Abstract
Intrinsically disordered regions (IDRs) and protein (IDPs) are highly flexible owing to their lack of well-defined structures. A subset of such proteins interacts with various substrates; including RNA; frequently adopting regular structures in the final complex. In this work; we have analysed a dataset of protein–RNA complexes undergoing disorder-to-order transition (DOT) upon binding. We found that DOT regions are generally small in size (less than 3 residues) for RNA binding proteins. Like structured proteins; positively charged residues are found to interact with RNA molecules; indicating the dominance of electrostatic and cation-π interactions. However, a comparison of binding frequency shows that interface hydrophobic and aromatic residues have more interactions in only DOT regions than in a protein. Further; DOT regions have significantly higher exposure to water than their structured counterparts. Interactions of DOT regions with RNA increase the sheet formation with minor changes in helix forming residues. We have computed the interaction energy for amino acids–nucleotide pairs; which showed the preference of His–G; Asn–U and Ser–U at for the interface of DOT regions. This study provides insights to understand protein–RNA interactions and the results could also be used for developing a tool for identifying DOT regions in RNA binding proteins.
1. Introduction
Intrinsically disordered proteins lack stable three-dimensional structures under physiological conditions and are known to perform important roles in several processes including signalling, enzymatic activity, and gene regulation [1,2]. To perform these functions, disordered regions interact with protein, RNA, DNA, and other small molecules to gain ordered structures [3,4]. Experimentally, interactions mediated by IDRs can be observed using NMR and X-ray crystallography. However, because of poor resolution, problems in crystallization, and high time and resource consumption, computational methods are necessary to identify disorder-mediated interactions [5,6].
Several methods have been developed for understanding the disorderness of proteins using sequence or structural information [7,8,9,10]. In addition, the transition of disorder-to-order regions in protein–protein interactions (PPI) is well studied experimentally and computationally [8,9,10,11,12]. For example, LMO4, a putative breast oncoprotein, interacts with various tandem LIM-domain containing proteins mediated by disordered regions [13]. BRCA1, a tumour suppressor protein, helps in binding with multiple protein and DNA partners by its central disorder region of ~1500 amino acids [14]. Recently, Papadakos et al. [15] showed that inducing intrinsic disorder in high-affinity protein–protein interactions reduces the affinity of binding.
Many proteins contain disordered regions and some of the regions attained ordered structures after binding to their cognate substrates, which are also known as MoRF (Molecular Recognition Features) segments [16,17]. Sugase et al. [18] have shown that folding and binding of IDPs or IDRs are coupled processes. Furthermore, binding partners are also shown to influence affinity and kinetics of binding. The flexibility of IDPs helps them to bind with multiple partners and have co-operative interactions [19]. Although induced fit and conformational selection processes are proposed explanations for the coupling of folding and binding, the exact model which is preferred by IDPs is not known [11,20].
The dynamics of the RNA molecule makes it more amenable to interact with disorder-mediated protein–RNA interactions [21]. The recognition of the protein–RNA complex has been experimentally studied using EMSA, yeast-3-hybrid assay, pull-down assay and CLIP [22,23]. On the other hand, plenty of tools have been developed to identify binding sites in RNA-binding proteins [24,25,26,27,28,29,30,31,32]. All these methods use the information in their sequence to compute the feature and/or evaluate the performance. Recently, Peng and Kurgan [33] developed a webserver for prediction of disorder-mediated interactions in RNA, DNA and protein–protein complexes. However, the knowledge for understanding the mechanisms or factors responsible for binding of disordered region with RNA has not yet been completely explored.
In this work, we constructed a dataset for protein–RNA complexes (provided in supplementary information), which are involved in disorder-to-order transitions. Utilizing the dataset, we analyzed the number and size of DOT regions in protein–RNA complexes, preference of residues involved in binding in DOT regions, secondary structure, solvent accessibility, pair preference at the interface, preference in different secondary structures of RNA, and interaction energy between protein and RNA DOT and non-DOT regions at the interface.
2. Results and Discussion
Our dataset contains a total of 23,452 and 2412 residues in non-ribosomal and ribosomal protein–RNA complexes. Among them, 1175 (5%) and 155 (6.4%) residues are found to be in DOT regions in non-ribosomal and ribosomal complexes, respectively. The residues binding with RNA are obtained by using 3.5 and 6 Å distance cut-offs and similar trends are obtained. Therefore, we have presented the results with 3.5 Å and those for 6 Å are shown in supplementary material.
2.1. Number of DOT Regions in Protein–RNA Complexes and Length of DOT Regions
The variation in the number of DOT regions in non-ribosomal and ribosomal complexes is shown in Figure 1. We observed that most of the complexes have less than three DOT regions (88% in non-ribosomal and 100% in ribosomal complexes). Most non-ribosomal proteins have one DOT region, whereas ribosomal proteins have mostly two or more DOT regions. In addition, at most eight DOT regions per complex are found in our dataset.
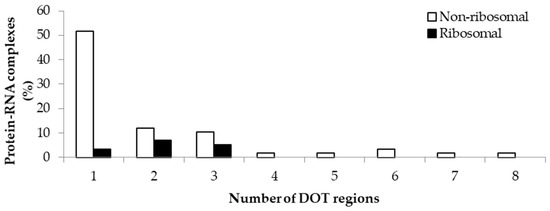
Figure 1.
Percentage of protein–RNA complexes containing different number of DOT regions.
Further, we analysed the length of each DOT region in non-ribosomal and ribosomal protein–RNA complexes, which shows that most DOT regions are short, as shown in Figure 2. In both non-ribosomal and ribosomal complexes, more than 70% of DOT regions have three to 10 residues and very few (only 5) regions have a length of more than 50 residues. This leads to a speculation that only a small conformational change might be required for bringing shape complementarity in protein–RNA complexes and these small DOT regions help in obtaining the same.
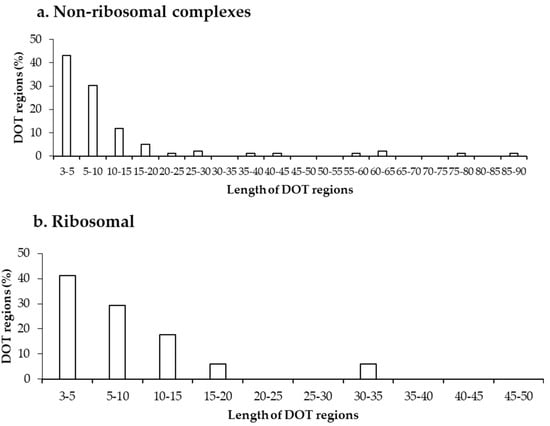
Figure 2.
Length distribution of DOT regions in protein–RNA complexes in (a) non-ribosomal and (b) ribosomal complexes.
2.2. Binding Frequency of Residues at DOT Regions
The binding frequencies of residues in DOT regions using 3.5 Å (NR3.5 and RB3.5) and 6 Å (NR6 and RB6) distance cut-offs are shown in Figure 3 and Figure S2, respectively. We observed that among all positively charged residues (Arg, Lys and His), Arg and Lys have high preference for binding in both NR3.5 (Figure 3a) and NR6 (Figure S2a) datasets. Interestingly, only eight and 13 among 20 residues are observed in binding DOT regions at RB3.5 (Figure 3b) and RB6 (Figure S2b) datasets, and Arg has the highest frequency of binding. Cys, Met, and Trp in DOT regions are not involved in binding with RNA, whereas in ordered complexes 0.97%, 4.52%, and 5.54% of Cys, Met, and Trp are involved in binding, respectively. The comparison of binding site residues in DOT regions and the whole protein showed an expected presence of 1.5% and 2.7% of Met and Trp, respectively, in the interface of the DOT region. These results showed that the non-occurrence of Met and Trp at the interface of the DOT regions is statistically significant.
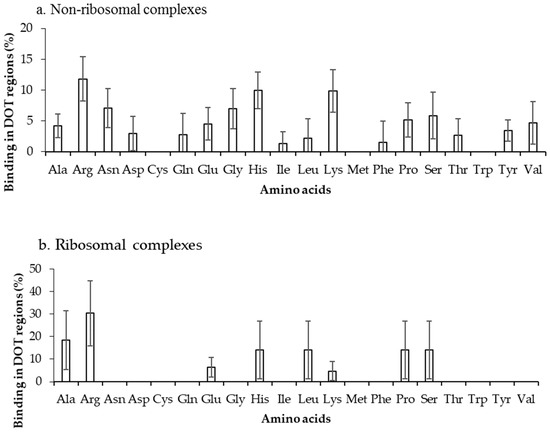
Figure 3.
Amino acid frequency of binding in the DOT region for (a) non-ribosomal and (b) ribosomal protein–RNA complexes.
We have computed the preference of binding of residues in DOT regions by dividing the number of residues in DOT regions with the total number of binding residues, and the results are presented in Figure 4 and Figure S3 for 3.5 Å (NR3.5 and RB3.5 datasets) and 6 Å (NR6 and RB6 datasets), respectively. In Figure 4a, high frequency of Arg, Gly, Lys, and Ser (z-score > 1) is observed for the NR3.5 dataset, which suggests that these residues are more probable to contact DOT regions with respect to all residues in contact with RNA. However, for the NR6 dataset (Figure S3a), the result is only consistent for Lys, and two other residues (Glu and Pro) show high binding frequency. In ribosomal protein complexes with 3.5 Å and 6 Å, Ala & Glu, and Glu & Tyr have high frequencies, respectively (Figure 4b and Figure S3b).
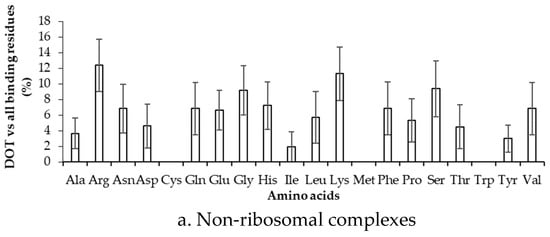
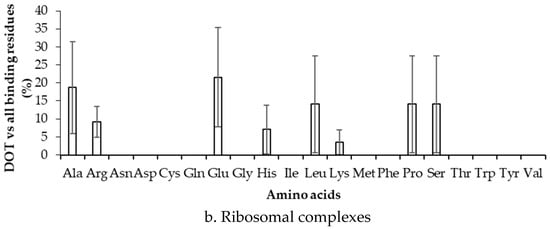
Figure 4.
Frequency of DOT regions by contact residues for (a) non-ribosomal and (b) ribosomal complexes.
2.3. Binding Propensity of Residues at DOT Region
Propensity is calculated by normalizing the binding frequency of residues in DOT regions with the overall frequency of the respective residues to be in a protein, using Equation (3). This can measure the bias in binding of residues in DOT regions, independent of their count in DOT regions. We have calculated the propensity of amino acids to be in DOT regions using distance cut-offs of 3.5 Å and 6 Å and the results are shown in Figure 5 and Figure S4, respectively. In the NR3.5 (Figure 5a) dataset, His, Arg, Asn, Gln, Phe, and Tyr have high propensity of binding, whereas in ribosomal proteins (RB3.5 dataset; Figure 5b), only His showed a high propensity. In the NR6 (Figure S4a), His has high propensity, whereas Asn, His and Tyr have high propensity in the RB6 (Figure S4b) dataset. Similarly, high propensity for binding is observed for positively charged residues along with Tyr and Phe in protein–RNA complexes [34]. On the other hand, among all charged residues only Arg has high tendency to bind with DOT regions in protein–protein complexes [35]. Furthermore, non-specific interactions occurred frequently in protein–protein complexes, which is not a common trend in the binding residues of DOT regions in protein–RNA complexes. Therefore, we can infer that the preferred residues at DOT regions are specific in protein–RNA complexes and, especially, charged interactions are important in DOT regions for binding with RNA.
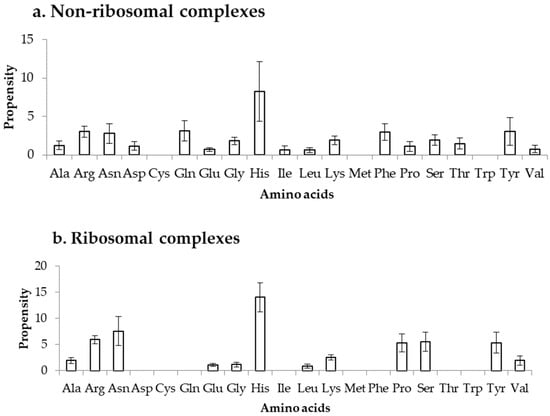
Figure 5.
Propensity for amino acids in (a) non-ribosomal and (b) ribosomal complexes.
2.4. Comparison of Frequency of Binding in the DOT Region and Other Residues of a Protein
To estimate the difference between binding in DOT regions and other part of proteins, we calculated the binding frequency of amino acids in these regions, as shown in Figure 6 and Figure S5. Amino acids significantly differ in their binding with RNA in DOT regions and in the complete protein (p-value for the mean is less than 0.01). In non-ribosomal proteins, when the 3.5 Å cut-off is considered, nonpolar and aromatic residues mostly have high frequency values in the DOT regions than in the overall protein. All the frequencies are observed to be significant when statistical analysis is performed for the bootstrapped sample of the frequencies (p-value is less than 0.01). Residues such as His, Phe, and Leu are found to have a more than 3-fold increase in the frequency of binding in the DOT regions than in other parts of the proteins. A similar trend is observed in the NR6 dataset (Figure S5).
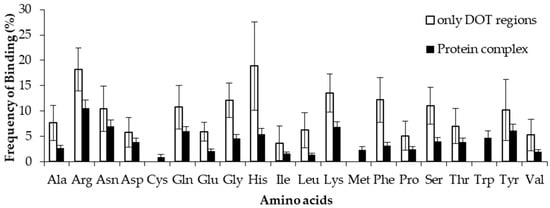
Figure 6.
Binding frequency of each amino acid in the DOT region and in the overall protein for non-ribosomal complexes using the 3.5 Å cut-off.
2.5. Amino Acid Contact Frequency with Nucleotides
We have also analysed amino acid contacts with each nucleotide in non-ribosomal complexes using 3.5 Å and 6 Å distance cut-offs for contacting residues and the results are shown in Figure 7 and Figure S6. In the 3.5 Å distance criterion, Arg and Lys have a high frequency to bind with nucleotides. Arg and Lys are observed to have the most and least binding frequencies with Guanine and Uracil, respectively. Whereas in the 6 Å criterion, almost the same frequency of binding is observed for Arg and Lys with Adenine, Guanine and Cytosine nucleotides; least binding was observed in the Uracil nucleotide. When compared with the results presented for ordered protein–DNA and protein–RNA complexes in our earlier works, Arg, Lys, Trp, and Tyr were favoured by RNA and Arg was selected by DNA-binding proteins together with Guanine in DNA and Uracil in RNA–protein complexes [36].

Figure 7.
Normalized amino acid nucleotide contact frequency in non-ribosomal protein–RNA complexes at 3.5 Å.
2.6. Secondary Structure of DOT and RNA-Interacting DOT Residues
The secondary structures of DOT residues are quantified to study the bias of residues to have a specific secondary structure in binding and non-binding regions and data are presented in Table 1 and Table S1 for NR3.5 and NR6 datasets, respectively. In the NR3.5 dataset, all the DOT residues have lower and higher preference in sheet (15%) and other structure class (8%), respectively. Interestingly, in DOT residues, binding with RNA molecules, strand-forming residues have a higher preference (15.2%) as compared to helical (8.6%) and other regions (8.9%).

Table 1.
Secondary structure of all DOT residues and residues binding with RNA in DOT regions in the NR3.5 dataset.
2.7. Relative Solvent Accessibility of DOT Residues
The spatial arrangement of DOT residues is further explored by solvent accessibility calculation and the result is shown in Table 2. Comparison of RASA of DOT regions and complete protein–RNA complex revealed that in DOT regions, solvent accessibility of every amino acid is more than that of other amino acids of a protein. As expected, charged residues have low fold difference (1.18 to 1.28) in RASA in DOT regions and the complete protein. However, most hydrophobic residues (Ala, Cys, Ile, Leu, Met, Phe, Tyr, and Val) have about 1.8 to 2 folds higher RASA in DOT regions than the complete protein, Met has the highest difference. On the other hand, the mean solvent accessibility of DOT regions of proteins is 44 Å2, which is similar to the average RASA of binding DOT regions (43 Å2) of protein–protein complexes [17].
Table 2.
Relative average solvent accessibility (RASA) of DOT residues and all residues in non-ribosomal protein–RNA complexes.
2.8. Number of Residues in Contact with Nucleotides in the DOT Region and in Entire Protein
Among 1175 residues in DOT regions in our dataset, only 96 (8.17%) and 268 (22.81%) are in contact with nucleotides in the NR3.5 and the NR6 dataset, respectively. Almost all the residues have a similar tendency of binding with nucleotides in proteins, ranging between 20% to 29%, as shown in Table 3 and Table S2. However, the number of nucleotides interacting with DOT residues is somewhat different, that is, the range of interaction is 18 to 33%. The DOT residues are more likely to bind with Guanine (20.4%), followed by Cytosine and Uracil, than to binding with Adenine (13.1%).
Table 3.
Number of interaction of nucleotides with DOT residues and with complete protein at 3.5 Å.
2.9. Secondary Structure of Nucleotides Interacting with DOT Residues
Further, we have classified the nucleotides based on location and contacts with DOT residues and preference of amino acids in a protein and the results are presented in Table 4 and Table S3. Among all secondary structures formed by nucleotides, unpaired bases are most likely to bind with DOT residues. Specifically, we observed that A and U in unpaired regions prefer to interact with DOT residues, whereas C and G in unpaired and base-paired positions interact with DOT residues with a similar preference. G and C also interact with DOT residues in pseudoknot secondary structure, whereas A and U are least likely to exist in pseudoknot form when bound to DOT regions.
Table 4.
Preference of nucleotides in different secondary structures to bind with DOT residues.
2.10. Interaction Energy of DOT Residues with Nucleotides
We have computed the interaction energy between amino acids and nucleotides in DOT and ordered regions at the binding interface and the results are presented in Table 5. Most of the amino acids have stronger interactions with nucleotides in ordered regions than DOT regions. However, we noticed that some combinations of amino acid–nucleotide pairs have favourable energy when interacting with DOT regions. For example, Arg, His, Ile, Leu, Val, and Phe interact with G, His, Ser, and Val with C, and Asn, Asp, Gly, Ile, Leu, and Ser with U. In addition, hydrophobic residues Ile, Leu, and Val have more favourable interactions with G at DOT regions than others. Since Arg and Lys are important for protein–RNA complex formation through electrostatic interactions these residues have stronger energies in ordered regions than DOT regions. On the other hand, His in the DOT region has favourable energy with G and C. These differences in energy could be important to understand the interactions between DOT regions and the RNA molecule, which might also be used to distinguish the RNA binding residues of proteins in DOT and other regions.
Table 5.
Interaction energy between amino acids and nucleotides in DOT regions.
We have compared the interaction energy of amino acid–nucleotide pairs in the interface of DOT and other regions and two typical examples are shown in Figure 8. We noticed a wide range of interactions such as stacking, cation-π, electrostatic, and van der Waals interactions at the interface. Most favourable energy is observed for Asn and His with U (−3.26 kcal/mol) and G (−5.44 kcal/mol), respectively, in DOT regions (Figure 8a). On the other hand, Arg and Phe have favourable energy with A (−8.49 kcal/mol) and C (−4.88 kcal/mol), respectively, in non-DOT regions.
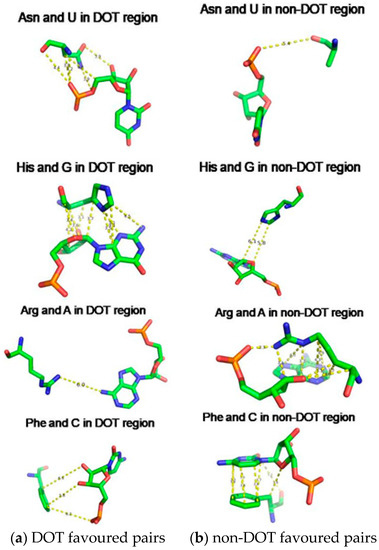
Figure 8.
Amino acid showing (a) strong interaction in DOT and weak interaction in non-DOT regions and (b)weak interaction in DOT and strong interaction in non-DOT regions.
3. Materials and Methods
We adopted the following protocol to obtain a set of protein–RNA complexes with disorder-to-order transition (DOT) regions: (i) Downloaded the protein–RNA complexes from PDB and NDB databases (www.rcsb.org) [37,38,39]; (ii) Clustered all the protein–RNA complexes with 30% sequence identity cut-off using CD-Hit suite [40]; (iii) Performed BLAST search (using 99% identity cut-off) of protein sequences to obtain free proteins corresponding to each protein–RNA complex [41,42]. The free proteins have the same sequences as the protein part of protein–RNA complexes but crystallized without RNA. Note that free proteins contain unique PDB IDs, which is distinct from the protein–RNA complex; (iv) Disordered residues are obtained from missing residues information in the protein–RNA complex and free protein pairs by locating “REMARK 465” statement in the protein structure file; (v) DOT residues are isolated by comparing the disorder residues of free and protein–RNA complex pairs such that the residue is ordered in the protein–RNA complex but disordered in free protein. Note that only the regions having 3 or more continuous DOT residues are considered. The final dataset contains 101 DOT regions in 52 proteins and complete data are given in supplementary information. The representation of DOT and ordered region in a typical protein–RNA complex (PDB ID: 4H4K) is shown in Figure 9.
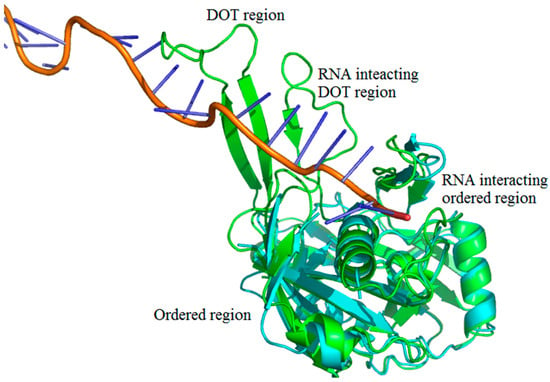
Figure 9.
Representation of disorder-to-order mediated interactions. Free protein, RNA, and complex (CRISPR-Cas RNA Silencing Cmr Complex) are shown in cyan, orange and green, respectively. The PDB IDs are 4H4K:A (free protein), 3XIL:I (RNA of protein–RNA complex) and 3XIL:B (RNA-bound protein). The disorder-to-order transition (DOT) region can be clearly seen in green with a missing overlapping region of free protein.
3.1. Number of DOT Regions and Their Lengths
The number of DOT regions and their lengths are obtained by counting the number of non-consecutive and consecutive residues, respectively, using custom build python scripts.
3.2. DOT Residues in Contact with RNA
The residues in contact with RNA molecules are obtained by using distance cut-offs mentioned in literature, that is, 3.5 Å and 6 Å [43,44,45]. Binding residues in DOT regions are obtained by taking common residues in the DOT dataset and RNA contacting residues. We have classified protein–RNA complexes in non-ribosomal and ribosomal classes because of the difference in their interaction pattern, number of interacting amino acids, and residue bias in them [46]. Therefore, using the type of complex and distance cut-off for interacting residues, we divided protein–RNA complexes into four different datasets: (1) NR3.5: non-ribosomal complex with a contact distance of 3.5 Å; (2) RB3.5: ribosomal complex with a contact distance of 3.5 Å; (3) NR6: non-ribosomal complex with a contact distance of 6 Å; and (4) RB6: ribosomal complex with a contact distance of 6 Å.
We computed the frequency of each DOT residue involved in binding using the Equation (1).
where Nib: number of ith residues binding in the DOT region and Nid: number of ith residues in DOT.
Moreover, the differences in the frequency of binding residues in DOT regions and in the protein complexes are obtained.
3.3. Frequency of Binding in DOT and Other Residues
We also computed the frequency of residues binding in DOT regions over all the binding residues by using Equation (2), an error bar is plotted using the bootstrap method by randomly re-sampling an equal sized data with a replacement 1000 times.
where Nibd: number of ith residues binding in DOT region; Nib is number of ith residues binding with RNA in complete protein.
3.4. Propensity of Binding Residues in DOT Region
The normalization of frequency of residues present in DOT regions by individual residue frequency provides the tendency of a residue in DOT regions. Accordingly, propensity values are calculated using the following equation:
where Propensity (I): propensity of ith residue; Nibd: number of ith residue binding in DOT region; Nid: number of ith residue in DOT regions; Nip: number of ith residue in protein; Np: number of residues in protein.
3.5. Boot Strap Sampling
To obtain the standard error in frequency and propensity calculations, bootstrap sampling is performed. In this technique all the protein–RNA complexes are sampled randomly and each sample contains complexes equal to the number of protein–RNA complexes. Therefore, each sample will have redundancy of some complexes and will be devoid of some complexes. In this manner, we have created 1000 samples on which the calculations are performed.
3.6. Relative Average Solvent Accessibility (RASA)
The DOT residues buriedness is analysed by the NACCESS [47] program and the RASA of each residue is calculated by using Equation (4).
where Aibd: RASA of ith residue binding with RNA in DOT region; n: number of DOT residues in a protein–RNA complex.
3.7. Secondary Structure of Protein and RNA
Secondary structure of both proteins and RNA molecules are analysed by DSSP and DSSR programs, respectively [48,49]. The DSSR program gives dot bracket notation of secondary structure of RNA as shown in Figure S1, in which “.” represents unpaired nucleotide, “(” or “)” represent paired bases, and “{” or “}” or “[” or “]” or “<” or “>” represent pseudoknot bases.
3.8. Binding Preference of Nucleotides for Amino Acids
The binding preference of nucleotide with DOT residues has been calculated by counting the occurrence of nucleotides–amino acid interacting pairs under the distance of 3.5 Å.
3.9. Interaction Energy between Amino Acids and Nucleotides at Binding Interface
The interaction energy of amino acids with nucleotides is computed using van der Waals and coulombs potential using AMBER force field [50]. It is given by
where, Aij = εij* (Rij*)12 and Bij = 2 εij* (Rij*)6; Rij* = (Ri* + Rj*); and εij* = (εi* εj*)1/2; R* and ε* van der Waals radius and well depth, respectively, and these parameters are obtained from Gromiha et al. [51]; qi and qj is the charge on atom i and j, respectively and Rij is the distance separating atom i and j.
4. Conclusions
The analysis of DOT regions in protein–RNA complexes revealed that in each complex these regions are generally small in size. Electrostatic interactions are found to be important, with the involvement of positively charged residues (Arg, Lys and His) in DOT regions. Among nucleotide–amino acid pairs, guanine–Arg and uracil–Lys pairs are identified to be the most and the least preferred ones at the interface, respectively. Generally, nucleotides prefer to bind DOT regions than other regions of protein. Further, DOT regions are significantly more exposed to solvent than other residues of protein–RNA complexes. Specifically, hydrophobic residues have higher difference in RASA of DOT regions and complete proteins. DOT regions are preferred to form coils, turns, and bends than regular secondary structures such as helices and strands. On the RNA side, DOT residues prefer to bind unpaired A and U and paired regions of C and G. In pseudoknot condition, mostly C and G interact with DOT residues. The interaction energy calculations revealed the types of interactions and preferred amino acid-nucleotide pairs at the interface based on energy.
The frequencies and propensities obtained in the present study could be used for discriminating DOT binding residues from other residues. Further, the location of DOT binding residues based on solvent accessibility and secondary structure of protein and RNA along with energy calculations may help to understand the recognition mechanism.
We obtained the DOT regions by comparing 3D coordinates of the missing residues in protein–RNA complexes and their respective free proteins. This might be an under representation of DOT regions since the structures solved by crystallization often stabilize the residues and reduce the native disorder. Hence, the disordered residues having 3D coordinates in free proteins are not considered. The current study can further be refined with the availability of more numbers of protein–RNA complexes and the improvements in structure determination techniques. In addition, development of disorder specific databases for protein–nucleic acid complexes with large datasets could enhance the confidence level of the result reported in the present study.
Supplementary Materials
Supplementary materials can be found at http://www.mdpi.com/1422-0067/19/6/1595/s1.
Author Contributions
M.M.G. and S.A. conceived the project and designed experiments. A.S. constructed the dataset and performed the analysis. M.M.G, S.A. and A.S took part in discussions. A.S. drafted the manuscript. M.M.G and S.A. edited and refined the manuscript.
Acknowledgments
We thank the Department of Biotechnology, Indian Institute of Technology Madras for computational facilities. A.S. thank Ministry of Human Resource and Development (MHRD) for the fellowship. This project is partially supported by the Council of Scientific & Industrial Research (CSIR), Government of India to M.M.G. and S.A. (grant numbers: 37(1694)/17/EMR-II) and 37(1695)/17/EMRII respectively). S.A. would like to acknowledge a grant from University for potential of excellence (UpoE-II) #270 and support DST-PURSE.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Toóth-Petroóczy, A.; Kraut, D.A.; Matouschek, A.T.; Lim, R.Y.; Xue, B.; Kurgan, L.; Uversky, V.N. Disordered proteinaceous machines. Chem. Rev. 2014, 114, 6806–6843. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.M.; Van der, L.R.; de Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struct. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signaling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Deller, M.C.; Kong, L.; Rupp, B. Protein stability: A crystallographer’s perspective. Acta Cryst. F 2016, 72, 72–95. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Xue, B.; Sickmeier, M.D.; Meng, J.; Cortese, M.S.; Oldfield, C.J.; Le Gall, T.; Dunker, A.K.; Uversky, V.N. High-throughput characterization of intrinsic disorder in proteins from the Protein Structure Initiative. J. Struct. Biol. 2012, 180, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2014, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Disfani, F.M.; Hsu, W.L.; Mizianty, M.J.; Oldfield, C.J.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics 2012, 28, i75–i83. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Uversky, V.N.; Kurgan, L. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell. Mol. Life Sci. 2017, 74, 3069–3090. [Google Scholar] [CrossRef] [PubMed]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Functional advantages of dynamic protein disorder. FEBS Lett. 2015, 589, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Söderquist, F.; Wallner, B. Proteus: A random forest classifier to predict disorder-to-order transitioning binding regions in intrinsically disordered proteins. J. Comput. Aided Mol. Des. 2017, 31, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Deane, J.E.; Ryan, D.P.; Sunde, M.; Maher, M.J.; Guss, J.M.; Visvader, J.E.; Matthews, J.M. Tandem LIM domains provide synergistic binding in the LMO4: Ldb1 complex. EMBO J. 2004, 23, 3589–3598. [Google Scholar] [CrossRef] [PubMed]
- Mark, W.Y.; Liao, J.C.; Lu, Y.; Ayed, A.; Laister, R.; Szymczyna, B.; Chakrabartty, A.; Arrowsmith, C.H. Characterization of segments from the central region of BRCA1: An intrinsically disordered scaffold for multiple protein–protein and protein–DNA interactions? J. Mol. Biol. 2005, 345, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Papadakos, G.; Sharma, A.; Lancaster, L.; Bowen, R.; Kaminska, R.; Leech, A.P.; Walker, D.; Redfield, C.; Kleanthous, C. Consequences of inducing intrinsic disorder in a high-affinity protein-protein interaction. J. Am. Chem. Soc. 2015, 137, 5252–5255. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, S.; Amemiya, T.; Sakamoto, S.; Nobe, Y.; Hosoda, K.; Kado, Y.; Murakami, S.D.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL in 2014 illustrates interaction networks composed of intrinsically disordered proteins and their binding partners. Nucleic Acids Res. 2014, 42, D320–D325. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Sugase, K.; Dyson, H.J.; Wright, P.E. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 2007, 447, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Shammas, S.L.; Travis, A.J.; Clarke, J. Allostery within a transcription coactivator is predominantly mediated through dissociation rate constants. Proc. Natl. Acad. Sci. USA 2014, 111, 12055–12060. [Google Scholar] [CrossRef] [PubMed]
- Shammas, S.L.; Crabtree, M.D.; Dahal, L.; Wicky, B.I.; Clarke, J. Insights into coupled folding and binding mechanisms from kinetic studies. J. Biol. Chem. 2016, 291, 6689–6695. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J. Roles of intrinsic disorder in protein–nucleic acid interactions. Mol. Biosyst. 2012, 8, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Dey, B.; Thukral, S.; Krishnan, S.; Chakrobarty, M.; Gupta, S.; Manghani, C.; Rani, V. DNA–protein interactions: Methods for detection and analysis. Mol. Cell. Biochem. 2012, 365, 279–299. [Google Scholar] [CrossRef] [PubMed]
- Popova, V.V.; Kurshakova, M.M.; Kopytova, D.V. Methods to study the RNA-protein interactions. Mol. Biol. 2015, 49, 472–481. [Google Scholar] [CrossRef]
- Walia, R.R.; Caragea, C.; Lewis, B.A.; Towfic, F.; Terribilini, M.; El-Manzalawy, Y.; Dobbs, D.; Honavar, V. Protein–RNA interface residue prediction using machine learning: An assessment of the state of the art. BMC Bioinform. 2012, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P. Prediction of RNA binding sites in a protein using SVM and PSSM profile. Proteins 2008, 71, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Terribilini, M.; Sander, J.D.; Lee, J.H.; Zaback, P.; Jernigan, R.L.; Honavar, V.; Dobbs, D. RNABindR: A server for analyzing and predicting RNA-binding sites in proteins. Nucleic Acids Res. 2007, 35, W578–W584. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brown, S.J. BindN: A web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 2006, 34, W243–W248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, Z.; Kurgan, L. Comprehensive review and empirical analysis of hallmarks of DNA-, RNA-and protein-binding residues in protein chains. Brief. Bioinform. 2017, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, C.; Yang, M.Q.; Yang, J.Y. BindN+ for accurate prediction of DNA and RNA-binding residues from protein sequence features. BMC Syst. Biol. 2010, 4, S3. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Friedrich, S.; Kurgan, L. A comprehensive comparative review of sequence-based predictors of DNA-and RNA-binding residues. Brief. Bioinform. 2015, 17, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Tuszynska, I.; Bujnicki, J.M. DARS-RNP and QUASI-RNP: New statistical potentials for protein–RNA docking. BMC Bioinform. 2011, 12, 348. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, Y.; Pu, X.; Li, M. A sequence-based computational method for prediction of MoRFs. RSC Adv. 2017, 7, 18937–18945. [Google Scholar] [CrossRef]
- Peng, Z.; Kurgan, L. High-throughput prediction of RNA, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res. 2015, 43, e121. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.T.; Yura, K.; Go, N. Amino acid residue doublet propensity in the protein–RNA interface and its application to RNA interface prediction. Nucleic Acids Res. 2006, 34, 6450–6460. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Kumagai, Y.; Standley, D.M.; Sarai, A.; Mizuguchi, K.; Ahmad, S. Prediction of dinucleotide-specific RNA-binding sites in proteins. BMC Bioinform. 2011, 12, S5. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; Green, R.K. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [PubMed]
- Berman, H.M.; Olson, W.K.; Beveridge, D.L.; Westbrook, J.; Gelbin, A.; Demeny, T.; Hsieh, S.H.; Srinivasan, A.R.; Schneider, B. The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J. 1992, 63, 751–759. [Google Scholar] [CrossRef]
- Coimbatore Narayanan, B.; Westbrook, J.; Ghosh, S.; Petrov, A.I.; Sweeney, B.; Zirbel, C.L.; Leontis, N.B.; Berman, H.M. The Nucleic Acid Database: New features and capabilities. Nucleic Acids Res. 2014, 42, D114–D122. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Schäffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct. 2012, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Gromiha, M.M. Protein Bioinformatics: From Sequence to Function; Academic Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Si, J.; Zhao, R.; Wu, R. An overview of the prediction of protein DNA-binding sites. Int. J. Mol. Sci. 2015, 16, 5194–5215. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, R.; Gromiha, M.M. Prediction of RNA binding residues: An extensive analysis based on structure and function to select the best predictor. PLoS ONE 2014, 9, e91140. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gallina, C.; Guerra, C. Analysis of interactions between ribosomal proteins and RNA structural motifs. BMC Bioinform. 2010, 11, S41. [Google Scholar] [CrossRef] [PubMed]
- NACCESS, V2.1.1. A Computer Program for Solvent Accessible Area Calculations; Department of Biochemistry and Molecular Biology, University College London: London, UK, 1993. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.J.; Bussemaker, H.J.; Olson, W.K. DSSR: An integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 2015, 43, e142. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Yokota, K.; Fukui, K. Understanding the recognition mechanism of protein–RNA complexes using energy based approach. Curr. Protein Pept. Sci. 2010, 11, 629–638. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).