Inhibition of Arachidonate 12/15-Lipoxygenase Improves α-Galactosidase Efficacy in iPSC-Derived Cardiomyocytes from Fabry Patients

Abstract
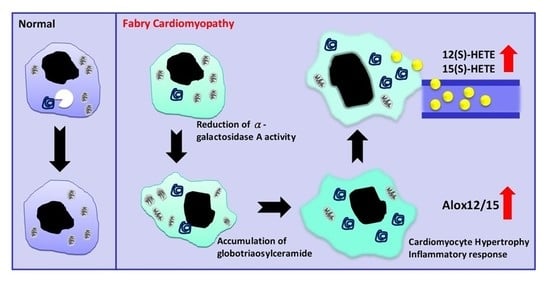
{kind=link}
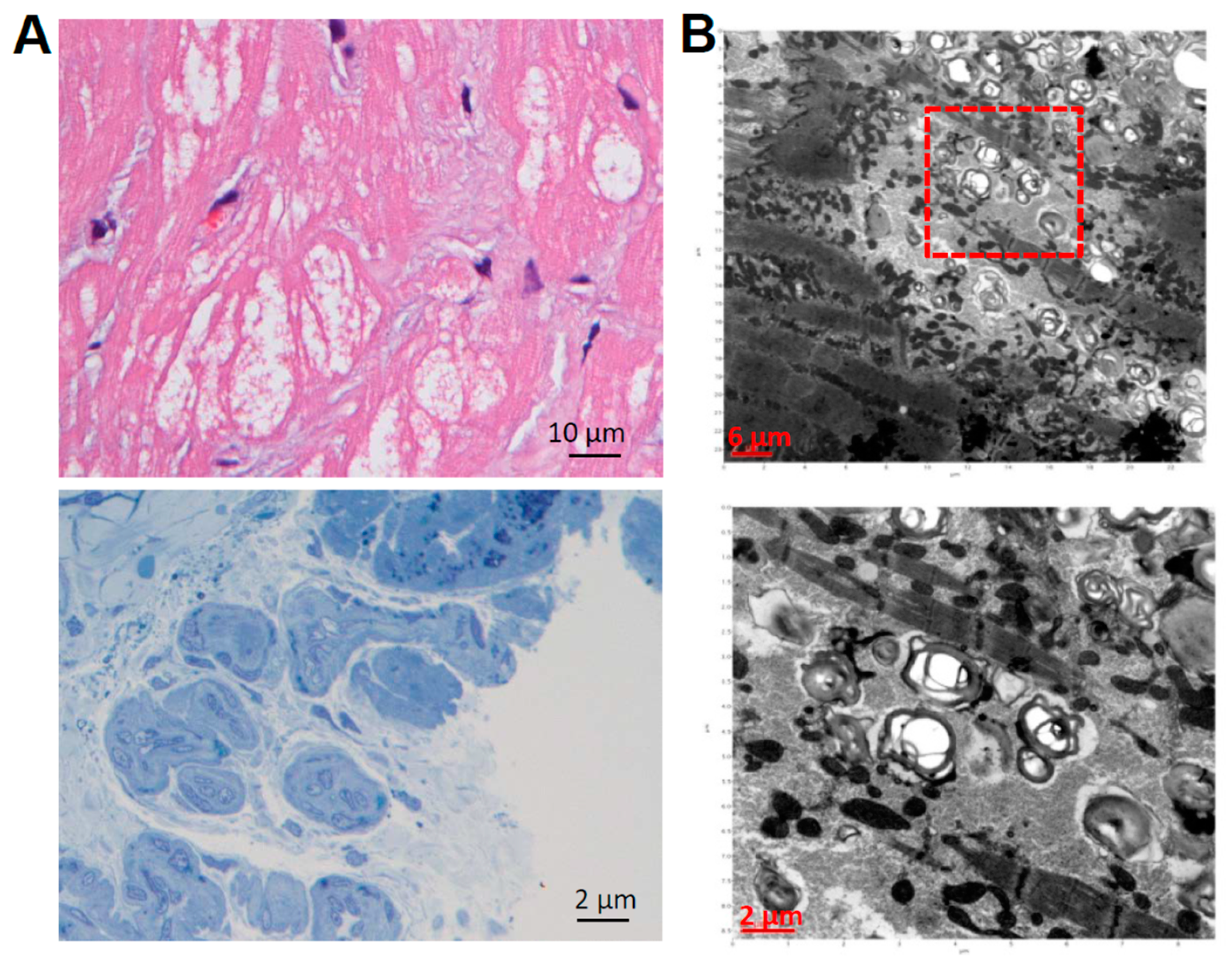{kind=link}
{kind=link}
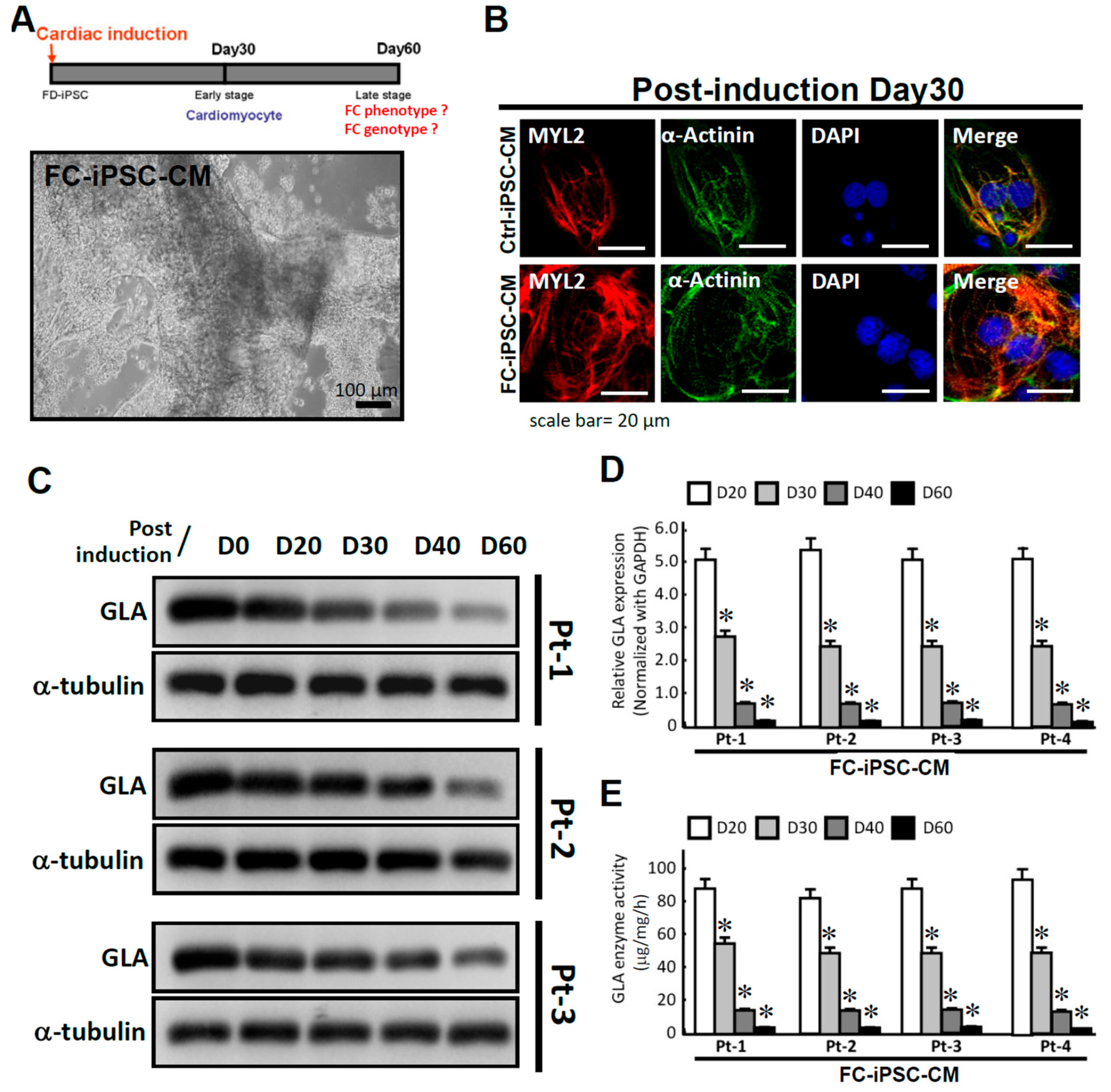{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Oct4/Sox2/Nanog/Glis1-Mediated Reprogramming of Fabry Patient-Derived Somatic Cells into iPSCs
2.2. Recapitulation of Cardiac Hypotrophy and GLA Enzymatic Decrease in FC Patient-Specific iPSC-Derived Cardiomyocytes (FC-iPSC-CMs)
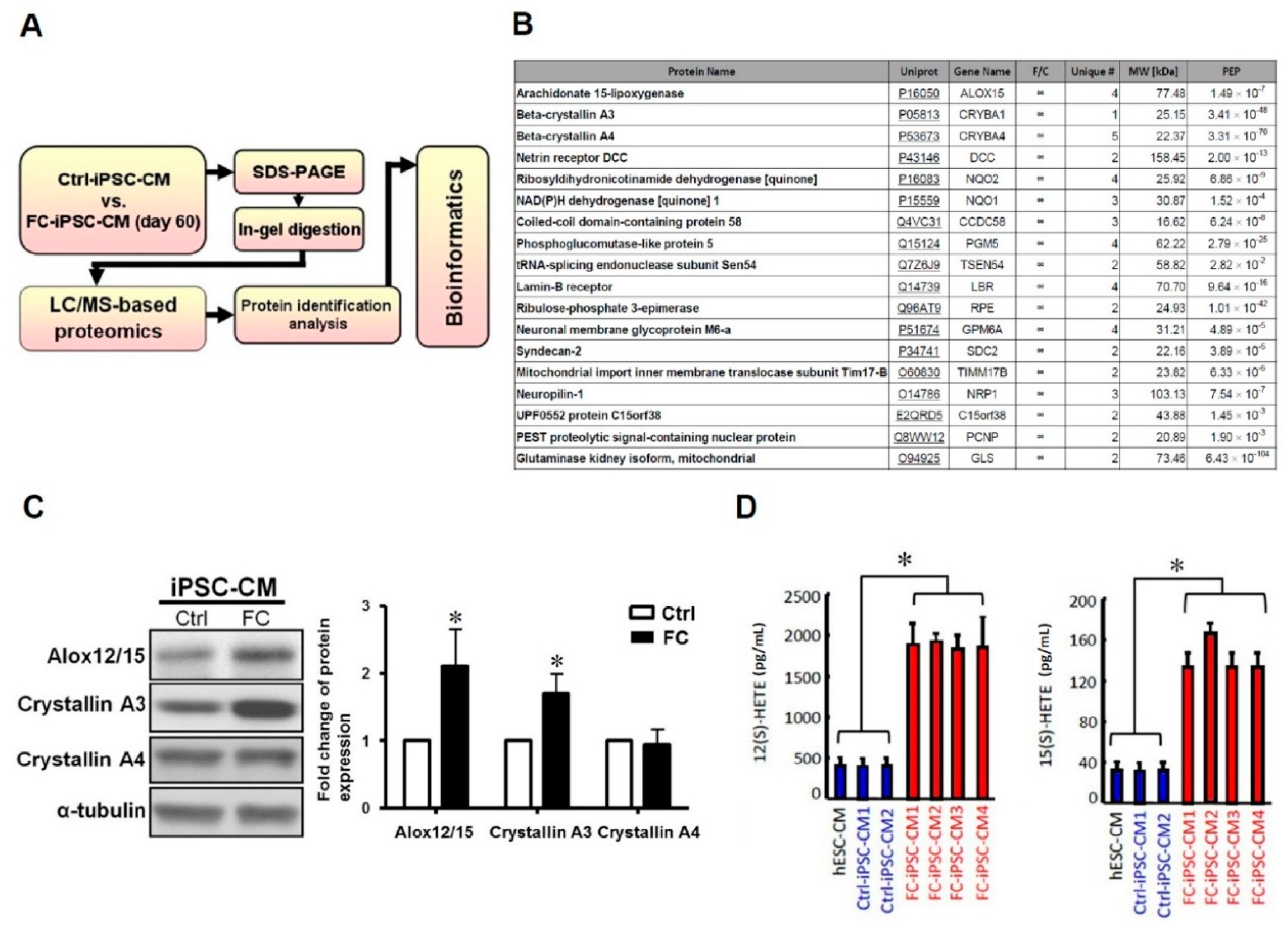
2.3. Upregulation of Cardiac Alox12/15 and Its Secretory Metabolites 12(S)-HETE and 15(S)-HETE in FC-iPSC-CMs
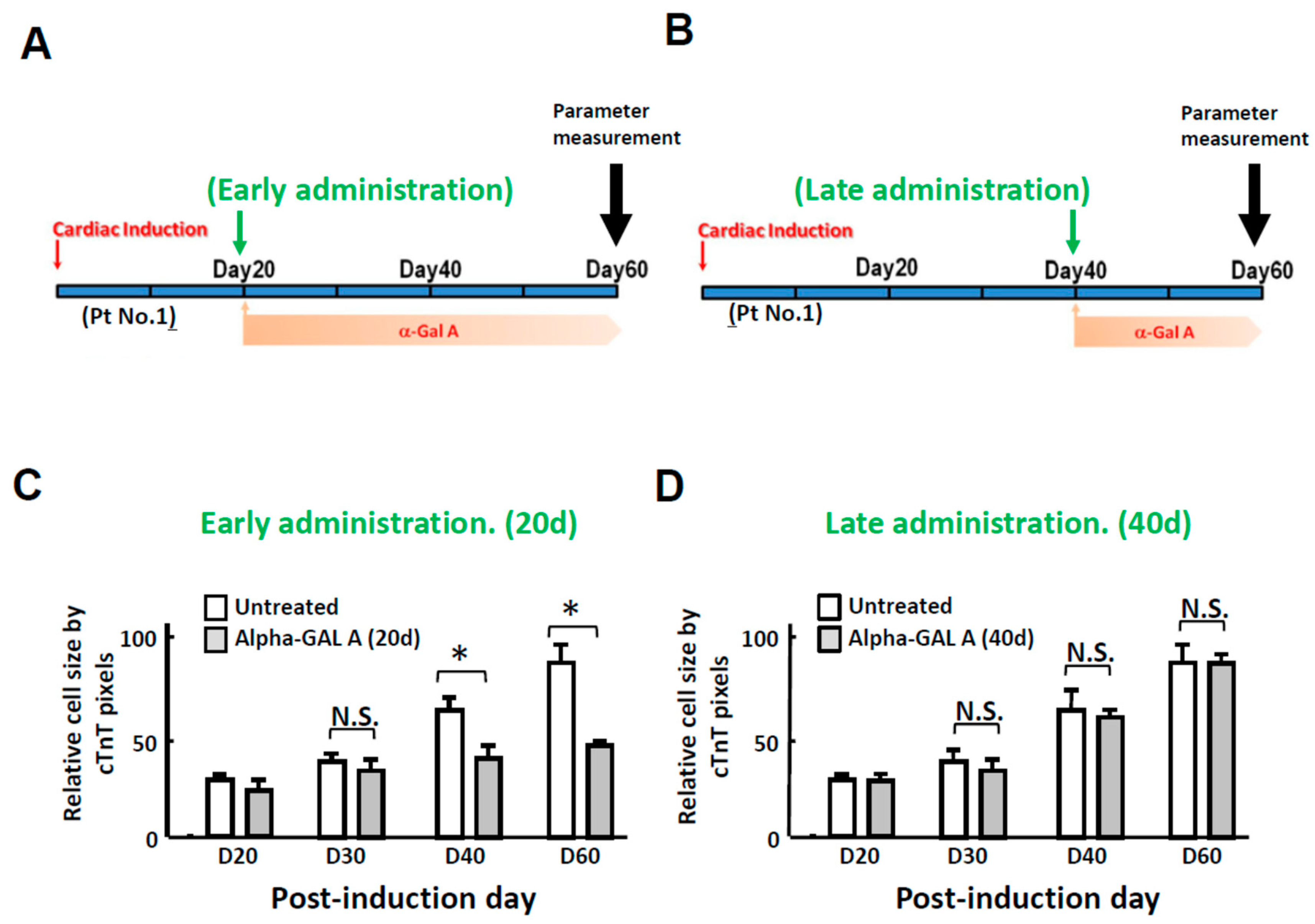
2.4. Time Dependency of α-Galactosidase Treatment Efficacy on Cardiomyocyte Hypertrophy in FC-iPSC-CMs
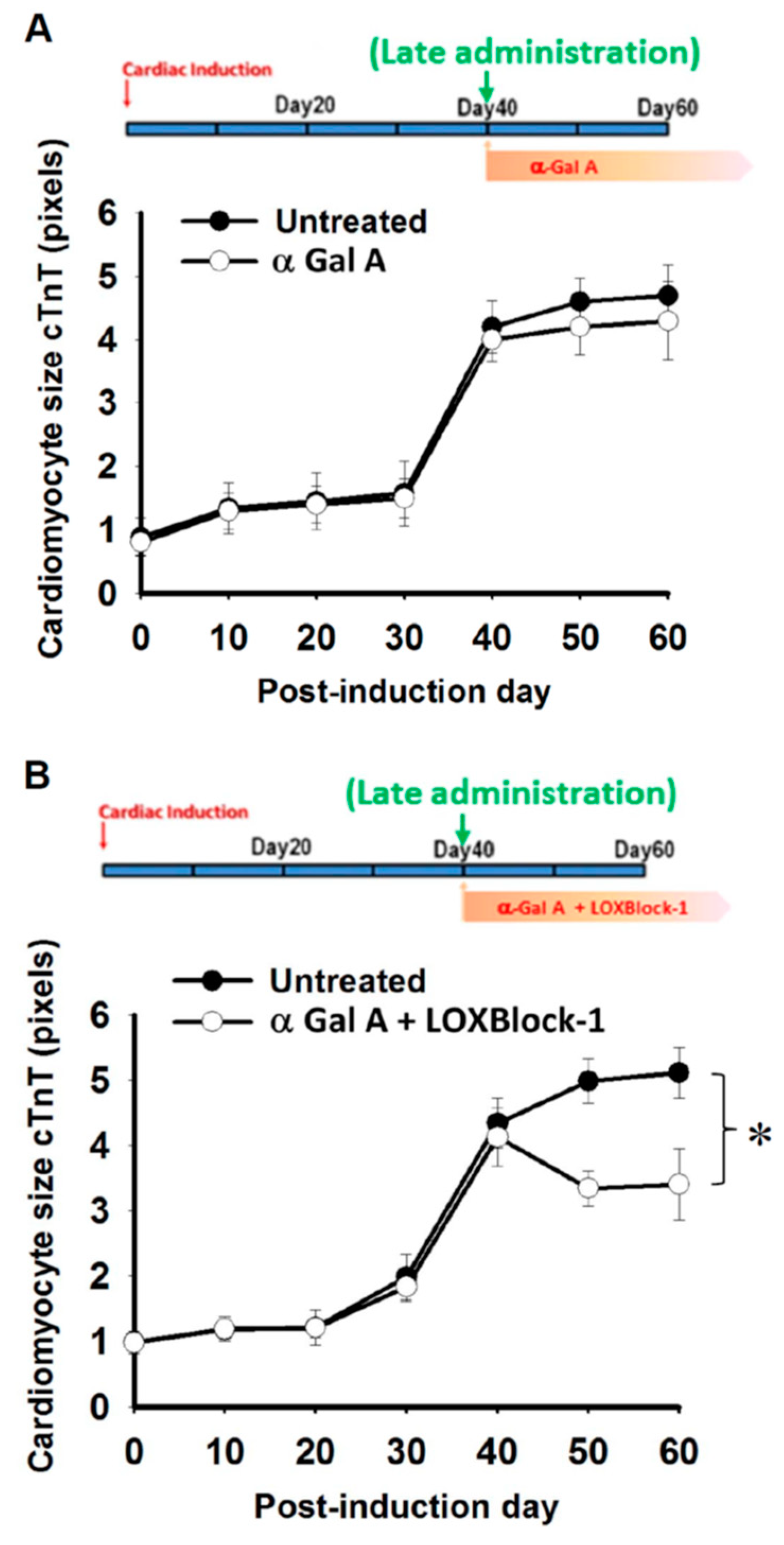
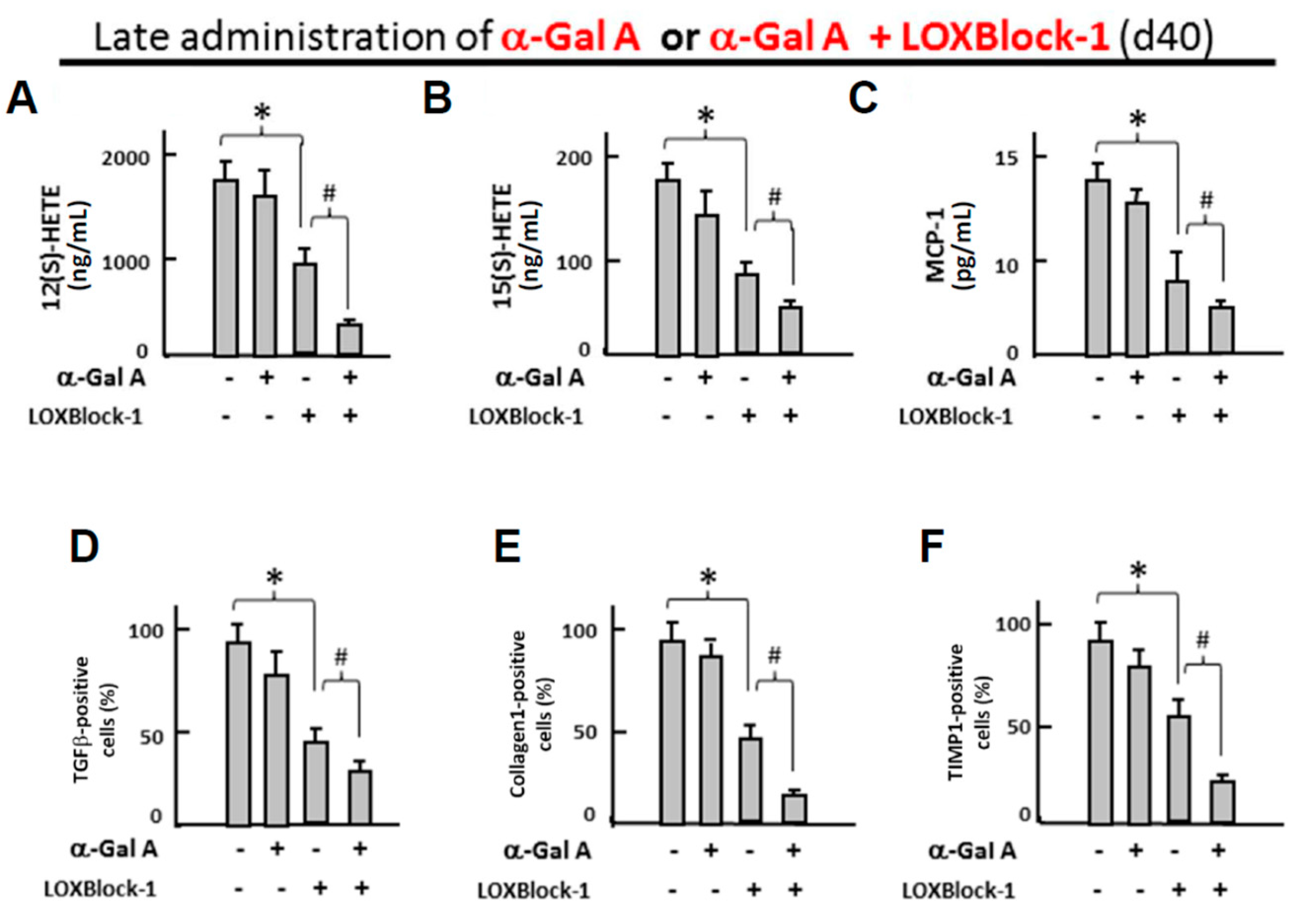
2.5. Simultaneous Alox12/15 Inhibition Improves the Treatment Efficacy of Late Administration of α-Galactosidase in FC-iPSC-CMs
3. Discussion
4. Materials and Methods
4.1. Generation of Patient-Specific iPSCs
4.2. In Vitro Differentiation of iPSCs
4.3. Quantitative PCR
4.4. Cardiac Differentiation from iPSCs
4.5. Western Blot Assay
4.6. Immunofluorescence Staining and the Quantification of the Cardiomyocyte Size
4.7. Transmission Electron Microscopy
4.8. LC-MS/MS Analysis
4.9. Measurement of α-Gal A Enzyme Activity
4.10. ELISA-Based 15(S)-HETE and 12(S)-HETE Measurements
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Eng, C.M.; Niehaus, D.J.; Enriquez, A.L.; Burgert, T.S.; Ludman, M.D.; Desnick, R.J. Fabry disease: Twenty-three mutations including sense and antisense CPG alterations and identification of a deletional hot-spot in the α-galactosidase a gene. Hum. Mol. Genet. 1994, 3, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.S.; Bishop, D.F.; Astrin, K.H.; Kornreich, R.; Eng, C.M.; Sakuraba, H.; Desnick, R.J. Fabry disease: Six gene rearrangements and an exonic point mutation in the α-galactosidase gene. J. Clin. Investig. 1989, 83, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and efficacy of recombinant human α-galactosidase a replacement therapy in fabry’s disease. N. Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Murray, G.J.; Treco, D.; Daniel, P.; Sellos-Moura, M.; Myers, M.; Quirk, J.M.; Zirzow, G.C.; Borowski, M.; Loveday, K.; et al. Infusion of α-galactosidase a reduces tissue globotriaosylceramide storage in patients with fabry disease. Proc. Natl. Acad. Sci. USA 2000, 97, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.R.; Sung, S.H.; Chang, F.P.; Yang, C.F.; Liu, H.C.; Lin, H.Y.; Huang, C.K.; Gao, H.J.; Huang, Y.H.; Liao, H.C.; et al. Endomyocardial biopsies in patients with left ventricular hypertrophy and a common chinese later-onset fabry mutation (IVS4+919 G>A). Orphanet J. Rare Dis. 2014, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Li, C.F.; Lin, H.Y.; Lin, C.H.; Liu, H.C.; Tsai, S.F.; Niu, D.M. High-throughput detection of common sequence variations of fabry disease in taiwan using DNA mass spectrometry. Mol. Genet. Metab. 2014, 111, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.C.; Chiang, C.C.; Niu, D.M.; Wang, C.H.; Kao, S.M.; Tsai, F.J.; Huang, Y.H.; Liu, H.C.; Huang, C.K.; Gao, H.J.; et al. Detecting multiple lysosomal storage diseases by tandem mass spectrometry–A national newborn screening program in taiwan. Clin. Chim. Acta 2014, 431, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.C.; Huang, Y.H.; Chen, Y.J.; Kao, S.M.; Lin, H.Y.; Huang, C.K.; Liu, H.C.; Hsu, T.R.; Lin, S.P.; Yang, C.F.; et al. Plasma globotriaosylsphingosine (lysogb3) could be a biomarker for fabry disease with a chinese hotspot late-onset mutation (IVS4+919 G>A). Clin. Chim. Acta 2013, 426, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chong, K.W.; Hsu, J.H.; Yu, H.C.; Shih, C.C.; Huang, C.H.; Lin, S.J.; Chen, C.H.; Chiang, C.C.; Ho, H.J.; et al. High incidence of the cardiac variant of fabry disease revealed by newborn screening in the taiwan chinese population. Circ. Cardiovasc. Genet. 2009, 2, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Lin, H.Y.; Yang, C.F.; Liao, H.C.; Hsu, T.R.; Lo, C.W.; Chang, F.P.; Huang, C.K.; Lu, Y.H.; Lin, S.P.; et al. Globotriaosylsphingosine (lyso-gb3) might not be a reliable marker for monitoring the long-term therapeutic outcomes of enzyme replacement therapy for late-onset fabry patients with the chinese hotspot mutation (IVS4 + 919 G>A). Orphanet J. Rare Dis. 2014, 9, 111. [Google Scholar] [CrossRef] [PubMed]
- Yousef, Z.; Elliott, P.M.; Cecchi, F.; Escoubet, B.; Linhart, A.; Monserrat, L.; Namdar, M.; Weidemann, F. Left ventricular hypertrophy in fabry disease: A practical approach to diagnosis. Eur. Heart J. 2013, 34, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Vedder, A.C.; Linthorst, G.E.; van Breemen, M.J.; Groener, J.E.; Bemelman, F.J.; Strijland, A.; Mannens, M.M.; Aerts, J.M.; Hollak, C.E. The dutch fabry cohort: Diversity of clinical manifestations and gb3 levels. J. Inherit. Metab. Dis. 2007, 30, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, P.D.; Calvin, J.; Hogg, S.; O’Driscoll, E.; Halsall, D.; Burling, K.; Maguire, G.; Wright, N.; Cox, T.M.; Meikle, P.J.; et al. Monitoring enzyme replacement therapy in fabry disease–Role of urine globotriaosylceramide. J. Inherit. Metab. Dis. 2005, 28, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.M.; Mostajo-Radji, M.A. How induced pluripotent stem cells are redefining personalized medicine. Gene 2013, 520, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long qt syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Josowitz, R.; Carvajal-Vergara, X.; Lemischka, I.R.; Gelb, B.D. Induced pluripotent stem cell-derived cardiomyocytes as models for genetic cardiovascular disorders. Curr. Opin. Cardiol. 2011, 26, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific ipscs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Limphong, P.; Zhang, H.; Christians, E.; Liu, Q.; Riedel, M.; Ivey, K.; Cheng, P.; Mitzelfelt, K.; Taylor, G.; Winge, D.; et al. Modeling human protein aggregation cardiomyopathy using murine induced pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, E.G.; Liang, P.; Lan, F.; Sanchez-Freire, V.; Simmons, C.; Gong, T.; Sharma, A.; Burridge, P.W.; Patlolla, B.; Lee, A.S.; et al. Screening drug-induced arrhythmia [corrected] using human induced pluripotent stem cell-derived cardiomyocytes and low-impedance microelectrode arrays. Circulation 2013, 128, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.L.; Shen, J.S.; Kawagoe, S.; Ohashi, T.; Brady, R.O.; Eto, Y. Induced pluripotent stem cells derived from mouse models of lysosomal storage disorders. Proc. Natl. Acad. Sci. USA 2010, 107, 7886–7891. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, S.; Higuchi, T.; Otaka, M.; Shimada, Y.; Kobayashi, H.; Ida, H.; Ohashi, T.; Okano, H.J.; Nakanishi, M.; Eto, Y. Morphological features of ips cells generated from fabry disease skin fibroblasts using sendai virus vector (SEVDP). Mol. Genet. Metab. 2013, 109, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Van Hoof, D.; Dormeyer, W.; Braam, S.R.; Passier, R.; Monshouwer-Kloots, J.; Ward-van Oostwaard, D.; Heck, A.J.; Krijgsveld, J.; Mummery, C.L. Identification of cell surface proteins for antibody-based selection of human embryonic stem cell-derived cardiomyocytes. J. Proteome Res. 2010, 9, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Rigbolt, K.T.; Blagoev, B. Quantitative phosphoproteomics to characterize signaling networks. Semin. Cell Dev. Biol. 2012, 23, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Cigna, D.; D’Anna, C.; Zizzo, C.; Francofonte, D.; Sorrentino, I.; Colomba, P.; Albeggiani, G.; Armini, A.; Bianchi, L.; Bini, L.; et al. Alteration of proteomic profiles in pbmc isolated from patients with fabry disease: Preliminary findings. Mol. Biosyst. 2013, 9, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Cuccurullo, M.; Beneduci, A.; Anand, S.; Mignani, R.; Cianciaruso, B.; Bachi, A.; Capasso, G. Fabry disease: Perspectives of urinary proteomics. J. Nephrol. 2010, 23 (Suppl. 16), S199–S212. [Google Scholar] [PubMed]
- Moore, D.F.; Krokhin, O.V.; Beavis, R.C.; Ries, M.; Robinson, C.; Goldin, E.; Brady, R.O.; Wilkins, J.A.; Schiffmann, R. Proteomics of specific treatment-related alterations in fabry disease: A strategy to identify biological abnormalities. Proc. Natl. Acad. Sci. USA 2007, 104, 2873–2878. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Yamaguchi, K.; Nakamura, T.; Shibukawa, R.; Kodanaka, I.; Ichisaka, T.; Kawamura, Y.; Mochizuki, H.; Goshima, N.; Yamanaka, S. Direct reprogramming of somatic cells is promoted by maternal transcription factor glis1. Nature 2011, 474, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Yamanaka, S. Glis1, a unique pro-reprogramming factor, may facilitate clinical applications of ipsc technology. Cell Cycle 2011, 10, 3613–3614. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.J.; Yu, W.C.; Chang, Y.L.; Chen, W.Y.; Chang, W.C.; Chien, Y.; Yen, J.C.; Liu, Y.Y.; Chen, S.J.; Wang, C.Y.; et al. Energy utilization of induced pluripotent stem cell-derived cardiomyocyte in fabry disease. Int. J. Cardiol. 2017, 232, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Chien, C.S.; Chiang, H.C.; Huang, W.L.; Chou, S.J.; Chang, W.C.; Chang, Y.L.; Leu, H.B.; Chen, K.H.; Wang, K.L.; et al. Interleukin-18 deteriorates fabry cardiomyopathy and contributes to the development of left ventricular hypertrophy in fabry patients with gla IVS4+919 G>A mutation. Oncotarget 2016, 7, 87161–87179. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Niemann, M.; Breunig, F.; Herrmann, S.; Beer, M.; Stork, S.; Voelker, W.; Ertl, G.; Wanner, C.; Strotmann, J. Long-term effects of enzyme replacement therapy on fabry cardiomyopathy: Evidence for a better outcome with early treatment. Circulation 2009, 119, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, M.; Camporeale, A.; Della Bona, R.; Sabini, A.; Cosmi, D.; Magnolfi, A.; Bolognese, L. Progression of fabry cardiomyopathy despite enzyme replacement therapy. Circulation 2013, 128, 1687–1688. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.; Niemann, M.; Stork, S.; Frantz, S.; Beer, M.; Ertl, G.; Wanner, C.; Weidemann, F. Relation of burden of myocardial fibrosis to malignant ventricular arrhythmias and outcomes in fabry disease. Am. J. Cardiol. 2014, 114, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Kayama, Y.; Minamino, T.; Toko, H.; Sakamoto, M.; Shimizu, I.; Takahashi, H.; Okada, S.; Tateno, K.; Moriya, J.; Yokoyama, M.; et al. Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. J. Exp. Med. 2009, 206, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kayama, Y.; Sakamoto, M.; Iuchi, H.; Shimizu, I.; Yoshino, T.; Katoh, D.; Nagoshi, T.; Tojo, K.; Minamino, T.; et al. Arachidonate 12/15-lipoxygenase-induced inflammation and oxidative stress are involved in the development of diabetic cardiomyopathy. Diabetes 2014, 64, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Itier, J.M.; Ret, G.; Viale, S.; Sweet, L.; Bangari, D.; Caron, A.; Le-Gall, F.; Benichou, B.; Leonard, J.; Deleuze, J.F.; et al. Effective clearance of gl-3 in a human ipsc-derived cardiomyocyte model of fabry disease. J. Inherit. Metab. Dis. 2014, 37, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D.; Keeney, D.S.; Oliw, E.H.; Boeglin, W.E.; Brash, A.R. Functional expression and cellular localization of a mouse epidermal lipoxygenase. J. Biol. Chem. 1996, 271, 23338–23344. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S. Mammalian lipoxygenases: Molecular structures and functions. Biochim. Biophys. Acta 1992, 1128, 117–131. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Mussell, R.; Kahook, K.; Tawfik, A.; Eladl, M.; Sarthy, V.; Nussbaum, J.; El-Marakby, A.; Park, S.Y.; Gurel, Z.; et al. Increased expression and activity of 12-lipoxygenase in oxygen-induced ischemic retinopathy and proliferative diabetic retinopathy: Implications in retinal neovascularization. Diabetes 2011, 60, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; O’Donnell, V.B. Inflammation and immune regulation by 12/15-lipoxygenases. Prog. Lipid Res. 2006, 45, 334–356. [Google Scholar] [CrossRef] [PubMed]
- Reilly, K.B.; Srinivasan, S.; Hatley, M.E.; Patricia, M.K.; Lannigan, J.; Bolick, D.T.; Vandenhoff, G.; Pei, H.; Natarajan, R.; Nadler, J.L.; et al. 12/15-lipoxygenase activity mediates inflammatory monocyte/endothelial interactions and atherosclerosis in vivo. J. Biol. Chem. 2004, 279, 9440–9450. [Google Scholar] [CrossRef] [PubMed]
- Bolick, D.T.; Orr, A.W.; Whetzel, A.; Srinivasan, S.; Hatley, M.E.; Schwartz, M.A.; Hedrick, C.C. 12/15-lipoxygenase regulates intercellular adhesion molecule-1 expression and monocyte adhesion to endothelium through activation of rhoa and nuclear factor-kappab. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2301–2307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Y.; Chen, M.; Su, X.; Yi, D.; Lu, P.; Zhu, D. 15-lo/15-hete mediated vascular adventitia fibrosis via p38 mapk-dependent TGF-β. J. Cell. Physiol. 2014, 229, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Schiller, M.; Javelaud, D.; Mauviel, A. TGF-β-induced smad signaling and gene regulation: Consequences for extracellular matrix remodeling and wound healing. J. Dermatol. Sci. 2004, 35, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Takawale, A.; Zhang, P.; Patel, V.B.; Wang, X.; Oudit, G.; Kassiri, Z. Tissue inhibitor of matrix metalloproteinase-1 promotes myocardial fibrosis by mediating cd63-integrin beta1 interaction. Hypertension 2017, 69, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Yigitkanli, K.; Pekcec, A.; Karatas, H.; Pallast, S.; Mandeville, E.; Joshi, N.; Smirnova, N.; Gazaryan, I.; Ratan, R.R.; Witztum, J.L.; et al. Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke. Ann. Neurol. 2013, 73, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.F.; Allard, J.; Avnur, Z.; Nikolcheva, T.; Rotstein, D.; Carlos, A.S.; Shea, M.; Waters, R.V.; Belknap, J.K.; Peltz, G.; et al. Regulation of bone mass in mice by the lipoxygenase gene alox15. Science 2004, 303, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Cyrus, T.; Pratico, D.; Zhao, L.; Witztum, J.L.; Rader, D.J.; Rokach, J.; FitzGerald, G.A.; Funk, C.D. Absence of 12/15-lipoxygenase expression decreases lipid peroxidation and atherogenesis in apolipoprotein e-deficient mice. Circulation 2001, 103, 2277–2282. [Google Scholar] [CrossRef] [PubMed]
- Bolick, D.T.; Srinivasan, S.; Whetzel, A.; Fuller, L.C.; Hedrick, C.C. 12/15 lipoxygenase mediates monocyte adhesion to aortic endothelium in apolipoprotein e-deficient mice through activation of rhoa and nf-kappab. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Schroen, B.; Vermeersch, P.; Milting, H.; Gao, F.; Kassner, A.; Gillijns, H.; Herijgers, P.; Flameng, W.; Carmeliet, P.; et al. Increased cardiac expression of tissue inhibitor of metalloproteinase-1 and tissue inhibitor of metalloproteinase-2 is related to cardiac fibrosis and dysfunction in the chronic pressure-overloaded human heart. Circulation 2005, 112, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, C.; Perrin, A.; Beck, M. Effectiveness of agalsidase alfa enzyme replacement in fabry disease: Cardiac outcomes after 10 years’ treatment. Orphanet J. Rare Dis. 2015, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Chien, Y.; Chen, Y.J.; Chen, S.F.; Chang, Y.L.; Chiang, C.H.; Jeng, S.Y.; Chang, C.M.; Wang, M.L.; Chen, L.K.; et al. Reprogramming induced pluripotent stem cells in the absence of c-myc for differentiation into hepatocyte-like cells. Biomaterials 2011, 32, 5994–6005. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating wnt/beta-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.P.; Chien, Y.; Chiou, G.Y.; Cherng, J.Y.; Wang, M.L.; Lo, W.L.; Chang, Y.L.; Huang, P.I.; Chen, Y.W.; Shih, Y.H.; et al. Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microrna145 with cationic polyurethane-short branch pei. Biomaterials 2012, 33, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.H.; Jiang, B.H.; Yu, Y.L.; Chou, S.J.; Tsai, P.H.; Chang, W.C.; Chen, L.K.; Chen, L.H.; Chien, Y.; Chiou, G.Y. Poly(adp-ribose) polymerase 1 regulates nuclear reprogramming and promotes ipsc generation without c-myc. J. Exp. Med. 2013, 210, 85–98. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chien, Y.; Chou, S.-J.; Chang, Y.-L.; Leu, H.-B.; Yang, Y.-P.; Tsai, P.-H.; Lai, Y.-H.; Chen, K.-H.; Chang, W.-C.; Sung, S.-H.; et al. Inhibition of Arachidonate 12/15-Lipoxygenase Improves α-Galactosidase Efficacy in iPSC-Derived Cardiomyocytes from Fabry Patients. Int. J. Mol. Sci. 2018, 19, 1480. https://doi.org/10.3390/ijms19051480
Chien Y, Chou S-J, Chang Y-L, Leu H-B, Yang Y-P, Tsai P-H, Lai Y-H, Chen K-H, Chang W-C, Sung S-H, et al. Inhibition of Arachidonate 12/15-Lipoxygenase Improves α-Galactosidase Efficacy in iPSC-Derived Cardiomyocytes from Fabry Patients. International Journal of Molecular Sciences. 2018; 19(5):1480. https://doi.org/10.3390/ijms19051480
Chicago/Turabian StyleChien, Yueh, Shih-Jie Chou, Yuh-Lih Chang, Hsin-Bang Leu, Yi-Ping Yang, Ping-Hsing Tsai, Ying-Hsiu Lai, Kuan-Hsuan Chen, Wei-Chao Chang, Shih-Hsien Sung, and et al. 2018. "Inhibition of Arachidonate 12/15-Lipoxygenase Improves α-Galactosidase Efficacy in iPSC-Derived Cardiomyocytes from Fabry Patients" International Journal of Molecular Sciences 19, no. 5: 1480. https://doi.org/10.3390/ijms19051480
APA StyleChien, Y., Chou, S.-J., Chang, Y.-L., Leu, H.-B., Yang, Y.-P., Tsai, P.-H., Lai, Y.-H., Chen, K.-H., Chang, W.-C., Sung, S.-H., & Yu, W.-C. (2018). Inhibition of Arachidonate 12/15-Lipoxygenase Improves α-Galactosidase Efficacy in iPSC-Derived Cardiomyocytes from Fabry Patients. International Journal of Molecular Sciences, 19(5), 1480. https://doi.org/10.3390/ijms19051480