá, C.; Abia, D.; Gámez, A.; Briones, P.; Artuch, R.; Desviat, L.R.; Ugarte, M.; Pérez, B. Expression analysis revealing destabilizing mutations in phosphomannomutase 2 deficiency (PMM2-CDG): Expression analysis of PMM2-CDG mutations. J. Inherit. Metab. Dis. 2011, 34, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Pedone, E.; Giordano, A.; Cubellis, M.V. Biochemical phenotype of a common disease-causing mutation and a possible therapeutic approach for the phosphomannomutase 2-associated disorder of glycosylation. Mol. Genet. Genom. Med. 2013, 1, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Yuste-Checa, P.; Gámez, A.; Brasil, S.; Desviat, L.R.; Ugarte, M.; Pérez-Cerdá, C.; Pérez, B. The Effects of PMM2-CDG-Causing Mutations on the Folding, Activity, and Stability of the PMM2 Protein. Hum. Mutat. 2015, 36, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Monti, M.C.; Citro, V.; Cubellis, M.V. Heterodimerization of two pathological mutants enhances the activity of human phosphomannomutase2. PLoS ONE 2015, 10, e0139882. [Google Scholar] [CrossRef] [PubMed]
- Dahl, R.; Bravo, Y.; Sharma, V.; Ichikawa, M.; Dhanya, R.-P.; Hedrick, M.; Brown, B.; Rascon, J.; Vicchiarelli, M.; Mangravita-Novo, A.; et al. Potent, Selective, and Orally Available Benzoisothiazolone Phosphomannose Isomerase Inhibitors as Probes for Congenital Disorder of Glycosylation Ia. J. Med. Chem. 2011, 54, 3661–3668. [Google Scholar] [CrossRef] [PubMed]
- Yuste-Checa, P.; Medrano, C.; Gámez, A.; Desviat, L.R.; Matthijs, G.; Ugarte, M.; Pérez-Cerdá, C.; Pérez, B. Antisense-mediated therapeutic pseudoexon skipping in TMEM165-CDG. Clin. Genet. 2015, 87, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Vega, A.I.; Perez-Cerda, C.; Desviat, L.R.; Matthijs, G.; Ugarte, M.; Pérez, B. Functional analysis of three splicing mutations identified in the PMM2 gene: Toward a new therapy for congenital disorder of glycosylation type IA. Hum. Mutat. 2009, 30, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Vuillaumier-Barrot, S.; Barnier, A.; Cuer, M.; Durand, G.; Grandchamp, B.; Seta, N. Characterization of the 415G>A (E139K) PMM2 mutation in carbohydrate-deficient glycoprotein syndrome type Ia disrupting a splicing enhancer resulting in exon 5 skipping. Hum. Mutat. 1999, 14, 543–544. [Google Scholar] [CrossRef]
- Vuillaumier-Barrot, S.; Le Bizec, C.; De Lonlay, P.; Madinier-Chappat, N.; Barnier, A.; Dupré, T.; Durand, G.; Seta, N. PMM2 intronic branch-site mutations in CDG-Ia. Mol. Genet. Metab. 2006, 87, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Bryson, T.E.; Anglin, C.M.; Bridges, P.H.; Cottle, R.N. Nuclease-Mediated Gene Therapies for Inherited Metabolic Diseases of the Liver. Yale J. Biol. Med. 2017, 90, 553–566. [Google Scholar] [PubMed]
- Malicdan, M.C.; Okada, T.; Sela, I.; Takeda, S.; Funato, F.; Nonaka, I.; Hayashi, Y.K.; Yakovlev, L.; Argov, Z.; Nishino, I.; et al. P4.46 Expression of human GNE through adeno-associated virus mediated therapy delays progression of myopathy in the DMRV/hIBM mouse model. Neuromuscul. Disord. 2011, 21, 718. [Google Scholar] [CrossRef]
- Tal-Goldberg, T.; Lorain, S.; Mitrani-Rosenbaum, S. Correction of the Middle Eastern M712T mutation causing GNE myopathy by trans-splicing. NeuroMol. Med. 2014, 16, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Meliani, A.; Boisgerault, F.; Fitzpatrick, Z.; Marmier, S.; Leborgne, C.; Collaud, F.; Simon Sola, M.; Charles, S.; Ronzitti, G.; Vignaud, A.; et al. Enhanced liver gene transfer and evasion of preexisting humoral immunity with exosome-enveloped AAV vectors. Blood Adv. 2017, 1, 2019–2031. [Google Scholar] [CrossRef] [PubMed]
- Phadke, A.P.; Jay, C.; Chen, S.J.; Haddock, C.; Wang, Z.; Yu, Y.; Nemunaitis, D.; Nemunaitis, G.; Templeton, N.S.; Senzer, N.; et al. Safety and in vivo expression of a GNE-transgene: A novel treatment approach for hereditary inclusion body myopathy-2. Gene Regul. Syst. Biol. 2009, 3, 89–101. [Google Scholar] [CrossRef]
- Nemunaitis, G.; Jay, C.M.; Maples, P.B.; Gahl, W.A.; Huizing, M.; Yardeni, T.; Tong, A.W.; Phadke, A.P.; Pappen, B.O.; Bedell, C.; et al. Hereditary Inclusion Body Myopathy: Single Patient Response to Intravenous Dosing of GNE Gene Lipoplex. Hum. Gene Ther. 2011, 22, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, G.; Maples, P.B.; Jay, C.; Gahl, W.A.; Huizing, M.; Poling, J.; Tong, A.W.; Phadke, A.P.; Pappen, B.O.; Bedell, C.; et al. Hereditary inclusion body myopathy: Single patient response to GNE gene Lipoplex therapy. J. Gene Med. 2010, 12, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Halldorson, J.; Kazi, Z.; Mekeel, K.; Kuo, A.; Hassanein, T.; Loomba, R.; Austin, S.; Valasek, M.A.; Kishnani, P.; Hemming, A.W. Successful combined liver/kidney transplantation from a donor with Pompe disease. Mol. Genet. Metab. 2015, 115, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Arnon, R.; Wasserstein, M.P.; Diaz, G.A. Liver transplantation for pediatric inherited metabolic disorders: Considerations for indications, complications, and perioperative management. Pediatr. Transplant. 2016, 20, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Dalle, J.-H.; de Latour, R.P. Allogeneic hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Int. J. Hematol. 2016, 103, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, L.; Zucker, N.; Frenckel, G.; Medalion, B.; Ben Gal, T.; Birk, E.; Mandel, H.; Nasser, N.; Morgenstern, S.; Zuckermann, A.; Lefeber, D.J.; et al. From discrete dilated cardiomyopathy to successful cardiac transplantation in congenital disorders of glycosylation due to dolichol kinase deficiency (DK1-CDG). Heart Fail. Rev. 2013, 18, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Klcovansky, J.; Mørkrid, L.; Möller, T. Heart transplantation in a child with congenital disorder of glycosylation. J. Hear. Lung Transplant. 2016, 35, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- Giannuzzi, V.; Devlieger, H.; Margari, L.; Odlind, V.L.; Ragab, L.; Bellettato, C.M.; D’Avanzo, F.; Lampe, C.; Cassis, L.; Cortès-Saladelafont, E.; et al. The ethical framework for performing research with rare inherited neurometabolic disease patients. Eur. J. Pediatr. 2017, 176, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, C.; Schaefer, A.M.; Elson, J.L.; Morava, E.; Bugiani, M.; Uziel, G.; Smeitink, J.A.; Turnbull, D.M.; McFarland, R. A scale to monitor progression and treatment of mitochondrial disease in children. Neuromuscul. Disord. 2006, 16, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Achouitar, S.; Mohamed, M.; Gardeitchik, T.; Wortmann, S.B.; Sykut-Cegielska, J.; Ensenauer, R.; De Baulny, H.O.; Õunap, K.; Martinelli, D.; De Vries, M.; et al. Nijmegen paediatric CDG rating scale: A novel tool to assess disease progression. J. Inherit. Metab. Dis. 2011, 34, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; De Diego, V.; Muchart, J.; Cuadras, D.; Felipe, A.; Macaya, A.; Velázquez, R.; Poo, M.P.; Fons, C.; O’Callaghan, M.M.; et al. Phosphomannomutase deficiency (PMM2-CDG): Ataxia and cerebellar assessment. Orphanet J. Rare Dis. 2015, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Serrano, N.L.; De Diego, V.; Cuadras, D.; Martinez Monseny, A.F.; Velázquez-Fragua, R.; López, L.; Felipe, A.; Gutiérrez-Solana, L.G.; Macaya, A.; Pérez-Dueñas, B.; et al. A quantitative assessment of the evolution of cerebellar syndrome in children with phosphomannomutase-deficiency (PMM2-CDG). Orphanet J. Rare Dis. 2017, 12, 10–15. [Google Scholar] [CrossRef] [PubMed]
- De Diego, V.; Martínez-Monseny, A.F.; Muchart, J.; Cuadras, D.; Montero, R.; Artuch, R.; Pérez-Cerdá, C.; Pérez, B.; Pérez-Dueñas, B.; Poretti, A.; et al. Longitudinal volumetric and 2D assessment of cerebellar atrophy in a large cohort of children with phosphomannomutase deficiency (PMM2-CDG). J. Inherit. Metab. Dis. 2017, 40, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Y.W.; Gadomski, T.; Kozicz, T.; Morava, E.; Beamer, L.J.; Honzik, T.; Mohamed, M.; Morava, E.; Wortmann, S.B.; Brocke Holmefjord, K.S.; et al. Defining the Phenotype and Assessing Severity in Phosphoglucomutase-1 Deficiency. J. Pediatr. 2016, 175, 130–136.e8. [Google Scholar] [CrossRef] [PubMed]
- Skrinar, A.M.; Argov, Z.; Caraco, Y.; Kolodny, E.; Lau, H.; Pestronk, A.; Shieh, P.; Bronstein, F.; Esposito, A.; Feinsod-Meiri, Y.; et al. P.3.1 GNE myopathy functional activity scale (GNEM-FAS): Development of a disease-specific instrument for measuring function and independence. Neuromuscul. Disord. 2013, 23, 755. [Google Scholar] [CrossRef]
- Mori-Yoshimura, M.; Oya, Y.; Yajima, H.; Yonemoto, N.; Kobayashi, Y.; Hayashi, Y.K.; Noguchi, S.; Nishino, I.; Murata, M. GNE myopathy: A prospective natural history study of disease progression. Neuromuscul. Disord. 2014, 24, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Mori-Yoshimura, M.; Hayashi, Y.K.; Yonemoto, N.; Nakamura, H.; Murata, M.; Takeda, S.; Nishino, I.; Kimura, E. Nationwide patient registry for GNE myopathy in Japan. Orphanet J. Rare Dis. 2014, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, A.Q.; Latham, L.L.; Celeste, F.; Ciccone, C.; Malicdan, M.C.; Goldspiel, B.; Terse, P.; Cradock, J.; Yang, N.; et al. Safety, pharmacokinetics and sialic acid production after oral administration of N-acetylmannosamine (ManNAc) to subjects with GNE myopathy. Mol. Genet. Metab. 2017, 122, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Argov, Z.; Caraco, Y.; Lau, H.; Pestronk, A.; Shieh, P.B.; Skrinar, A.; Koutsoukos, T.; Ahmed, R.; Martinisi, J.; Kakkis, E. Aceneuramic Acid Extended Release Administration Maintains Upper Limb Muscle Strength in a 48-week Study of Subjects with GNE Myopathy: Results from a Phase 2, Randomized, Controlled Study. J. Neuromuscul. Dis. 2016, 3, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Sparks, S.; Rakocevic, G.; Joe, G.; Manoli, I.; Shrader, J.; Harris-Love, M.; Sonies, B.; Ciccone, C.; Dorward, H.; Krasnewich, D.; et al. Intravenous immune globulin in hereditary inclusion body myopathy: A pilot study. BMC Neurol. 2007, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Home—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 6 January 2018).
- Clinical Trials Register. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search (accessed on 9 January 2018).
- Wang, Y.; Schachter, H.; Marth, J.D. Mice with a homozygous deletion of the Mgat2 gene encoding UDP-N-acetylglucosamine:α-6-d-mannoside β1,2-N-acetylglucosaminyltransferase II: A model for congenital disorder of glycosylation type IIa. Biochim. Biophys. Acta Gen. Subj. 2002, 1573, 301–311. [Google Scholar] [CrossRef]
- Lühn, K.; Marquardt, T.; Harms, E.; Vestweber, D. Discontinuation of fucose therapy in LADII causes rapid loss of selectin ligands and rise of leukocyte counts. Blood 2001, 97, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.-L. E.; O’Mara, T.A.; Glubb, D.M. Enhancing the Promise of Drug Repositioning through Genetics. Front. Pharmacol. 2017, 8, 896. [Google Scholar] [CrossRef] [PubMed]
- EURORDIS. Eurordis Breaking the Access Deadlock to Leave No One Behind; A Reflection Paper by EURORDIS and Its Members; EURORDIS: Paris, France, 2017; pp. 1–47. [Google Scholar]
- Zhao, M.; Wei, D.-Q. Rare Diseases: Drug Discovery and Informatics Resource. Interdiscip. Sci. Comput. Life Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zamami, Y.; Imanishi, M.; Takechi, K.; Ishizawa, K. Pharmacological approach for drug repositioning against cardiorenal diseases. J. Med. Investig. 2017, 64, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Leaf, D.E.; Goldfarb, D.S. Mechanisms of action of acetazolamide in the prophylaxis and treatment of acute mountain sickness. J. Appl. Physiol. 2007, 102, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.H.; Ahn, S.J.; Lim, H.W.; Lee, B.R. The effect of oral acetazolamide on cystoid macular edema in hydroxychloroquine retinopathy: A case report. BMC Ophthalmol. 2017, 17, 124. [Google Scholar] [CrossRef] [PubMed]
- Finer, G.; Price, H.E.; Shore, R.M.; White, K.E.; Langman, C.B. Hyperphosphatemic familial tumoral calcinosis: Response to acetazolamide and postulated mechanisms. Am. J. Med. Genet. Part A 2014, 164, 1545–1549. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.S. Epidemiology of Cerebellar Diseases and Therapeutic Approaches. Cerebellum 2017, 17, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Westphal, V.; Kjaergaard, S.; Schollen, E.; Martens, K.; Grunewald, S.; Schwartz, M.; Matthijs, G.; Freeze, H.H. A frequent mild mutation in ALG6 may exacerbate the clinical severity of patients with congenital disorder of glycosylation Ia (CDG-Ia) caused by phosphomannomutase deficiency. Hum. Mol. Genet. 2002, 11, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Sabry, S.; Vuillaumier-Barrot, S.; Mintet, E.; Fasseu, M.; Valayannopoulos, V.; Héron, D.; Dorison, N.; Mignot, C.; Seta, N.; Chantret, I.; et al. A case of fatal Type I congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity. Orphanet J. Rare Dis. 2016, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- PubMed—NCBI. Available online: https://www.ncbi.nlm.nih.gov/pubmed/ (accessed on 21 January 2018).







{kind=link}
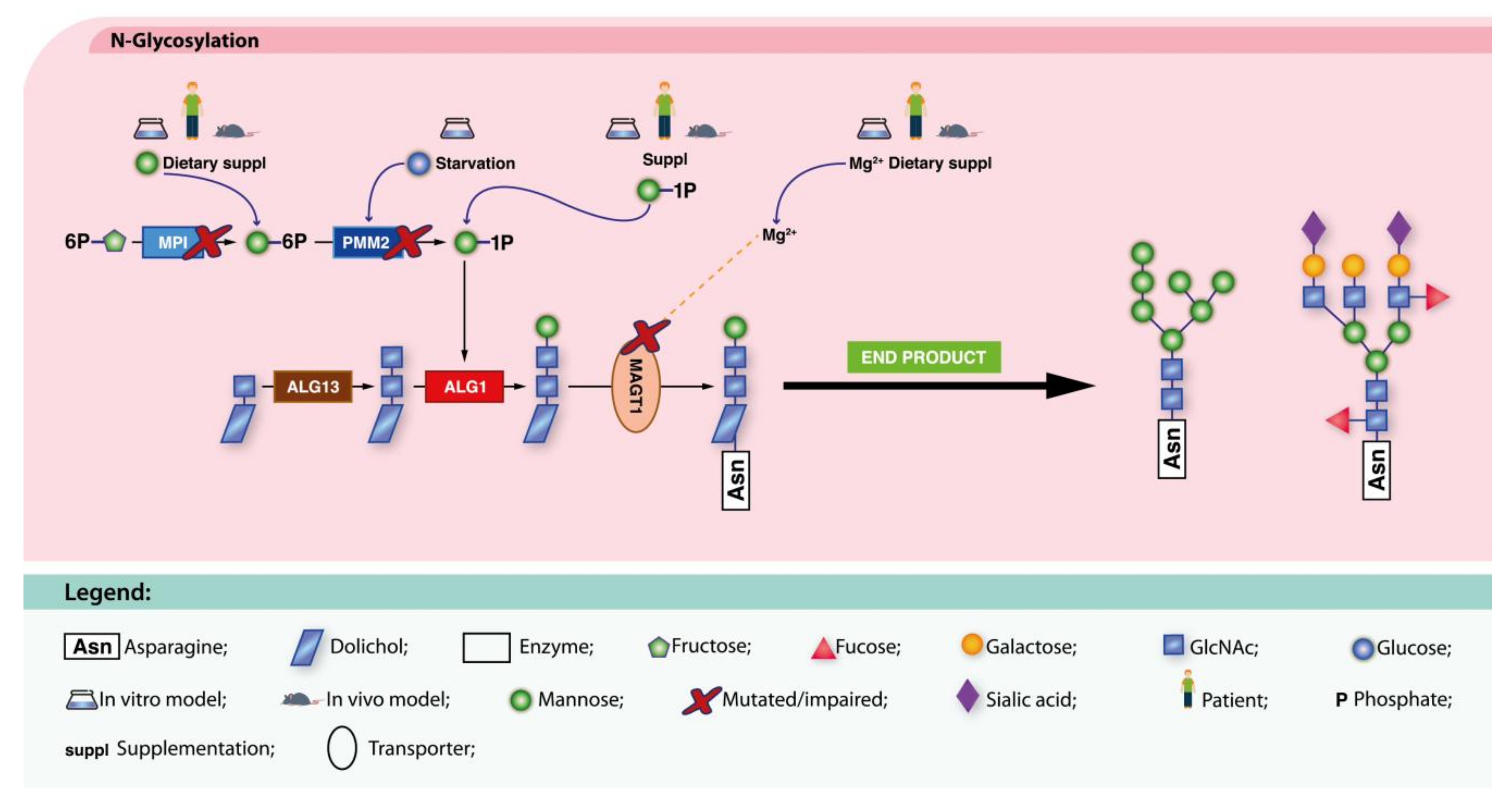{kind=link}
{kind=link}
{kind=link}
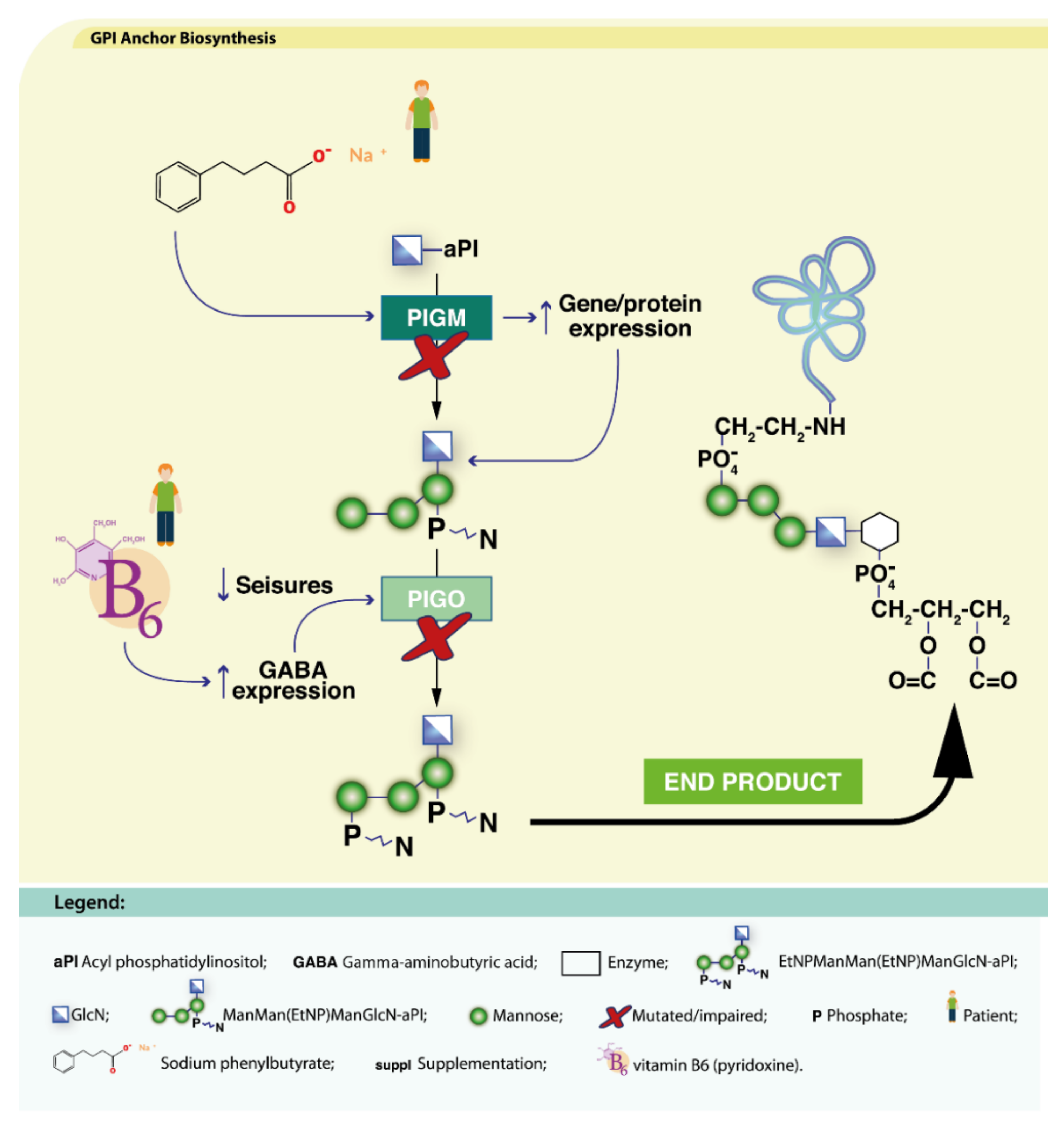{kind=link}
{kind=link}
{kind=link}
| Defect | CDG/Protein | Cell/Organism/Animal | Model | Major Findings/Phenotype | Reference |
|---|---|---|---|---|---|
| Protein N-glycosylation | ALG1-CDG/Chitobiosyldiphosphodolichol β-mannosyltransferase | Saccharomyces cerevisiae | Alg1 mutants (K57-6C strain) | -Thermosensitive. -Increased levels of GDP-Man by Mannose-1-phosphate guanylyltransferase (MPG1) gene overexpression restores defects in mannosylation, in contrast to Man supplementation. | [24,34,41] |
| S. cerevisiae | Alg1 mutants (PRY56 strain) | -Thermosensitive. | [31] | ||
| ALG6-CDG/α-1,3-glucosyltransferase | S. cerevisiae | Alg6 mutants | -Unable to transfer glucose from dolichol phosphoglucose in the Lipid-linked oligosaccharides (LLO) synthesis leading to the accumulation of Man9GlcNAc. -Shorter LLO glucose chain causes growth defects. -Alg6 mutants stopped growing completely. | [87,88] | |
| Chinese hamster (CHO) cell line | MI8-5 cells (Alg6−/−) | -Unable to synthetize glucosylated oligosaccharides. | [89] | ||
| MAGT1-CDG/Magnesium transporter 1 | Human embryonic kidney (HEK293) cell line | Magt1 knockdown by siRNA | -Decreased Mg2+ uptake. -Combined Magt1 and TUSC3 overexpression raised cellular Mg2+ content. | [90] | |
| S. cerevisiae | Alr1∆ strain | -Inability to proliferate in Mg2+ free medium is overcome by magt1 complementation. | [90] | ||
| Dario renio (zebrafish) | (a) Zygotic Magt1 knockdown (b) Maternal and zygotic Magt1 knockdown | (a) Embryonic lethality. Profound developmental abnormalities. (b) Inability to hatch. MgCl2 recovered lethality. | [90] | ||
| Protein N-glycosylation | MPI-CDG/Mannose-6-phosphate isomerase | Human colon adenocarcinoma (HT-29) cell line | Mpi knockdown by siRNA | -Inhibiting Mpi by 50–70% increases Man incorporation into proteins. | [59] |
| Mus musculus (mouse) | Mpi (Mpi−/−) knockout | -Normal glycosylation profile. -High level of embryonic lethality that was exacerbated by Man supplementation due to Man-6-P accumulation and a decrease in adenosine triphosphate (ATP) levels. | [91] | ||
| Mouse | Hypomorphic MpiY255C/Y255C | -Reduced in utero mortality which was increased by mannose supplementation to the pregnant dams. The surviving offspring presented severe ocular defects. -No phenotypic presentations. | [72] | ||
| Zebrafish | Mpi mutant with 13% of enzymatic activity | -MPI-CDG biochemical and phenotypic presentations. -Addition of mannose to the fish water rescued mpi morphants phenotype but only if provided prior to 24 h post fecundation (hpf). | [92] | ||
| PMM2-CDG/Phosphomannomutase 2 | Human induced pluripotent stem cells (iPSC) | (a) Hypomorphic PMM2422G>A/357C>A-iPSC (b) Hypomorphic PMM2422G>A/357C>A-iPSC with additional knockdown by shRNA | -Reduced PMM activity, accumulation of shorter glycan structures and reduced mannosylation (b) with a more severe phenotype. | [93] | |
| Mouse | Knockout | -Incompatible with life. | [75] | ||
| Protein N-glycosylation | PMM2-CDG/Phosphomannomutase 2 | Mouse | (a) Homozygous Pmm2R137H/R137H (b) Homozygous Pmm2F118L/F118L (c) Heterozygous Pmm2R137H/F118L | (a) Embryonic lethality. (b) Viable with no phenotype. (c) Embryonic lethality recovered by oral Man administration to pregnant dams. | [77] |
| Mouse | Heterozygous Pmm2R137H/F115L | -Prenatal lethality which could not be restored with Man supplementation. -Survival mice presented delayed growth and impaired general protein glycosylation. | [94] | ||
| Zebrafish | Pmm2 knockout | -Reduced Pmm2 enzymatic activity, decreased LLO levels and Man-6-P accumulation. -Impaired motility and altered craniofacial cartilage development. | [95] | ||
| Drosophila melanogaster | Pmm2-null mutant using CRISPR/Cas9 | -Reduced lifespan, psychomotor retardation and impairment of the synaptic matrix metalloproteinase (MMP) pathway. | [96] | ||
| D. melanogaster | Pmm2 knockdown adults using RNAi | -Severe ataxia, loss of coordination and inability to fly. | [96] | ||
| Multiple and other glycosylation pathways | ATP6AP1-CDG/Accessory subunit of the vacuolar (V)-ATPase protein pump | S. cerevisiae | voa1::H vma21QQ strain | -Voa1 is the yeast homologue for human ATPase H+ transporting accessory protein 1 (ATP6AP1). -E356K and Y313C mutations compromise cell growth. | [97] |
| Zebrafish | Homozygous Atp6ap1ba82/a82 | -Pigmentation defects. | [98] | ||
| Multiple and other glycosylation pathways | ATP6AP1-CDG/Accessory subunit of the vacuolar (V)-ATPase protein pump | Zebrafish | (a) Zygotic Atp6ap1b knockdown (b) Maternal and zygotic Atp6ap1b knockdown | (a) Eye abnormalities at 3–5 days of development. (b) Reduced development of precursor cells (DFCs) which resulted in smaller Kupffer’s vesicle (KV) organ size, due to reduced KV cell number. Defects in the development of ciliated organs, spaw and heart laterality defects were also observed. Loss of Atp6ap1b led to V-ATPase mislocalization and affected DFCs pH. | [98] |
| Mouse | Chimeric model with reduced Atp6ap1 (ac45) expression | -One chimeric female that died approximately 6 weeks after birth. | [99] | ||
| CAD-CDG/Enzyme complex (ATase, CPSase, ATCase and DHOase) | CHO cell line | CHO-G9C CAD-deficient cells | -Reduced levels of uridine diphosphate- N-acetylglucosamine (UDP-GlcNAc), UDP-N-acetylgalactosamine (UDP-GalNAc), UDP-galactose (UDP-Gal), UDP-glucose (UDP-Glc), uridine triphosphate (UTP) and cytosine triphosphate (CTP) (restored by addition of exogenous uridine) as well as less aspartate incorporation into nucleic acids which led to defective growth. | [100] | |
| Caenorhabditis elegans | pyr-1(cu8) mutant | -Maternal effect (mother environment and genotype influence) lethality. -Defective pharynx development. Cytoskeletal organization defects. | [101] | ||
| D. melanogaster | Rudimentary mutant | -High sterility levels in homozygous females. -Reduced viability and defective wing morphogenesis. -Increased survival after uridine, orotic acid, free uracil and carbamoyl aspartic acid supplementation. | [102] | ||
| Multiple and other glycosylation pathways | CAD-CDG/Enzyme complex (ATase, CPSase, ATCase and DHOase) | Zebrafish | Perplexed (plxa52) mutant | -Impaired retinal, tectal fin and jaw morphogenesis. -Phenotype rescue by pyrimidine treatment. | [103] |
| Zebrafish | Transgenic Tg(p2xr3.2:gfp)sl23 mutant | -Cranial sensory circuit malformation. -Small eyes and deformed jaws. -Orotic acid or uridine treatment failed to rescue phenotype. | [104] | ||
| CCDC115-CDG/Coiled-coil domain-containing protein 115 | S. cerevisiae | HY13 (vma22∆::LEU2) KHY34 (vma22∆::LEU2 pep4-3) KHY38 (vma22∆::URA3) KHY39 (vma22∆::URA3 pep4-3) | -No vacuolar ATPase activity. -Normal production of V1 subunits, but lack of association to the vacuolar membrane. -Destabilization of Vph1p and Vma3p/Vma11p subunits. | [105] | |
| HeLa cell line | CCDC115 knockdown using CRISPR/Cas9 | -Impaired iron(II) prolyl hydroxylase (PHD) activity and hypoxia-inducible factor 1 (HIF1α) activation. | [106] | ||
| DOLK-CDG/Dolichol kinase | S. cerevisiae | Sec59 mutant | -Sec59, the homologue of human dolichol kinase (DOLK), catalyzes the phosphorylation of the dolichol lipid carrier. -Thermosensitive. | [18,29] | |
| Multiple and other glycosylation pathways | GNE-CDG/UDP-GlcNAc 2-epimerase/ManNAc kinase | CHO cell line | Gne-deficient lec3 mutant | -Reduced NCAM polysialic acid content. -Lec3 cells are defective in GNE activity. -Sialylation defects were rescued by ManNAc and manosamine complementation. -Expression of Gne mutation M712T is responsible for less celular and glycoprotein-linked sialic acid content (HIBM) whereas R-236L and R266Q produce higher amounts than WT (sialuria). | [19,26,28,107,108] |
| HEK293 cell line | (a) D176V-Gne mutant (b) V572L-Gne mutant (c) Gne knockdown by shRNA | -(a), (b) and (c) have decreasingly levels of sialylation of membrane and cytosolic protein, restored by supplementation with Neu5Ac (SA) and ManNAc. -β-integrin hyposialylation leads to increased cell adhesion. | [39,109] | ||
| Human promyelocytic leukemia (HL60) cell line | HL60-I clone | -Increased overall surface SA content by supplementation with ManNAc and ManNProp. | [110,111] | ||
| Spodoptera frugiperda (Sf9) cell line | M712T Gne | -Reduced activity but not overall sialylation, indicating that disease is not directly caused by lack of SA. | [40] | ||
| Embryonic stem cells (ESC) | Mice GNE−/− ESC | -Impaired proliferation, differentiation and altered genetic expression. -Smaller embryonic bodies size which was corrected by the presence of SA. | [74,112,113,114,115] | ||
| Multiple and other glycosylation pathways | GNE-CDG/UDP-GlcNAc 2-epimerase/ ManNAc kinase | Mouse | Gne−/− | -Incompatible with life. | [74] |
| Mouse | Gne−/+ | -No disease phenotype. | [116] | ||
| Mouse | GneM712T/M712T in a C57BL/6J background | -High lethality within the first 72 h after birth, which was prevented by oral administration of ManNAc to the pregnant and nursing dams. -Muscle hyposialylation at an adult (4 to 6 months) age. -Renal phenotype. | [81,117] | ||
| Mouse | GneM712T/M712T in a mixed genetic background (129Sv/ICR) | -High survival rate. -No renal phenotype. -No muscle deterioration up to 18 months of age. | [76] | ||
| Mouse | Transgenic mouse (Gne(−/−)hGNED176V-Tg) | -Decreased levels of SA in different organs. -Adult onset with muscle pathology. -β-amyloid deposits in the muscles, but not in the CNS. | [118,119] | ||
| Mouse | GneV572L/V572L | -Renal phenotype that was corrected by SA administration. | [79] | ||
| Mouse | Transgenic FVBN-GNR-R263L | -Sialuria with elevated SA in urine. -Increased neural cell adhesion molecule (NCAM) polysialylation. -Increased cell surface sialylation in leucocytes. | [120] | ||
| Multiple and other glycosylation pathways | GNE-CDG/UDP-GlcNAc 2-epimerase/ManNAc kinase | Zebrafish | Gne knockout | -High mortality levels. -Impaired muscle structure and development with consequent decreased locomotor activity. -Slightly reduced GNE enzymatic activity. | [121] |
| NANS-CDG/CMP-N-acetylneuraminic acid synthetase | Zebrafish | Nansa and Nansb knockdown | -Nansb disruption did not generated a disease phenotype. -Nansa morphant embryos displayed a small head with a complex phenotype, pericardial edema and skeletal developmental impairment. -Addition of SA to the zebrafish water resulted in partial rescue of the skeletal phenotype but only if added 24 hpf. | [122] | |
| PGM1-CDG/Phosphogluco-mutase 1 | Hela cell line | Pgm1 and LDB3 two hybrid system | -DC-related LDB3 mutations impair binding of PGM1 to Z-band alternatively spliced PDZ-motif (ZASP)/Cypher. -PGM/PMM domain IV of PGM1 is essential for recruitment of ZASP/Cypher. | [123] | |
| PGM3-CDG/Phosphogluco-mutase 3 | Mouse | (a) Hypomorphic (Pgm3mld1) (b) Null (Pgm3gt) | (a) Embryonic lethality. (b) Reduced viability with no major alteration in general protein glycosylation, except for the testis-specific isoform of angiotensin-converting enzyme (ACE). Reduced size, mild anemia, splenomegaly, thrombocytopenia, glomerulonephritis and low B- and T-cell numbers. | [124] | |
| Multiple and other glycosylation pathways | SLC35A1-CDG/CMP-sialic acid transporter | CHO cell line | Lec2 (Slc35a1) mutants | -Asialo phenotype at the cell membrane and unable to translocate CMP-SA to the lumen of the Golgi. -When combined with large overexpression, the α-DG is functionally glycosylated. | [17,25,26,38,125] |
| CHO cell line | MAR-11 mutant | -Decreased levels of surface SA. | [126] | ||
| Near-haploid human (HAP1) cell line | Slc35a1 knockout using transcription activator-like effector nucleases (TALEN) | -SLC35A1 is required for α-DG mannosylation, independently from sialylation. | [32] | ||
| SLC35A2-CDG/UDP-galactose transporter | CHO cell line | Lec8(Slc35a2) deficient | -Defective galactosylation. -Gal treatment slightly increased galactosylation. | [33] | |
| MDCK-RCAr cell line | Slc35a2 deficient | -Defective galactosylation. -UGT1 and UGT2 are localized in the Golgi the ER, respectively. -UGT forms complexes with NGT and Mgats. | [20,21,33] | ||
| C. elegans | Srf-3 mutants | -Reduced O- and N-linked glycans. | [127] | ||
| SCL35C1-CDG/GDP-fucose transporter | CHO cell line | Slc35c1 knockout (CHO-gmt5) derived from MAR-11 mutants. | -Asialylated and afucosylated proteins due to absence of functional CMP-sialic acid and GDP-fucose transporter. | [128,129] | |
| CHO cell line | Slc35c1 disruption by zinc fingers, TALEN and CRISPR (CHO-gmt3) | -Lack of functional GDP-fucose transporter. -Fucose free glycoproteins. | [130] | ||
| ESC cell line | Slc35c1 knockout | -Abolishment of N- and O-glycoproteins fucosylation. -Ricin resistant. | [131] | ||
| Multiple and other glycosylation pathways | SCL35C1-CDG/GDP-fucose transporter | Mouse | Slc35c1 (Slc35c1−/−) knockout | -Elevated postnatal mortality, severe growth retardation and immune system affectation. -Significant reduction of fucosylated selectin ligans as well as severe impairment of P-, E- and L-selectin ligand function. -Defective neutrophil migration to the inflamed peritoneum, reduced leukocyte rolling to inflamed muscle venules and absent lymphocyte homing to lymph nodes but normal homing of lymphocytes to the spleen. Normal very long chain polyunsaturated fatty acids (VLC-PUFAs) levels. | [132,133,134] |
| Zebrafish | Slytherin (srn) mutant with a point mutation | -Bent tail that became progressively more severe. -Reduced protein fucosylation in CNS and other tissues. -Reduced number of neurons and glia cells. -Signaling reduction in the Notch-Delta pathway leading to defective neuromuscular synaptogenesis. | [135,136] | ||
| Zebrafish | Slc35c1 protein over-expression | -Increased N-linked fucosylation and disruption of embryonic patterning. -Negative regulation of Wnt signaling. | [137] | ||
| SLC39A8-CDG/Solute carrier family 39 (zinc transporter), member 8—ZIP8 | Mouse | Slc39a8−/− knockout | -Impaired cardiovascular function, absence of sternum, small chest cavity and a small liver. | [85] | |
| Mouse | Hypomorphic Slc39a8(neo/neo) | -Reduced mRNA and protein levels of the ZIP8 Zn2+/(HCO3-)2 symporter in several tissues of the neonate mutants. -Reduced zinc and iron levels. -Embryonic and neonatal lethality. -Surviving offspring was pale, presented growth arrest, severe anemia, hypoplastic spleen, hypoplasia of liver, kidney, lung and lower extremities. | [138,139] | ||
| SRD5A3-CDG/Steroid 5 α-reductase 3 | S. cerevisiae | Dfg10-100 | -SRD5A3 is the human ortholog of yeast dfg10. -Defective filamentous growth and carboxypeptidase Y (CPY) hypoglycosylation. -Defective metabolism of polyprenol to dolichol. | [140] | |
| Multiple and other glycosylation pathways | SRD5A3-CDG/Steroid 5 α-reductase 3 | Mouse | Homozygous (Srd5a3Gt/Gt) | -Complete embryonic lethality beyond E12.5. -Homozygous embryos were smaller and displayed dilated hearts and open neural tubes. -Transcriptomic analysis revealed an up-regulation of the unfolded protein response (UPR) and a downregulation of genes involved in general cellular metabolic processes and specific embryonic development. -Elevated polyprenol levels. | [140] |
| TMEM165-CDG/Transmembrane protein 165 | S. cerevisiae | Gdt1∆ | -Growth defect and defective glycosylation in high Ca2+ concentration which are supressed by Mn2+ administration. -Gdt1p controls cellular calcium stores and respond to osmotic shock. | [30,141,142] | |
| HEK293 cell line | TMEM165 knockout by CRISPR | -TMEM165 degradation in lysosomes upon Mn2+ exposure. | [36] | ||
| HEK293 cell line | TMEM165 knockdown by shRNA | -Impaired Golgi Mn2+ homeostasis. -Mn2+ rescues impaired Golgi glycosylation. | [142] | ||
| HeLa cell line | TMEM165 knockdown by shRNA | -Impaired Golgi Mn2+ homeostasis. | [142] | ||
| Multiple and other glycosylation pathways | TMEM165-CDG/Transmembrane protein 165 | Zebrafish | Homozygous null tmem165 (tmem165−/−) | -Dysfunctional N-glycosylation, reduced osteoblast differentiation and altered craniofacial cartilage development due to defects in chondrocyte maturation. | [143] |
| TMEM199-CDG/Transmembrane protein 199 | S. cerevisiae | DJY62/DJY102 (pep4–3 vma12∆::LEU2) DJY63 (vma12∆::LEU2) | -Decreased stability of 100-kDa V0 subunit (Vph1p) of V-ATPase. | [105,144] | |
| HeLa cell line | TMEM199 knockdown by CRISPR/Cas9 | -Lethality after three weeks and accumulation of HIF1α. | [106] | ||
| Lipid glycosylation and glycosylphosphatidyl inositol (GPI) synthesis | PIGA-CDG/Phosphatidylinositol N-acetylglucosaminyl-transferase (subunit A) | iPSC | (a) Hypomorphic (PIGAc.1234C>T) (b) PIGA null | (a) Permissive for hematopoiesis with neuronal proliferation, differentiation, maturation and presynaptic defects. (b) Non-permissive for hematopoiesis and differentiation. | [145] |
| Mouse | Piga-deficient chimeric mice | -Chimeric surface expression of GPI-anchored proteins. | [146] | ||
| Mouse | Partial exon 2 excision mediated by loxP | -Viable mosaic mice with lack of GPI-linked proteins on a proportion of circulating blood cells. -Increased sensitivity toward complement mediated lysis and a decreased life span in circulation. | [147] | ||
| PIGM-CDG/GPI α-1,4-mannosyltransferase I | Ramos517 cell line | PIGM-deficient | -Cloning of human homologues PfPIG-M (Plasmodium falciparum) and GPI14 (S. cerevisiae) but only PfPIG-M restored cell-surface expression of GPI proteins. | [148] | |
| PIGM-CDG/GPI α-1,4-mannosyltransferase I | S. cerevisiae | Gpi14 (PIGM homolog) -deficient | -Glucosaminyl(acyl)phosphatidylinositol accumulation. -Growth lethality. | [149] | |
| Lipid glycosylation and GPI synthesis | PIGO-CDG/GPI ethanolamine phosphate transferase 3 | CHO cell line | PIGO-deficient cells | -Impaired levels of CD59 and urokinase receptor (uPAR). | [35,37] |
| HEK293 cell line | PIGO knockout using CRISPR/Cas9 | -Impaired GPI-AP expression. | [35] | ||
| O-mannosyl-glycan synthesis | ISPD-CDG/2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase | HEK293 cell line | Ispd knockout using CRISPR/Cas9 | -Reduced α-DG glycosylation. -ISPD synthesize CDP-ribitol required for α-DG glycosylation. | [68] |
| HAP1 cell line | Ispd-disrupted cells by a CRISPR/Cas9 deletion causing a frameshift | -Supplementation with CDP-Rbo restored α-DG glycosylation. | [150] | ||
| Mouse | Homozygous IspdL79*/L79* | -No embryonic lethality, but did not survived beyond birth due to apparent respiratory failure. -Normal total dystroglycan protein levels, but severely reduced levels of α-DG and laminin binding activity in brain extracts. -Lose of dystroglycan glycosylation in cortex extracts. | [151] | ||
| O-mannosyl-glycan synthesis | ISPD-CDG/2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase | Mouse | Ispd conditional knockout using Cas9 nickase (Cas9n) and a single guide RNA (sgRNA) | -No phenotype available. | [152] |
| Zebrafish | Ispd knock-out | -Incomplete brain folding in the majority of the embryos as well as hydrocephalus, reduced eye size, muscle fiber degeneration and impaired motility. -Protein hypoglycosylation, especially for α-DG. | [153] |
| CDG/Protein | Biomarker | Sample * | Major Findings | Detection Technique | Reference |
|---|---|---|---|---|---|
| CDG-I/CDG-II | Transferrin | Serum/plasma | Altered glycosylation pattern | IEF, high performance liquid chromatography (HPLC), capilary zone electrophoresis (CZE) | [7,156] |
| CDG-I/CDG-II | α1-antitrypsin | Serum/plasma | Altered glycosylation pattern | 2-Dimensional difference gel electrophoresis (2D DIGE), IEF | [156] |
| CDG-II | Lipoprotein ApoCIII | Serum/plasma | Altered profile of the three protein isoforms: apoCIII0, apoCIII1 and apoCIII2 (hypoglycosylation) | IEF | [4,7,157] |
| ALG1-CDG/β-1,4-mannosyl-transferase | Tetrasaccharide (NeuAc-Gal-GlcNAc2) | Serum/plasma TF | Increased levels | Liquid chromatography—mass spectrometry (LC/MS) and enzymatic digestions | [34,176] |
| ALG6-CDG/Glucosyl-Transferase | Man9GlcNAc2-P-P-dolichol | Fibroblasts | Increased levels | 2-[3H]mannose labeling and HPLC analysis | [177] |
| ALG1-CDG/β-1,4-mannosyl-transferase, MPI-CDG/Phospho-mannose isomerase, PMM2-CDG/Phosphomannomutase 2 | N-tetrasaccharide (Neu5Ac_2,6Gal_1,4-GlcNAc_1,4GlcNAc) | Sera, plasma and fibroblasts | Increased levels compared to control | LC-MS/MS Matrix assisted laser desorption/ionization-time of flight-mass spectrometry (MALDI-TOF-MS) | [178] |
| CDG/Protein | Biomarker | Sample * | Major Findings | Detection Technique | Reference |
|---|---|---|---|---|---|
| CDG-I | Glyc-ER-GFP | Fibroblasts and iPSCs | Fluorescence | Flow Cytometry | [179] |
| CDG-I | Thyroxine binding globulin | Serum | Abnormal glycosylation | IEF | [180] |
| CDG-II | Glyc-ER-GFP | Fibroblasts | No fluorescence | Flow Cytometry | [179] |
| CDG-II | (a) α1-acid glycoprotein (b) Ceruloplasmin | Serum | (a) Extra isoform with higher pI value (b) Subtle pI change | 2D DIGE | [155] |
| GNE-CDG/UDP-GlcNAc 2-epimerase/ManNAc kinase | GM3 and GD3 gangliosides | Human embryonic kidney (HEK AD293) cells Muscle of GneM712T/M712T mouse model | Increased levels | Flow cytometry HPLC | [181,182] |
| NCAM | Brain solubilisates from heterozygous GNE-deficient mice Serum obtained from patients and GneM712T/M712T mouse | Hypolysialylated | WB | [116,183] | |
| Thomsen-Friedenreich (T)-antigen | Plasma | Increased ratio of T-antigen (Gal-GalNAc-) to ST (sialylated)- antigen (core 1 SA-Gal-GalNAc-) | MS | [184] | |
| PMM2-CDG/Phosphomannomutase 2 | Band 3 and glycophorin A | Erythrocytes | Underglycosylated | SDS-PAGE | [185] |
| Glycosphingolipids (Gb3, GM2, GD3 and GD1a) | Fibroblasts | Increased levels compared to control | Radiolabeling followed by HPTLC | [186] | |
| PMM2-CDG/Phosphomannomutase 2 | (a) α1-acid glycoprotein (b) Ceruloplasmin (c) α1-antichymotrypsin (d) α1B-glycoprotein (e) Haptoglobin | Serum | (a) Lower MW isoform (b) Profound pI change (c) Abnormal profile (d) Abnormal profile (e) Absence of stainable protein | 2D DIGE | [155,187] |
| TSH and TF4 | Serum | Increased TSH and decreased TF4 levels | Immune-based method | [4,188] | |
| ALG6-CDG/Glucosyl-Transferase, PMM2-CDG/Phosphomannomutase 2 | β-trace protein | Cerebrospinal fluid | Abnormal glycosylation profile | SDS-PAGE and immunoblotting | [180,189] |
| ALG6-CDG/Glucosyl-Transferase, MPI-CDG/Phospho-mannose isomerase, PMM2-CDG/Phosphomannomutase 2 | Aspartylglucosaminidase (AGA) | Plasma | Increased levels | Enzymatic activity | [166] |
| ALG13-CDG/UDP-GlcNAc transferase, MPI-CDG/Phospho-mannose isomerase, PMM2-CDG/Phosphomannomutase 2 | ICAM-1 | (a) Lec9 CHO cells and fibroblasts (b) Mesenteric endothelial cells of Mpi−/− mouse | Decreased levels | (a) LC-MS/MS SDS-PAGE and WB IF staining Flow Cytometry (b) Immuno- histochemistry | [174,175,190] |
| Study Identifier | Status | Study Title | Condition | Intervention | Study Characteristics | Study Type |
|---|---|---|---|---|---|---|
| NCT02089789§ | Active, recruiting | Clinical and basic investigations into known and suspected Congenital Disorders of Glycosylation | CDG | N.A | N.A | Observational |
| NCT02503267§ | Active, recruiting | Incidence and consequences of Disorders of Glycosylation in patients with conotruncal and septal heart defects (CARDIoG) | CDG | N.A | N.A | Observational |
| NCT03250728§ | Active, not recruiting | Role of the endothelium in stroke-like episode among CDG Patients (PECDG) | CDG | N.A | N.A | Interventional Peripheral blood puncture |
| NCT02955264§ [208] | Active, recruiting | Using d-Galactose as a food supplement in Congenital Disorders of Glycosylation | CDG | Drug: d-galactose (dietary supplement) Administration: Oral | Open label, single group | Interventional Phase 2 |
| NCT02346461§ | Active, not recruiting | An open label Phase 2 study of ManNAc in subjects with GNE Myopathy | -GNE-CDG | Drug: ManNAc Administration: Oral | Open label, Non-randomized | Interventional Phase 2 |
| NCT01634750§ [264] | Completed | Phase I clinical trial of ManNAc in patients with GNE Myopathy or Hereditary Inclusion Body Myopathy (HIBM) | -GNE-CDG -Hereditary Inclusion Body Myopathy (HIBM) | Drug: ManNAc Administration: Oral | Randomized, double-blind, placebo-controlled | Interventional Phase 1 |
| NCT02736188§∞ 2016-000360-42 *∞ | Active, not recruiting | Study to evaluate the safety and efficacy of Ace-ER Tablets in patients with GNE Myopathy or Hereditary Inclusion Body Myopathy | -GNE-CDG -HIBM -Quadriceps Sparing Myopathy -Distal Myopathy With Rimmed Vacuoles | Drug: Aceneuramic Acid Extended-Release Tablets (Ace-ER) Administration: Oral | Open label, single group | Interventional Phase 3 |
| NCT02731690§≈ 2015-004553-41 *≈ | Active, not recruiting | A study to evaluate the safety of Aceneuramic Acid Extended Release (Ace-ER) tablets in GNE Myopathy (GNEM) (Also Known as Hereditary Inclusion Body Myopathy (HIBM)) patients with severe ambulatory impairment | -GNE-CDG | Drug: Aceneuramic Acid Extended-Release tablets (Ace-ER) Administration: Oral | Open label, single group | Interventional Phase 2 |
| NCT01517880§ [265] | Completed | A phase 2 study to evaluate the dose and pharmacodynamic efficacy of Sialic Acid-Extended Release (SA-ER) tablets in patients with GNE Myopathy or Hereditary Inclusion Body Myopathy (HIBM) | -GNE-CDG -HIBM | Drug: Sialic Acid Extended Release (SA-ER) Administration: Oral | Randomized, double-blind, placebo-controlled | Interventional Phase 2 |
| NCT01830972§ | Completed | An open label phase 2 extension study of higher dose Sialic Acid (ER Tablets + IR Capsules) in patients with GNE Myopathy | -GNE-CDG | Drug: Sialic Acid Extended Release (SA-ER) Sialic Acid Immediate Release (SA-IR) Administration: Oral | Open label, non-randomized | Interventional Phase 2 |
| NCT01359319§ [265] | Completed | Safety and pharmacokinetics of Sialic Acid tables in patients With Hereditary Inclusion Body Myopathy (HIBM) | -GNE-CDG -HIBM | Drug: Sialic Acid Extended Release (SA-ER) tablets Administration: Oral | Open label, non-randomized, single group | Interventional Phase 1 |
| NCT02377921§ | Completed | Phase 3 randomized, double-blind, placebo-controlled study to evaluate Sialic Acid in patients with GNE Myopathy or Hereditary Inclusion Body Myopathy (HIBM) | -GNECDG -HIBM | Drug: Sialic Acid Tablets (UX001) Administration: Oral | Randomized, double-blind, placebo-controlled | Interventional Phase 3 |
| NCT00195637§ [266] | Completed | Intravenous immune globulin to treat Hereditary Inclusion Body Myopathy | -GNE-CDG -HIBM | Drug: Immune Globulin Administration: intravenous | Pilot study with 4 participants | Interventional Phase 1 |
| NCT01236898§ | Completed | Pharmacokinetic study on N-acetylneuraminic Acid | -GNE-CDG -HIBM | Drug: N-acetylneuraminic acid (anhydride) (NPC-09) Administration: Oral | Open label, non-randomized, single group | Interventional Phase 1 |
| NCT01784679§ | Active, recruiting | GNE-Myopathy disease monitoring program (GNEM-DMP): a registry and prospective observational natural history study to assess GNE Myopathy or Hereditary Inclusion Body Myopathy (HIBM) | -GNE-CDG -HIBM | N.A | N.A | Observational |
| NCT02196909§ | Active, not recruiting | Clinical, biological and NMR outcome measures study for Hereditary Inclusion Body Myopathy due to mutation of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene (GNE) (ClinBio-GNE) | -GNE-CDG -HIBM | N.A | N.A | Interventional Parallel assignment with blood and urine collection |
| NCT01902940§ | Completed | Natural history in CCFDN and IBM syndromes | -GNE-CDG -HIBM | N.A | N.A | Observational |
| NCT01417533§ | Active, recruiting | A natural history study of patients with GNE Myopathy | -GNE-CDG -HIBM | N.A | N.A | Observational |
| NCT03173300§ | Active, recruiting | Natural history study protocol in PMM2-CDG (CDG-Ia) | PMM2-CDG | N.A | N.A | Observational |
| 2017-000810-44 * | Active | Phase II clinical trial to evaluate the effectiveness and safety of acetazolamide in the treatment of cerebellar syndrome in patients with PMM2-CDG deficiency | PMM2-CDG | Drug: Acetazolamide Administration: Oral | Randomized, open labeled | Interventional Phase 2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brasil, S.; Pascoal, C.; Francisco, R.; Marques-da-Silva, D.; Andreotti, G.; Videira, P.A.; Morava, E.; Jaeken, J.; Dos Reis Ferreira, V. CDG Therapies: From Bench to Bedside. Int. J. Mol. Sci. 2018, 19, 1304. https://doi.org/10.3390/ijms19051304
Brasil S, Pascoal C, Francisco R, Marques-da-Silva D, Andreotti G, Videira PA, Morava E, Jaeken J, Dos Reis Ferreira V. CDG Therapies: From Bench to Bedside. International Journal of Molecular Sciences. 2018; 19(5):1304. https://doi.org/10.3390/ijms19051304
Chicago/Turabian StyleBrasil, Sandra, Carlota Pascoal, Rita Francisco, Dorinda Marques-da-Silva, Giuseppina Andreotti, Paula A. Videira, Eva Morava, Jaak Jaeken, and Vanessa Dos Reis Ferreira. 2018. "CDG Therapies: From Bench to Bedside" International Journal of Molecular Sciences 19, no. 5: 1304. https://doi.org/10.3390/ijms19051304
APA StyleBrasil, S., Pascoal, C., Francisco, R., Marques-da-Silva, D., Andreotti, G., Videira, P. A., Morava, E., Jaeken, J., & Dos Reis Ferreira, V. (2018). CDG Therapies: From Bench to Bedside. International Journal of Molecular Sciences, 19(5), 1304. https://doi.org/10.3390/ijms19051304