Growth Hormone Receptor Mutations Related to Individual Dwarfism


Abstract
:1. Introduction
2. GHR Function and Process
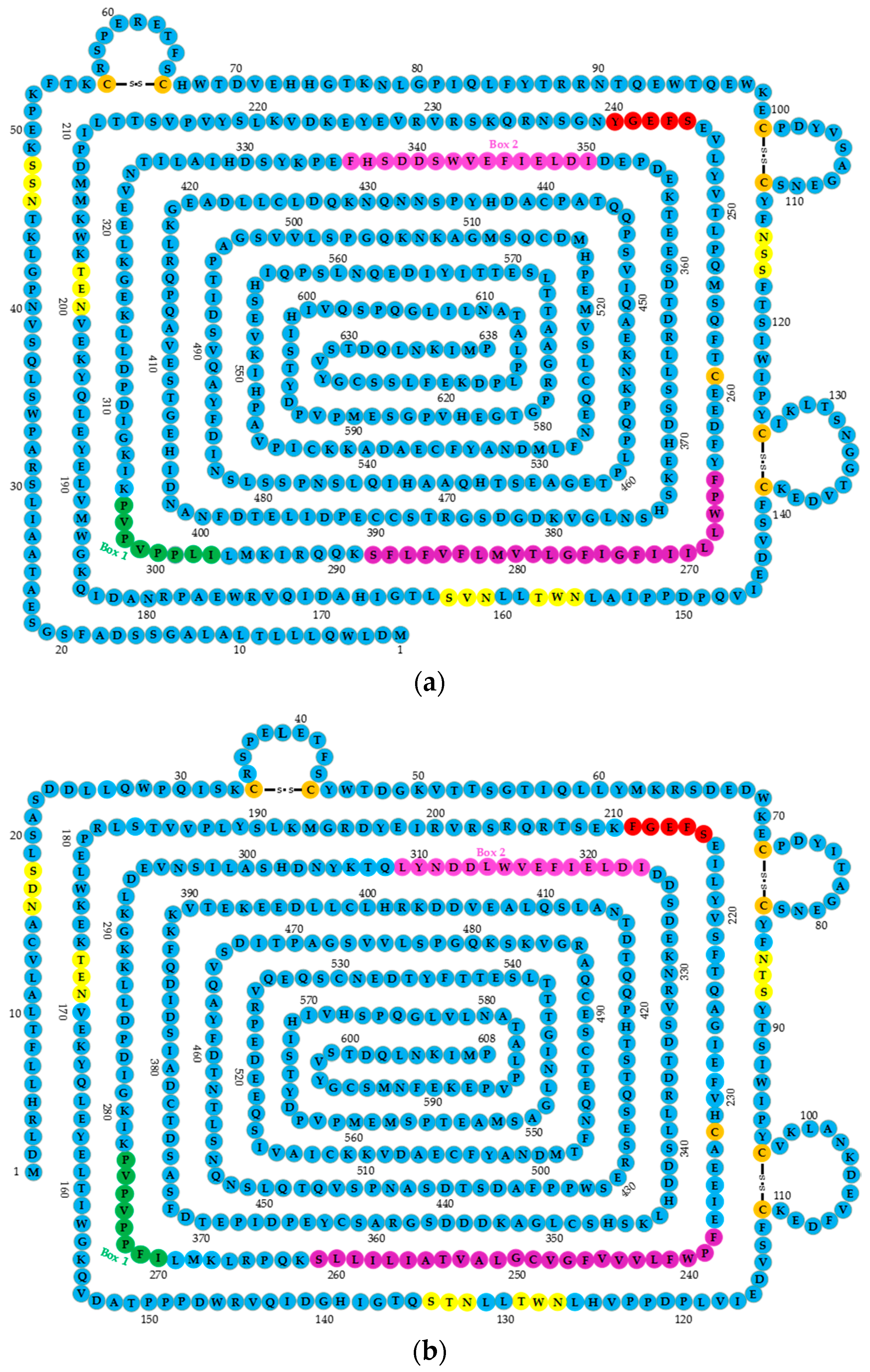
2.1. GHR Structure
2.2. GHR Signal Transduction Mechanism
2.2.1. GH and GHR Recognition Event
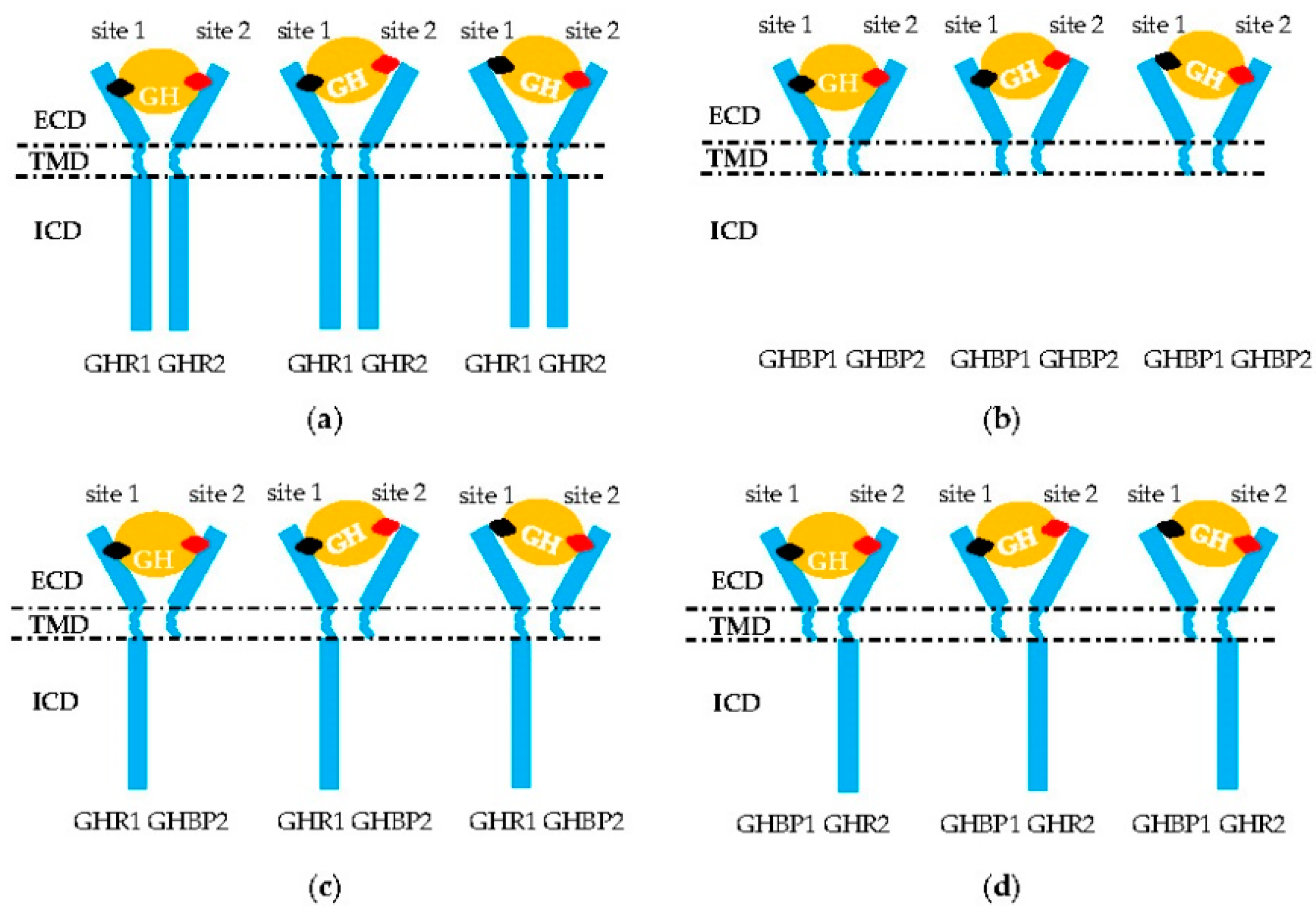
2.2.2. GHR Dimerization
2.2.3. GH Binding to GHR Dimer
2.2.4. GHR Mediating GH to Janus Activating Tyrosine Kinase (JAK)/STAT Signature Transduction
3. GHR Polymorphisms and Individual Dwarfism
3.1. GHR Gene Mutations that Causing Aberrant GH–GHR Binding
3.2. GHR Gene Mutations Causing Aberrant GHR Dimerization
3.3. GHR Mutations Causing GHR Failure of GH Delivery to Downstream Genes
3.4. Mutations Resulting in GHR Expression Failure
3.5. GHR Regulates Development of Both Bone and Muscle Fiber
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GH | Growth hormone |
| GHR | GH receptor |
| GHBP | GH binding protein |
| IGF | Insulin-like growth factor |
| LS | Laron syndrome |
| SLD | Sex-linked dwarf |
| ECD | Extracellular domain |
| TMD | Transmembrane domain |
| ECD | Intracellular domain |
| JAK | Janus kinase |
| STAT | Signal transducers and activators of transcription |
References
- Conway-Campbell, B.L.; Brooks, A.J.; Robinson, P.J.; Perani, M.; Waters, M.J. The extracellular domain of the growth hormone receptor interacts with coactivator activator to promote cell proliferation. Mol. Endocrinol. 2008, 22, 2190–2202. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Li, H.; Mu, H.; Luo, W.; Li, Y.; Jia, X.; Wang, S.; Jia, X.; Nie, Q.; Li, Y.; et al. Let-7b regulates the expression of the growth hormone receptor gene in deletion-type dwarf chickens. BMC Genom. 2012, 13, 306. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.J. The growth hormone receptor. Growth Horm. IGF Res. 2016, 28, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Moreno, C.; Calderón-Vallejo, D.; Harvey, S.; Arámburo, C.; Quintanar, J. Growth Hormone (GH) and Gonadotropin-Releasing Hormone (GnRH) in the Central Nervous System: A Potential Neurological Combinatory Therapy? Int. J. Mol. Sci. 2018, 19, 375. [Google Scholar] [CrossRef] [PubMed]
- Reh, C.S.; Geffner, M.E. Somatotropin in the treatment of growth hormone deficiency and Turner syndrome in pediatric patients: A review. Clin. Pharmacol. 2010, 2, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Soendergaard, C.; Young, J.; Kopchick, J. Growth Hormone Resistance—Special Focus on Inflammatory Bowel Disease. Int. J. Mol. Sci. 2017, 18, 1019. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.; Giordano, M.; Frechette, R.; Bellone, S.; Polychronakos, C.; Legault, L.; Deal, C.L.; Goodyer, C.G. Genetic variations at the humangrowth hormone receptor (GHR) gene locus are associated with idiopathic short stature. J. Cell. Mol. Med. 2017, 21, 2985–2999. [Google Scholar] [CrossRef] [PubMed]
- Porto, W.F.; Marques, F.A.; Pogue, H.B.; de Oliveira Cardoso, M.T.; Do Vale, M.G.R.; Da Silva Pires, Á.; Franco, O.L.; de Alencar, S.A.; Pogue, R. Computational Investigation of Growth Hormone Receptor Trp169Arg Heterozygous Mutation in a Child With Short Stature. J. Cell. Biochem. 2017, 118, 4762–4771. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, A.L. A half-century of studies of growth hormone insensitivity/Laron syndrome: A historical perspective. Growth Horm. IGF Res. 2016, 28, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Arman, A.; Ozon, A.; Isguven, P.S.; Coker, A.; Peker, I.; Yordam, N. Novel splice site mutation in the growth hormone receptor gene in Turkish patients with Laron-type dwarfism. J. Pediatr. Endocrinol. Metab. 2008, 21, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Hull, K.L.; Marsh, J.A.; Harvey, S. A missense mutation in the GHR gene of Cornell sex-linked dwarf chickens does not abolish serum GH binding. J. Endocrinol. 1999, 161, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Hui, H.N.; Metherell, L.A.; Ng, K.L.; Savage, M.O.; Camacho-Hubner, C.; Clark, A.J. Novel growth hormone receptor mutation in a Chinese patient with Laron syndrome. J. Pediatr. Endocrinol. Metab. 2005, 18, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Pagani, S.; Petkovic, V.; Messini, B.; Meazza, C.; Bozzola, E.; Mullis, P.; Bozzola, M. Heterozygous GHR gene mutation in a child with idiopathic short stature. J. Pediatr. Endocrinol. Metab. 2014, 27, 329–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadagurski, M.; Landeryou, T.; Cady, G.; Kopchick, J.J.; List, E.O.; Berryman, D.E.; Bartke, A.; Miller, R.A. Growth hormone modulates hypothalamic inflammation in long-lived pituitary dwarf mice. Aging Cell 2015, 14, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, Y.; Liu, H.; Zhang, X.; Liu, C.; Tian, K.; Bolund, L.; Dou, H.; Yang, W.; Yang, H.; et al. Transgenic Wuzhishan minipigs designed to express a dominant-negative porcine growth hormone receptor display small stature and a perturbed insulin/IGF-1 pathway. Transgenic Res. 2015, 24, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Jiang, Q.; Wu, D.; Lan, G.; Fan, J.; Guo, Y.; Chen, B.; Yang, X.; Jiang, H. Correlation analysis between expression levels of hepatic growth hormone receptor, janus kinase 2, insulin-like growth factor-i genes and dwarfism phenotype in Bama minipig. J. Nanosci. Nanotechnol. 2015, 15, 1789–1792. [Google Scholar] [CrossRef] [PubMed]
- Boegheim, I.; Leegwater, P.; van Lith, H.A.; Back, W. Current insights into the molecular genetic basis of dwarfism in livestock. Vet. J. 2017, 224, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; Cogburn, L.A.; Burnside, J. Dysfunctional growth hormone receptor in a strain of sex-linked dwarf chicken: Evidence for a mutation in the intracellular domain. J. Endocrinol. 1994, 142, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Hull, K.L.; Harvey, S. Growth hormone resistance: Clinical states and animal models. J. Endocrinol. 1999, 163, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Lin, S.; Li, G.; Nie, Q.; Zhang, X. Integrative analyses of miRNA-mRNA interactions reveal let-7b, miR-128 and MAPK pathway involvement in muscle mass loss in sex-linked dwarf chickens. Int. J. Mol. Sci. 2016, 17, 276. [Google Scholar] [CrossRef] [PubMed]
- Fassone, L.; Corneli, G.; Bellone, S.; Camacho-Hubner, C.; Aimaretti, G.; Cappa, M.; Ubertini, G.; Bona, G. Growth hormone receptor gene mutations in two Italian patients with Laron Syndrome. J. Endocrinol. Investig. 2007, 30, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Gennero, I.; Edouard, T.; Rashad, M.; Bieth, E.; Conte-Aurio, F.; Marin, F.; Tauber, M.; Salles, J.P.; El, K.M. Identification of a novel mutation in the human growth hormone receptor gene (GHR) in a patient with Laron syndrome. J. Pediatr. Endocrinol. Metab. 2007, 20, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Derr, M.A.; Aisenberg, J.; Fang, P.; Tenenbaum-Rakover, Y.; Rosenfeld, R.G.; Hwa, V. The growth hormone receptor (GHR) c.899dupC mutation functions as a dominant negative: Insights into the pathophysiology of intracellular GHR defects. J. Clin. Endocrinol. Metab. 2011, 96, E1896–E1904. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.J.; Brooks, A.J. Growth hormone receptor: Structure function relationships. Horm. Res. Paediatr. 2011, 76, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Langenheim, J.F.; Wang, X.; Jiang, J.; Chen, W.Y.; Frank, S.J. Activation of growth hormone receptors by growth hormone and growth hormone antagonist dimers: Insights into receptor triggering. Mol. Endocrinol. 2008, 22, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Baudet, M.L.; Harvey, S. Small chicken growth hormone (scGH) variant in the neural retina. J. Mol. Neurosci. 2007, 31, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Barclay, J.L.; Kerr, L.M.; Arthur, L.; Rowland, J.E.; Nelson, C.N.; Ishikawa, M.; D’Aniello, E.M.; White, M.; Noakes, P.G.; Waters, M.J. In vivo targeting of the growth hormone receptor (GHR) Box1 sequence demonstrates that the GHR does not signal exclusively through JAK2. Mol. Endocrinol. 2010, 24, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A. Binding in the growth hormone receptor complex. Proc. Natl. Acad. Sci. USA 1996, 93, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Behncken, S.N.; Waters, M.J. Molecular recognition events involved in the activation of the growth hormone receptor by growth hormone. J. Mol. Recognit. 1999, 12, 355–362. [Google Scholar] [CrossRef]
- De Vos, A.M.; Ultsch, M.; Kossiakoff, A.A. Human growth hormone and extracellular domain of its receptor: Crystal structure of the complex. Science 1992, 255, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, A.L. Physiology and disorders of the growth hormone receptor (GHR) and GH-GHR signal transduction. Endocrine 2000, 12, 107–119. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, J.; Lepik, B.; Zhang, Y.; Zinn, K.R.; Frank, S.J. Subdomain 2, not the transmembrane domain, determines the dimerization partner of growth hormone receptor and prolactin receptor. Endocrinology 2017, 158, 3235–3248. [Google Scholar] [CrossRef] [PubMed]
- Huo, S.; Massova, I.; Kollman, P.A. Computational alanine scanning of the 1:1 human growth hormone-receptor complex. J. Comput. Chem. 2002, 23, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Gent, J.; van Kerkhof, P.; Roza, M.; Bu, G.; Strous, G.J. Ligand-independent growth hormone receptor dimerization occurs in the endoplasmic reticulum and is required for ubiquitin system-dependent endocytosis. Proc. Natl. Acad. Sci. USA 2002, 99, 9858–9863. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.J.; Dai, W.; O’Mara, M.L.; Abankwa, D.; Chhabra, Y.; Pelekanos, R.A.; Gardon, O.; Tunny, K.A.; Blucher, K.M.; Morton, C.J.; et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science 2014, 344, 1249783. [Google Scholar] [CrossRef] [PubMed]
- Smit, L.S.; Meyer, D.J.; Billestrup, N.; Norstedt, G.; Schwartz, J.; Carter-Su, C. The role of the growth hormone (GH) receptor and JAK1 and JAK2 kinases in the activation of Stats 1, 3, and 5 by GH. Mol. Endocrinol. 1996, 10, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Arman, A.; Yuksel, B.; Coker, A.; Sarioz, O.; Temiz, F.; Topaloglu, A.K. Novel growth hormone receptor gene mutation in a patient with Laron syndrome. J. Pediatr. Endocrinol. Metab. 2010, 23, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, F.T.; Fridman, C.; Pinto, E.M.; Guevara-Aguirre, J.; Shevah, O.; Rosembloom, A.L.; Hwa, V.; Cassorla, F.; Rosenfeld, R.G.; Lins, T.S.; et al. The E180splice mutation in the GHR gene causing Laron syndrome: Witness of a Sephardic Jewish exodus from the Iberian Peninsula to the new world? Am. J. Med. Genet. Part A 2014, 164A, 1204–1208. [Google Scholar] [CrossRef] [PubMed]
- Ostrer, H. The origin of the p.E180 growth hormone receptor gene mutation. Growth Horm. IGF Res. 2016, 28, 51–52. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Li, F.; Li, Q.; Li, J.; Zhao, Y.; Hu, X.; Zhang, R.; Li, N. Generation of a miniature pig disease model for human Laron syndrome. Sci. Rep. 2015, 5, 15603. [Google Scholar] [CrossRef] [PubMed]
- Sobrier, M.L.; Dastot, F.; Duquesnoy, P.; Kandemir, N.; Yordam, N.; Goossens, M.; Amselem, S. Nine novel growth hormone receptor gene mutations in patients with Laron syndrome. J. Clin. Endocrinol. Metab. 1997, 82, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, F.; Dai, Y.; Bao, X.; Jin, Y. A novel mutation of the growth hormone receptor gene (GHR) in a Chinese girl with Laron syndrome. J. Pediatr. Endocrinol. Metab. 2003, 16, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Putzolu, M.; Meloni, A.; Loche, S.; Pischedda, C.; Cao, A.; Moi, P. A homozygous nonsense mutation of the human growth hormone receptor gene in a Sardinian boy with Laron-type dwarfism. J. Endocrinol. Investig. 1997, 20, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Riedl, S.; Amselem, S.; Pratt, K.L.; Little, B.M.; Haeusler, G.; Hwa, V.; Frisch, H.; Rosenfeld, R.G. Primary growth hormone (GH) insensitivity and insulin-like growth factor deficiency caused by novel compound heterozygous mutations of the GH receptor gene: Genetic and functional studies of simple and compound heterozygous states. J. Clin. Endocrinol. Metab. 2007, 92, 2223–2231. [Google Scholar] [CrossRef] [PubMed]
- Moia, S.; Tessaris, D.; Einaudi, S.; de Sanctis, L.; Bona, G.; Bellone, S.; Prodam, F. Compound heterozygosity for two GHR missense mutations in a patient affected by Laron Syndrome: A case report. Ital. J. Pediatr. 2017, 43, 94. [Google Scholar] [CrossRef] [PubMed]
- Bonioli, E.; Taro, M.; Rosa, C.L.; Citana, A.; Bertorelli, R.; Morcaldi, G.; Gastaldi, R.; Coviello, D.A. Heterozygous mutations of growth hormone receptor gene in children with idiopathic short stature. Growth Horm. IGF Res. 2005, 15, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Duriez, B.; Sobrier, M.L.; Duquesnoy, P.; Tixier-Boichard, M.; Decuypere, E.; Coquerelle, G.; Zeman, M.; Goossens, M.; Amselem, S. A naturally occurring growth hormone receptor mutation: In vivo and in vitro evidence for the functional importance of the WS motif common to all members of the cytokine receptor superfamily. Mol. Endocrinol. 1993, 7, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Ipek, M.; Keth, A.; Minnemann, T.; von Mach, M.A.; Weise, A.; Ittner, J.R.; Nawroth, P.P.; Plöckinger, U.; Stalla, G.K.; et al. Short stature and decreased insulin-like growth factor I (IGF-I)/growth hormone (GH)-ratio in an adult GH-deficient patient pointing to additional partial GH insensitivity due to a R179C mutation of the growth hormone receptor. Growth Horm. IGF Res. 2007, 17, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Kim, O.S.; Kim, J.H.; Lee, S.K.; Park, Y.J.; Baik, H.W. A novel mutation of exon 7 in growth hormone receptor mRNA in a patient with growth hormone insensitivity syndrome and neurofibromatosis type I. Int. J. Mol. Med. 2012, 30, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Diniz, E.T.; Jorge, A.A.; Arnhold, I.J.; Rosenbloom, A.L.; Bandeira, F. Novel nonsense mutation (p.Y113X) in the human growth hormone receptor gene in a Brazilian patient with Laron syndrome. Arq. Bras. Endocrinol. Metabol. 2008, 52, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.E.; Lima, C.H.; Ogino, L.L.; Kasuki, L.; Gadelha, M.R. Growth hormone receptor exon 3 isoforms may have no importance in the clinical setting of multiethnic Brazilian acromegaly patients. Pituitary 2016, 19, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, A.L.; Guevara-Aguirre, J. Lessons from the genetics of laron syndrome. Trends Endocrinol. Metab. 1998, 9, 276–283. [Google Scholar] [CrossRef]
- El, K.M.; Mella, P.; Rashad, M.; Buzi, F.; Meazza, C.; Zahra, S.; Elsedfy, H.H. Growth hormone/IGF-I axis and growth hormone receptor mutations in idiopathic short stature. Horm. Res. Paediatr. 2011, 76, 300–306. [Google Scholar] [CrossRef]
- Ying, Y.Q.; Wei, H.; Cao, L.Z.; Lu, J.J.; Luo, X.P. Clinical features and growth hormone receptor gene mutations of patients with Laron syndrome from a Chinese family. Zhongguo Dang Dai Er Ke Za Zhi 2007, 9, 335–338. [Google Scholar] [PubMed]
- Tiulpakov, A.; Rubtsov, P.; Dedov, I.; Peterkova, V.; Bezlepkina, O.; Chrousos, G.P.; Hochberg, Z. A novel C-terminal growth hormone receptor (GHR) mutation results in impaired GHR-STAT5 but normal STAT-3 signaling. J. Clin. Endocrinol. Metab. 2005, 90, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Meacham, L.R.; Brown, M.R.; Murphy, T.L.; Keret, R.; Silbergeld, A.; Laron, Z.; Parks, J.S. Characterization of a noncontiguous gene deletion of the growth hormone receptor in Laron’s syndrome. J. Clin. Endocrinol. Metab. 1993, 77, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, J.Y.; Lai, C.C.; Lin, H.C.; Yeh, G.C.; Hsu, H.H. Clinical, biochemical and molecular investigations of three Taiwanese children with Laron syndrome. J. Pediatr. Endocrinol. Metab. 2004, 17, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, J.; Berg, M.A.; Esposito, N.; Geffner, M.E.; Sakati, N.; Reiter, E.O.; Dower, S.; Francke, U.; Postel-Vinay, M.C.; Finidori, J. Four contiguous amino acid substitutions, identified in patients with Laron syndrome, differently affect the binding affinity and intracellular trafficking of the growth hormone receptor. J. Clin. Endocrinol. Metab. 1998, 83, 4481–4489. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Katoh-Fukui, Y.; Nakamura, A.; Matsubara, K.; Kamimaki, T.; Tanaka, H.; Dateki, S.; Adachi, M.; Muroya, K.; Yoshida, S.; et al. Next generation sequencing-based mutation screening of 86 patients with idiopathic short stature. Endocr. J. 2017, 64, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.A.; Souza, S.C.; Arnhold, I.J.; Mendonca, B.B. The first homozygous mutation (S226I) in the highly-conserved WSXWS-like motif of the GH receptor causing Laron syndrome: Supression of GH secretion by GnRH analogue therapy not restored by dihydrotestosterone administration. Clin. Endocrinol. 2004, 60, 36–40. [Google Scholar] [CrossRef]
- Hopp, M.; Rosenbloom, A.L.; Griffiths, J.; Kgwete, S.; Vaccarello, M.A. Growth hormone receptor deficiency (Laron syndrome) in black African siblings. S. Afr. Med. J. 1996, 86, 268–270. [Google Scholar] [PubMed]
- Akinci, A.; Rosenfeld, R.G.; Hwa, V. A novel exonic GHR splicing mutation (c.784G > C) in a patient with classical growth hormone insensitivity syndrome. Horm. Res. Paediatr. 2013, 79, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Vairamani, K.; Merjaneh, L.; Casano-Sancho, P.; Sanli, M.E.; David, A.; Metherell, L.A.; Savage, M.O.; Del, P.J.; Backeljauw, P.F.; Rosenfeld, R.G.; et al. Novel dominant-negative GH receptor mutations expands the spectrum of GHI and IGF-I deficiency. J. Endocr. Soc. 2017, 1, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Billestrup, N.; Bouchelouche, P.; Allevato, G.; Ilondo, M.; Nielsen, J.H. Growth hormone receptor C-terminal domains required for growth hormone-induced intracellular free Ca2+ oscillations and gene transcription. Proc. Natl. Acad. Sci. USA 1995, 92, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Klammt, J.; Shen, S.; Kiess, W.; Kratzsch, J.; Stobbe, H.; Vogel, M.; Luo, F.; Pfaffle, R. Clinical and biochemical consequences of an intragenic growth hormone receptor (GHR) deletion in a large Chinese pedigree. Clin. Endocrinol. 2015, 82, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Feigerlova, E.; Swinyard, M.; Derr, M.A.; Farnsworth, J.; Andrew, S.F.; Rosenfeld, R.G.; Hwa, V. A novel GHR intronic variant, c.266+83G>T, activates a cryptic 5’ splice site causing severe GHR deficiency and classical GH insensitivity syndrome. Horm. Res. Paediatr. 2013, 80, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Takahashi, Y.; Kaji, H.; Nose, O.; Okimura, Y.; Abe, H.; Chihara, K. Growth hormone (GH) insensitivity syndrome with high serum GH-binding protein levels caused by a heterozygous splice site mutation of the GH receptor gene producing a lack of intracellular domain. J. Clin. Endocrinol. Metab. 1998, 83, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Moffat, J.G.; Edens, A.; Talamantes, F. Structure and expression of the mouse growth hormone receptor/growth hormone binding protein gene. J. Mol. Endocrinol. 1999, 23, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Elzein, S.; Goodyer, C.G. Regulation of human growth hormone receptor expression by microRNAs. Mol. Endocrinol. 2014, 28, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Dastot, F.; Sobrier, M.L.; Duquesnoy, P.; Duriez, B.; Goossens, M.; Amselem, S. Alternatively spliced forms in the cytoplasmic domain of the human growth hormone (GH) receptor regulate its ability to generate a soluble GH-binding protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10723–10728. [Google Scholar] [CrossRef] [PubMed]
- Amselem, S.; Duquesnoy, P.; Duriez, B.; Dastot, F.; Sobrier, M.L.; Valleix, S.; Goossens, M. Spectrum of growth hormone receptor mutations and associated haplotypes in Laron syndrome. Hum. Mol. Genet. 1993, 2, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Shapiro, L.; Rose, S.J.; Mushtaq, T.; Clayton, P.E.; Ten, S.; Bhangoo, A.; Kumbattae, U.; Dias, R.; Savage, M.O.; et al. Phenotypic spectrum and responses to recombinant human IGF1 (rhIGF1) therapy in patients with homozygous intronic pseudoexon growth hormone receptor mutation. Eur. J. Endocrinol. 2018, 178, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Singhal, V.; Goh, B.C.; Bouxsein, M.L.; Faugere, M.C.; DiGirolamo, D.J. Osteoblast-restricted disruption of the growth hormone receptor in mice results in sexually dimorphic skeletal phenotypes. Bone Res. 2013, 1, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Isaksson, O. Regulation of skeletal growth and mineral acquisition by the GH/IGF-1 axis: Lessons from mouse models. Growth Horm IGF Res. 2016, 28, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Mohan, S.; Yakar, S. Does the GH/IGF-1 axis contribute to skeletal sexual dimorphism? Evidence from mouse studies. Growth Horm. IGF Res. 2016, 27, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Wit, J.M.; Camacho-Hubner, C. Endocrine regulation of longitudinal bone growth. Endocr. Dev. 2011, 21, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.H.; Clausen, N.M.; Schjerling, P.; Larsen, J.O.; Martinussen, T.; List, E.O.; Kopchick, J.J.; Kjaer, M.; Heinemeier, K.M. Chronic alterations in growth hormone/insulin-like growth factor-I signaling lead to changes in mouse tendon structure. Matrix Biol. 2014, 34, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Gevers, E.F.; van der Eerden, B.C.; Karperien, M.; Raap, A.K.; Robinson, I.C.; Wit, J.M. Localization and regulation of the growth hormone receptor and growth hormone-binding protein in the rat growth plate. J. Bone Miner. Res. 2002, 17, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Mavalli, M.D.; DiGirolamo, D.J.; Fan, Y.; Riddle, R.C.; Campbell, K.S.; van Groen, T.; Frank, S.J.; Sperling, M.A.; Esser, K.A.; Bamman, M.M.; et al. Distinct growth hormone receptor signaling modes regulate skeletal muscle development and insulin sensitivity in mice. J. Clin. Investig. 2010, 120, 4007–4020. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, A.; Buffin, N.J.; Gallagher, E.J.; Blank, J.; Wu, Y.; Yakar, S.; LeRoith, D. Deletion of growth hormone receptors in postnatal skeletal muscle of male mice does not alter muscle mass and response to pathological injury. Endocrinology 2013, 154, 3776–3783. [Google Scholar] [CrossRef] [PubMed]
- Lucy, M.C. Growth hormone regulation of follicular growth. Reprod. Fertil. Dev. 2012, 24, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Ranke, M.B.; Wit, J.M. Growth hormone—Past, present and future. Nat. Rev. Endocrinol. 2018, 14, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Werner, H.; Rosen, C. Insulin-like growth factors: Actions on the skeleton. J. Mol. Endocrinol. 2018. [Google Scholar] [CrossRef]
- Lee, S.S.; Han, A.; Ahn, M.B.; Kim, S.H.; Cho, W.K.; Cho, K.S.; Park, S.H.; Jung, M.H.; Suh, B. Growth without growth hormone in combined pituitary hormone deficiency caused by pituitary stalk interruption syndrome. Ann. Pediatr. Endocrinol. Metab. 2017, 22, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Walters, T.D.; Griffiths, A.M. Mechanisms of growth impairment in pediatric Crohn’s disease. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.B.; Pashankar, F.; Camacho-Hubner, C.; Savage, M.O.; Clark, A.J. Analysis of the intracellular signalling domain of the human growth hormone receptor in children with idiopathic short stature. Clin. Endocrinol. 2000, 52, 463–469. [Google Scholar] [CrossRef]
- Hujeirat, Y.; Hess, O.; Shalev, S.; Tenenbaum-Rakover, Y. Growth hormone receptor sequence changes do not play a role in determining height in children with idiopathic short stature. Horm. Res. 2006, 65, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Kijas, J.M.; Wales, R.; Tornsten, A.; Chardon, P.; Moller, M.; Andersson, L. Melanocortin receptor 1 (MC1R) mutations and coat color in pigs. Genetics 1998, 150, 1177–1185. [Google Scholar] [PubMed]
- Lau, J.S.; Yip, C.W.; Law, K.M.; Leung, F.C. Cloning and characterization of chicken growth hormone binding protein (cGHBP). Domest. Anim. Endocrinol. 2007, 33, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; Cogburn, L.A.; Burnside, J. Comparison of gene expression in normal and growth hormone receptor-deficient dwarf chickens reveals a novel growth hormone regulated gene. Biochem. Biophys. Res. Commun. 1995, 206, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, S.; An, L.; Ma, J.; Qiu, F.; Jia, R.; Nie, Q.; Zhang, D.; Luo, Q.; Li, T.; et al. Chicken GHR natural antisense transcript regulates GHR mRNA in LMH cells. Oncotarget 2016, 7, 73607–73617. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Location | Base Mutation | Defect | Mutation Type | Mechanism | References |
|---|---|---|---|---|---|
| Exon 2 | c.1 A > G 1 | M18V | Missense | Inhibits the correct expression of GHR | [45] |
| c.1 A > T | M18L | Missense | [50] | ||
| c.12 G > A | W15X | Nonsense | Introduces a premature termination codon that leads to a truncated non-functioning receptor | ||
| Exon 3 | c.101 G > A | W16X | Nonsense | ||
| 71–136 del 2 | 7–22 del | Deletion 7–22 | [37] | ||
| del | exon 3del | Frameshift | d3-GHR, represented by a 532-bp fragment, forms a nonfunctional receptor that terminates the signal transmission in advance | [37,51] | |
| Exon 4 | c.162 delC | 36delC | Frameshift | May interfere with GH binding activity | [41] |
| c.166 T > A | C38S | Missense | May affect GH binding activity | [52] | |
| c.168 C > A | C38X | Nonsense | |||
| c.173 C > T | S40L | Missense | May interfere with GH binding activity | [10] | |
| c.180 G > A | E42K | Missense | Impairs the GHR binding affinity to GH | [42] | |
| c.181 C > T | R43X | Nonsense | Causes undetectable GH binding protein (GHBP) | [43] | |
| c.192_193delTT | 46delTT | Frameshift | [52] | ||
| c.202 T > C | W50R | Missense | May affect the GH binding activity | [41] | |
| G62V | Be associated with idiopathic short stature | [53] | |||
| c.247 C > T | Q65X | Nonsense | Could interfere with GH binding activity | [41] | |
| c.249 T > C | S65H | Lowers serum levels of IGF-1, IGFBP-3, and GHBP compared to normal controls | [54] | ||
| c.266 G > A | R71K | Missense | [52] | ||
| del(5) p11–p13.1 | del exon 4–10 | Nonsense | [50] | ||
| Exon 5 | c.293 G > A | W80X | Nonsense | Could interfere with GH binding activity | [41] |
| c.303 C > A | C83X | Nonsense | Leads to a lack of GHR expression as a result of mRNA decay or defect in cell membrane anchoring | [55] | |
| c.310 T > G | Y86D | Missense | Located in one of the two cysteine-rich regions of GHR | [50] | |
| c.335 G > C | C94S | Missense | Loses the ability of binding to GH | [44] | |
| c.337 T > C | C94C | Synonymous | Delays bone age | [46] | |
| c.341 T > C | F96S | Missense | Interferes with GHR intracellular transport to the cell membrane | [52] | |
| c.338dupA 3 | Y97X | Nonsense | Causes truncated GHR and a loss in receptor function because it lacks amino acids comprising the transmembrane and intracellular regions of the GHR protein | [50] | |
| 307 G > A | D103N | Responsible for impaired GH binding that alters receptor functionality | [45] | ||
| W104R | [37] | ||||
| c.421_422dupTT | 422insTT | Frameshift | Results in a frameshift that introduces a premature termination codon that leads to a truncated receptor | [22] | |
| c.428 T > C | V125A | Missense | [10] | ||
| ~1.2 kb deletion | Deletion | Skipping the truncated exon 5 leads to a frameshift and a premature termination codon | [50] | ||
| ~4 kb deletion | Deletion | Leads to a stop codon in exon 6 that predicts a truncated, non-functional GHR protein | |||
| del | exon 5del | Frameshift | Forms a nonfunctional receptor that terminates the signal transmission in advance | [56] | |
| Exons 5 and 6 | ~19 kb deletion | Deletion | Deletion of a large portion of the ECD, hormone binding domain of GHR | [50] |
| Location | Base Mutation | Defect | Mutation Type | Mechanism | References |
|---|---|---|---|---|---|
| Exon 6 | P131Q | Missense | [52] | ||
| c.476 T > A 1 | L141X | Nonsense | Introduces a premature termination codon that leads to a truncated non-functioning receptor | [50] | |
| c.484 G > A | V144I | Associated with idiopathic short stature | [13] | ||
| c.484 G > T | V144F | [46] | |||
| c.485 T > A | V144D | Missense | [52] | ||
| c.485 T > C | V144A | [46] | |||
| c.504 T > G | H150Q | Missense | [44] | ||
| c.508 G > C | D152H | Missense | Disrupts the expression, dimerization, and signaling of GHR | [52] | |
| D152G | Missense | [57] | |||
| c.512 T > C | I153T | Missense | Mainly affects intracellular trafficking and binding affinity of the receptor | [50,58] | |
| c.515 A > C | Q154P | Missense | Leads to severe defects both at the cell surface and in total particulate membrane fractions | ||
| c.518 T > G | V155G | Missense | Affects intracellular trafficking and binding affinity of GHR | ||
| c.524 G > A | W157X | Nonsense | Produces a truncated GHR, lacking part of exons 6 and 7–10, defective in both cell membrane anchoring and GH binding | [41] | |
| c.535 C > T | R161C | Missense | Causes low serum GHBP concentrations | [52] | |
| c.558 A > G | G168G | Deletes the ECD and forms a nonfunctional receptor that terminates the signal transmission in advance | [10] | ||
| c.559 T > C | W169R | Since Trp169 plays an important role in the stabilization of the GH–GHR interaction, this mutation in chain 1, which binds to GH site 1, showed a decreased affinity for GH, affecting the interaction in the complex | [8] | ||
| c.591 C > T | R179C | [48] | |||
| c.594 G > T | E180X | Nonsense | Deletes the ECD and forms a nonfunctional receptor, which terminates the signal transmission in advance | [21,44] | |
| c.594 A > G | E180sp 2 | Splice site | Causes deletion of residues 181–188 in the dimerization functional region | [44] | |
| c.601 G > T | E183X | Nonsense | [50] | ||
| del 3 | deletion of exon 6 | Frameshift | Results in a deletion of a large portion of the ECD of GHR | [50] | |
| Exon 7 | M207 fs. X8 | Deletion | Results in premature termination, which decreases GH binding affinity | [49] | |
| c.677 A > G | Y208C | Missense | Prevents normal interactions in the membrane proximal domain of the extracellular part of the receptor | [50] | |
| c.685 C > G | R211G | Missense | [52] | ||
| c.703 C > T | R217X | Nonsense | Deletes the ECD and forms a nonfunctional receptor, which terminates the signal transmission in advance | [21] | |
| c.723 C > T | G223G | Splice site | May interfere with GH binding activity | [41] | |
| c.724 G > T | G224X | Nonsense | [52] | ||
| 656 C > T | p.S219L 4 | [59] | |||
| c.731 G > T | S226I | Missense | Occurs in WSXWS-like motif of GHR causing GH insensitivity | [60] | |
| R229H | [23] | ||||
| a nucleotide del | del203AT or TA | Frameshift | Results in high GH levels and low levels of IGF-1, IGF-2, IGFBP 3, and GHBP | [61] | |
| c.743_744 del AT | 230delAT | Nonsense | [52] | ||
| c.766 C > T | G236sp | Splice site | Activates the cryptic splice donor site within exon 7 | ||
| c.784 G > C | D244N | Missense | Induces functional loss of the native intron 7 donor splice site, leading to a frame shift and predicted early protein termination | [62] | |
| Exon 8 | c.875 G > C | R274T | Splice site | Generates a truncated protein | [52] |
| S473S | [37] |
| Location | Base Mutation | Defect | Mutation Type | Mechanism | References |
|---|---|---|---|---|---|
| Exon 5 | c.335 T > C 1 | F122S | Missense | Causes a large decrease in GH binding activity | [11] |
| c.352+2 T > C | Splice site | Reduces GH binding activity | [11] | ||
| Exon 7 | c. 679 G > T | S226I | Missense | Protein not expressed on the surface of hepatocytes | [10,47] |
| Exon 10 and 3’ UTR 2 | c.1773 del 3 | Frameshift | Lacks the target site for microRNA let-7b, which down-regulates the GHR expression level | [2,18] |
| Location | Base Mutation | Defect | Mutation Type | Mechanism | References |
|---|---|---|---|---|---|
| Exon 9 | removal of exon 9 | truncated GHR 1–279 | Frameshift | Removal of 26 base pairs of exon 9 does not have direct signaling function, but can form a long–short heterodimer with full-length GHR, inhibiting the STAT5 signal of full-length GHR and may therefore play a significant role in regulating the function of wild-type GHR | [6] |
| c.889_911del 1,2 | 889_911del | Results in an intracellular trafficking of GHR | [50] | ||
| c.899dupC 3 | Influences the critical JAK2-binding Box 1 region of the GHR ICD; the duplication predicts early protein termination | [23] | |||
| G920_921insTCTCAAAGATTACA | truncated | Robustly expressed as truncated, fails to activate STAT5B signaling | [63] | ||
| c.945 + 2 T > C | lost Box 1 | Excision of exon 9 that can form a long–short heterodimer | |||
| Exon 10 | c.964dupG | truncated | Robustly expressed as truncated, fails to activate STAT5B signaling | ||
| c.981delC | 309delC | Causes the production of 20 novel amino acids (310–329) instead of the wild-type sequence, premature termination at codon 330, and the subsequent deletion of the C terminal portion of the intracellular domain | [52] | ||
| c.1342_1345del | GHR (1–499) | Truncated after Box 1, which results in the isolated failure of STAT 5 signal transduction | [50] | ||
| c.1734delG | 1776delG | Lower STAT5-mediated transcriptional activation | [55] | ||
| I544L | Deletes the extracellular domain and forms a nonfunctional receptor, which terminates the signal transmission in advance | [21] |
| Location | Base Mutation | Defect | Mutation Type | Mechanism | References |
|---|---|---|---|---|---|
| Exon 2-intron 2 | GT > GGT | Splice site | Results in an immature stop codon in exon 3 | [10] | |
| Intron 2 | c.70 + 1 G > A 1 | 70 + 1 G > A | May interfere with GH binding activity | [37] | |
| Intron 4 | c.266 + 1 G > A | 71 + 1 G > A | Destroys the splice donor and acceptor invariant sequences of consensus sites for mRNA processing | [52,71] | |
| c.266 + 83 G > T | Results in retention of 81 intronic nucleotides in the GHR mRNA that leads to early protein termination | [65] | |||
| Intron 5 | c.440 − 1 G > C | IVS5 − 1 G > C | Destroys the splice donor and acceptor invariant sequences of consensus sites for mRNA processing | [71] | |
| Intron 6 | c.618 + 18kb A > G | ψ6 | Leads to recognition of the pseudoexon and inclusion of an additional 108 bases between exons 6 and 7 that adds 36 amino acids in the GHR ECD | [72] | |
| c.619 + 1 G > A | IVS6 + 1 G > A | Leads to the skipping of exon 6 and premature termination of the mRNA | [12,44] | ||
| c.619 − 1 G > T | 189 − 1 G > T | [71] | |||
| c.619 − 1 G > C | 189 − 1 G > C | [50] | |||
| c.619 − 25 A > G | IVS6 − 25 A > G | ||||
| Intron 7 | c. 785 − 1 G > T | 785 − 1 G > T | Results in a truncated protein that retains GH binding activity and is probably no longer anchored in the cell membrane, affecting signal transmission | ||
| Intron 8 | c.876 − 1 C > G | GHR (1–277) | Truncates the ICD of the GHR, which could form a heterodimer with the wild-type GHR, the activity of which is inhibited in a dominant-negative manner | [6,52] | |
| Intron 9 | c.945 + 1 G > A | GHR (1–277) | Produces a truncated protein with deletion of 98% of the ICD of the GHR, including Boxes 1 and 2, resulting in failure of GH signal transduction and GHR internalization | [52,67] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.; Li, C.; Li, C.; Zhang, X. Growth Hormone Receptor Mutations Related to Individual Dwarfism. Int. J. Mol. Sci. 2018, 19, 1433. https://doi.org/10.3390/ijms19051433
Lin S, Li C, Li C, Zhang X. Growth Hormone Receptor Mutations Related to Individual Dwarfism. International Journal of Molecular Sciences. 2018; 19(5):1433. https://doi.org/10.3390/ijms19051433
Chicago/Turabian StyleLin, Shudai, Congjun Li, Charles Li, and Xiquan Zhang. 2018. "Growth Hormone Receptor Mutations Related to Individual Dwarfism" International Journal of Molecular Sciences 19, no. 5: 1433. https://doi.org/10.3390/ijms19051433
APA StyleLin, S., Li, C., Li, C., & Zhang, X. (2018). Growth Hormone Receptor Mutations Related to Individual Dwarfism. International Journal of Molecular Sciences, 19(5), 1433. https://doi.org/10.3390/ijms19051433