Endogenous Antiangiogenic Factors in Chronic Kidney Disease: Potential Biomarkers of Progression


Abstract
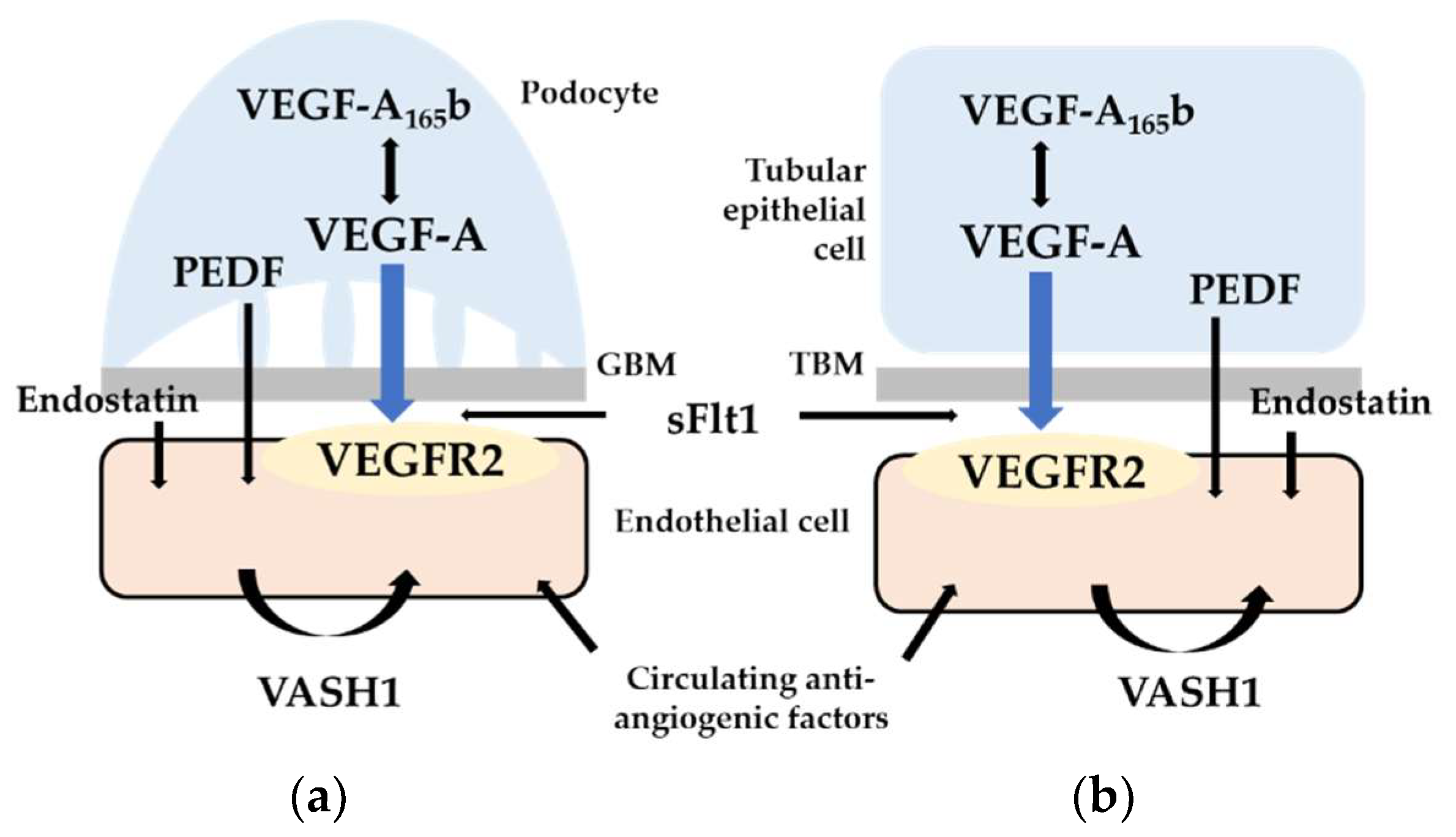
:1. Introduction
2. Soluble Fms-Related Tyrosine Kinase 1 (Flt-1)
3. Soluble Endoglin
4. Pigment Epithelium-Derived Factor (PEDF)
5. VEGF-A165b
6. Endostatin
7. Vasohibins
8. Conclusions
Funding
Conflicts of Interest
References
- Dimke, H.; Sparks, M.A.; Thomson, B.R.; Frische, S.; Coffman, T.M.; Quaggin, S.E. Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J. Am. Soc. Nephrol. 2015, 26, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. Vegf inhibition and renal thrombotic microangiopathy. New Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Veron, D.; Reidy, K.J.; Bertuccio, C.; Teichman, J.; Villegas, G.; Jimenez, J.; Shen, W.; Kopp, J.B.; Thomas, D.B.; Tufro, A. Overexpression of vegf-a in podocytes of adult mice causes glomerular disease. Kidney Int. 2010, 77, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Hakroush, S.; Moeller, M.J.; Theilig, F.; Kaissling, B.; Sijmonsma, T.P.; Jugold, M.; Akeson, A.L.; Traykova-Brauch, M.; Hosser, H.; Hahnel, B.; et al. Effects of increased renal tubular vascular endothelial growth factor (vegf) on fibrosis, cyst formation, and glomerular disease. Am. J. Pathol. 2009, 175, 1883–1895. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Maeshima, Y.; Sato, Y.; Wada, J. Antiangiogenic therapy for diabetic nephropathy. BioMed Res. Int. 2017, 2017, 5724069. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Wada, J. Vegf-targeting strategies against diabetic nephropathy: Obsolete or still promising? Biomed. J. Sci. Tech. Res. 2018, 2, 000758. [Google Scholar] [CrossRef]
- Avihingsanon, Y.; Benjachat, T.; Tassanarong, A.; Sodsai, P.; Kittikovit, V.; Hirankarn, N. Decreased renal expression of vascular endothelial growth factor in lupus nephritis is associated with worse prognosis. Kidney Int. 2009, 75, 1340–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sflt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. New Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Cerdeira, A.S.; Redman, C.; Vatish, M. Meta-analysis and systematic review to assess the role of soluble fms-like tyrosine kinase-1 and placenta growth factor ratio in prediction of preeclampsia: The sappphire study. Hypertension 2018, 71, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, A.; Attini, R.; Nuzzo, A.M.; Piazzese, A.; Parisi, S.; Ferraresi, M.; Todros, T.; Piccoli, G.B. Chronic kidney disease may be differentially diagnosed from preeclampsia by serum biomarkers. Kidney Int. 2013, 83, 177–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.H.; Oh, J.H.; Seo, J.A.; Lee, K.W.; Kim, S.G.; Choi, K.M.; Baik, S.H.; Choi, D.S.; Kang, Y.S.; Han, S.Y.; et al. Vascular endothelial growth factor (vegf) and soluble vegf receptor flt-1 in diabetic nephropathy. Kidney Int. 2005, 67, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Theilade, S.; Lajer, M.; Jorsal, A.; Tarnow, L.; Parving, H.H.; Rossing, P. Evaluation of placental growth factor and soluble fms-like tyrosine kinase 1 as predictors of all-cause and cardiovascular mortality in patients with type 1 diabetes with and without diabetic nephropathy. Diabet. Med. J. Br. Diabet. Assoc. 2012, 29, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.H.; White, K.E.; Dei Cas, A.; Hayward, A.; Webster, Z.; Bilous, R.; Marshall, S.; Viberti, G.; Gnudi, L. Inducible overexpression of sflt-1 in podocytes ameliorates glomerulopathy in diabetic mice. Diabetes 2008, 57, 2824–2833. [Google Scholar] [CrossRef] [PubMed]
- Bus, P.; Scharpfenecker, M.; Van Der Wilk, P.; Wolterbeek, R.; Bruijn, J.A.; Baelde, H.J. The vegf-a inhibitor sflt-1 improves renal function by reducing endothelial activation and inflammation in a mouse model of type 1 diabetes. Diabetologia 2017, 60, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Sison, K.; Li, C.; Tian, R.; Wnuk, M.; Sung, H.K.; Jeansson, M.; Zhang, C.; Tucholska, M.; Jones, N.; et al. Soluble flt1 binds lipid microdomains in podocytes to control cell morphology and glomerular barrier function. Cell 2012, 151, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, T.; Nakayama, T.; Li, Q.; Chiodo, V.A.; Zhang, L.; Campbell-Thompson, M.; Grant, M.; Croker, B.P.; Nakagawa, T. Soluble flt-1 gene therapy ameliorates albuminuria but accelerates tubulointerstitial injury in diabetic mice. Am. J. Physiol. Ren. Physiol. 2010, 298, F609–F616. [Google Scholar] [CrossRef] [PubMed]
- Chapal, M.; Neel, M.; Le Borgne, F.; Meffray, E.; Carceles, O.; Hourmant, M.; Giral, M.; Foucher, Y.; Moreau, A.; Fakhouri, F. Increased soluble flt-1 correlates with delayed graft function and early loss of peritubular capillaries in the kidney graft. Transplantation 2013, 96, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Edelbauer, M.; Kshirsagar, S.; Riedl, M.; Billing, H.; Tonshoff, B.; Haffner, D.; Dotsch, J.; Wechselberger, G.; Weber, L.T.; Steichen-Gersdorf, E. Soluble vegf receptor 1 promotes endothelial injury in children and adolescents with lupus nephritis. Pediatr. Nephrol. 2012, 27, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.L.; Zhu, L.; Shi, S.F.; Liu, L.J.; Lv, J.C.; Zhang, H. Elevated soluble vegf receptor sflt-1 correlates with endothelial injury in iga nephropathy. PLoS ONE 2014, 9, e101779. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Reuter, S.; Hillebrand, U.; Amler, S.; Konig, M.; Larger, E.; Oberleithner, H.; Brand, E.; Pavenstadt, H.; Brand, M. The soluble vegf receptor sflt1 contributes to endothelial dysfunction in ckd. J. Am. Soc. Nephrol. 2009, 20, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Kentrup, D.; Reuter, S.; Mayer, A.B.; Golle, L.; Tiemann, K.; Fobker, M.; Engelbertz, C.; Breithardt, G.; Brand, E.; et al. Soluble flt-1 links microvascular disease with heart failure in ckd. Basic Res. Cardiol. 2015, 110, 30. [Google Scholar] [CrossRef] [PubMed]
- Ky, B.; French, B.; Ruparel, K.; Sweitzer, N.K.; Fang, J.C.; Levy, W.C.; Sawyer, D.B.; Cappola, T.P. The vascular marker soluble fms-like tyrosine kinase 1 is associated with disease severity and adverse outcomes in chronic heart failure. J. Am. Coll. Cardiol. 2011, 58, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Onoue, K.; Uemura, S.; Takeda, Y.; Somekawa, S.; Iwama, H.; Imagawa, K.; Nishida, T.; Morikawa, Y.; Takemoto, Y.; Asai, O.; et al. Reduction of circulating soluble fms-like tyrosine kinase-1 plays a significant role in renal dysfunction-associated aggravation of atherosclerosis. Circulation 2009, 120, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Takeda, Y.; Uemura, S.; Matsumoto, T.; Seno, A.; Onoue, K.; Tsushima, H.; Morimoto, K.; Soeda, T.; Okayama, S.; et al. Suppressed soluble fms-like tyrosine kinase-1 production aggravates atherosclerosis in chronic kidney disease. Kidney Int. 2014, 85, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Bellon, T.; Corbi, A.; Lastres, P.; Cales, C.; Cebrian, M.; Vera, S.; Cheifetz, S.; Massague, J.; Letarte, M.; Bernabeu, C. Identification and expression of two forms of the human transforming growth factor-beta-binding protein endoglin with distinct cytoplasmic regions. Eur. J. Immunol. 1993, 23, 2340–2345. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Sorensen, L.K.; Brooke, B.S.; Urness, L.D.; Davis, E.C.; Taylor, D.G.; Boak, B.B.; Wendel, D.P. Defective angiogenesis in mice lacking endoglin. Science 1999, 284, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Jerkic, M.; Rodriguez-Barbero, A.; Prieto, M.; Toporsian, M.; Pericacho, M.; Rivas-Elena, J.V.; Obreo, J.; Wang, A.; Perez-Barriocanal, F.; Arevalo, M.; et al. Reduced angiogenic responses in adult endoglin heterozygous mice. Cardiovasc. Res. 2006, 69, 845–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawinkels, L.J.; Kuiper, P.; Wiercinska, E.; Verspaget, H.W.; Liu, Z.; Pardali, E.; Sier, C.F.; ten Dijke, P. Matrix metalloproteinase-14 (mt1-mmp)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010, 70, 4141–4150. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pena, A.; Eleno, N.; Duwell, A.; Arevalo, M.; Perez-Barriocanal, F.; Flores, O.; Docherty, N.; Bernabeu, C.; Letarte, M.; Lopez-Novoa, J.M. Endoglin upregulation during experimental renal interstitial fibrosis in mice. Hypertension 2002, 40, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Scharpfenecker, M.; Floot, B.; Russell, N.S.; Ten Dijke, P.; Stewart, F.A. Endoglin haploinsufficiency reduces radiation-induced fibrosis and telangiectasia formation in mouse kidneys. Radiother. Oncol J. Eur. Soc. Ther. Radiol. Oncol. 2009, 92, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Docherty, N.G.; Lopez-Novoa, J.M.; Arevalo, M.; Duwel, A.; Rodriguez-Pena, A.; Perez-Barriocanal, F.; Bernabeu, C.; Eleno, N. Endoglin regulates renal ischaemia-reperfusion injury. Nephrol. Dial. Transpl. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2006, 21, 2106–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oujo, B.; Munoz-Felix, J.M.; Arevalo, M.; Nunez-Gomez, E.; Perez-Roque, L.; Pericacho, M.; Gonzalez-Nunez, M.; Langa, C.; Martinez-Salgado, C.; Perez-Barriocanal, F.; et al. L-endoglin overexpression increases renal fibrosis after unilateral ureteral obstruction. PLoS ONE 2014, 9, e110365. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Felix, J.M.; Perez-Roque, L.; Nunez-Gomez, E.; Oujo, B.; Arevalo, M.; Ruiz-Remolina, L.; Cuesta, C.; Langa, C.; Perez-Barriocanal, F.; Bernabeu, C.; et al. Overexpression of the short endoglin isoform reduces renal fibrosis and inflammation after unilateral ureteral obstruction. Biochim. Biophys. Acta 2016, 1862, 1801–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Remolina, L.; Ollauri-Ibanez, C.; Perez-Roque, L.; Nunez-Gomez, E.; Perez-Barriocanal, F.; Lopez-Novoa, J.M.; Pericacho, M.; Rodriguez-Barbero, A. Circulating soluble endoglin modifies the inflammatory response in mice. PLoS ONE 2017, 12, e0188204. [Google Scholar] [CrossRef] [PubMed]
- Ikemoto, T.; Hojo, Y.; Kondo, H.; Takahashi, N.; Hirose, M.; Nishimura, Y.; Katsuki, T.; Shimada, K.; Kario, K. Plasma endoglin as a marker to predict cardiovascular events in patients with chronic coronary artery diseases. Heart Vessel. 2012, 27, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Blazquez-Medela, A.M.; Garcia-Ortiz, L.; Gomez-Marcos, M.A.; Recio-Rodriguez, J.I.; Sanchez-Rodriguez, A.; Lopez-Novoa, J.M.; Martinez-Salgado, C. Increased plasma soluble endoglin levels as an indicator of cardiovascular alterations in hypertensive and diabetic patients. BMC Med. 2010, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Charytan, D.M.; Helfand, A.M.; MacDonald, B.A.; Cinelli, A.; Kalluri, R.; Zeisberg, E.M. Circulating endoglin concentration is not elevated in chronic kidney disease. PLoS ONE 2011, 6, e23718. [Google Scholar] [CrossRef] [PubMed]
- Tombran-Tink, J.; Chader, G.G.; Johnson, L.V. Pedf: A pigment epithelium-derived factor with potent neuronal differentiative activity. Exp. Eye Res. 1991, 53, 411–414. [Google Scholar] [CrossRef]
- Pina, A.L.; Kubitza, M.; Brawanski, A.; Tombran-Tink, J.; Kloth, S. Expression of pigment-epithelium-derived factor during kidney development and aging. Cell Tissue Res. 2007, 329, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Abramson, L.P.; Browne, M.; Stellmach, V.; Doll, J.; Cornwell, M.; Reynolds, M.; Arensman, R.M.; Crawford, S.E. Pigment epithelium-derived factor targets endothelial and epithelial cells in wilms’ tumor. J. Pediatr. Surg. 2006, 41, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.W.; Volpert, O.V.; Gillis, P.; Crawford, S.E.; Xu, H.; Benedict, W.; Bouck, N.P. Pigment epithelium-derived factor: A potent inhibitor of angiogenesis. Science 1999, 285, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Duh, E.; Gehlbach, P.; Ando, A.; Takahashi, K.; Pearlman, J.; Mori, K.; Yang, H.S.; Zack, D.J.; Ettyreddy, D.; et al. Pigment epithelium-derived factor inhibits retinal and choroidal neovascularization. J. Cell. Physiol. 2001, 188, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, S.S.; Barnstable, C.J.; Tombran-Tink, J. Pedf induces apoptosis in human endothelial cells by activating p38 map kinase dependent cleavage of multiple caspases. Biochem. Biophys. Res. Commun. 2006, 348, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Jiang, W.G.; Grant, M.B.; Boulton, M. Pigment epithelium-derived factor inhibits angiogenesis via regulated intracellular proteolysis of vascular endothelial growth factor receptor 1. J. Biol. Chem. 2006, 281, 3604–3613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.X.; Wang, J.J.; Gao, G.; Parke, K.; Ma, J.X. Pigment epithelium-derived factor downregulates vascular endothelial growth factor (vegf) expression and inhibits vegf-vegf receptor 2 binding in diabetic retinopathy. J. Mol. Endocrinol. 2006, 37, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rychli, K.; Huber, K.; Wojta, J. Pigment epithelium-derived factor (pedf) as a therapeutic target in cardiovascular disease. Expert Opin. Ther. Targets 2009, 13, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Zhang, S.X.; Lu, K.; Chen, Y.; Mott, R.; Sato, S.; Ma, J.X. Decreased expression of pigment epithelium-derived factor is involved in the pathogenesis of diabetic nephropathy. Diabetes 2005, 54, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Zhang, S.X.; Mott, R.; Knapp, R.R.; Cao, W.; Lau, K.; Ma, J.X. Salutary effect of pigment epithelium-derived factor in diabetic nephropathy: Evidence for antifibrogenic activities. Diabetes 2006, 55, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Zhang, S.X.; Mott, R.; Chen, Y.; Knapp, R.R.; Cao, W.; Ma, J.X. Anti-inflammatory effects of pigment epithelium-derived factor in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2008, 294, F1166–F1173. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Matsui, T.; Takeuchi, M.; Yoshida, Y.; Yamakawa, R.; Fukami, K.; Yamagishi, S. Pigment epithelium-derived factor (pedf) inhibits proximal tubular cell injury in early diabetic nephropathy by suppressing advanced glycation end products (ages)-receptor (rage) axis. Pharmacol. Res. 2011, 63, 241–248. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Cheng, R.; Park, K.; Benyajati, S.; Moiseyev, G.; Sun, C.; Olson, L.E.; Yang, Y.; Eby, B.K.; Lau, K.; et al. Pigment epithelium-derived factor, a noninhibitory serine protease inhibitor, is renoprotective by inhibiting the wnt pathway. Kidney Int. 2017, 91, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, K.; Ogata, N.; Matsuoka, M.; Shima, C.; Wada, M.; Jo, N.; Matsumura, M. Relationship between pigment epithelium-derived factor (pedf) and renal function in patients with diabetic retinopathy. Mol. Vis. 2008, 14, 992–996. [Google Scholar] [PubMed]
- Chen, H.; Zheng, Z.; Li, R.; Lu, J.; Bao, Y.; Ying, X.; Zeng, R.; Jia, W. Urinary pigment epithelium-derived factor as a marker of diabetic nephropathy. Am. J. Nephrol. 2010, 32, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.; Yeung, C.Y.; Lee, P.C.; Woo, Y.C.; Fong, C.H.; Chow, W.S.; Xu, A.; Lam, K.S. Elevated circulating pigment epithelium-derived factor predicts the progression of diabetic nephropathy in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E2169–E2177. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.J.; Fu, D.; Azar, M.; Stoner, J.A.; Kaufman, D.G.; Zhang, S.; Klein, R.L.; Lopes-Virella, M.F.; Ma, J.X.; Lyons, T.J. Clinical correlates of serum pigment epithelium-derived factor in type 2 diabetes patients. J. Diabetes Its Complicat. 2014, 28, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiga, Y.; Miura, S.; Mitsutake, R.; Yamagishi, S.; Saku, K. Significance of plasma levels of pigment epithelium-derived factor as determined by multidetector row computed tomography in patients with mild chronic kidney disease and/or coronary artery disease. J. Int. Med. Res. 2011, 39, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Gitay-Goren, H.; Soker, S.; Vlodavsky, I.; Neufeld, G. The binding of vascular endothelial growth factor to its receptors is dependent on cell surface-associated heparin-like molecules. J. Biol. Chem. 1992, 267, 6093–6098. [Google Scholar] [PubMed]
- Park, J.E.; Keller, G.A.; Ferrara, N. The vascular endothelial growth factor (vegf) isoforms: Differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound vegf. Mol. Biol. Cell 1993, 4, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. Vegf165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar] [PubMed]
- Cebe Suarez, S.; Pieren, M.; Cariolato, L.; Arn, S.; Hoffmann, U.; Bogucki, A.; Manlius, C.; Wood, J.; Ballmer-Hofer, K. A vegf-a splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through vegfr-2. Cell. Mol. Life Sci. 2006, 63, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Magnussen, A.L.; Rennel, E.S.; Hua, J.; Bevan, H.S.; Long, N.B.; Lehrling, C.; Gammons, M.; Floege, J.; Harper, S.J.; Agostini, H.T.; et al. Vegf-a165b is cytoprotective and antiangiogenic in the retina. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4273–4281. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Neal, C.R.; Salmon, A.H.J.; Bates, D.O.; Harper, S.J.; Oltean, S. Vegf-a165 b protects against proteinuria in a mouse model with progressive depletion of all endogenous vegf-a splice isoforms from the kidney. J. Physiol. 2017, 595, 6281–6298. [Google Scholar] [CrossRef] [PubMed]
- Bevan, H.S.; van den Akker, N.M.; Qiu, Y.; Polman, J.A.; Foster, R.R.; Yem, J.; Nishikawa, A.; Satchell, S.C.; Harper, S.J.; Gittenberger-de Groot, A.C.; et al. The alternatively spliced anti-angiogenic family of vegf isoforms vegfxxxb in human kidney development. Nephron Physiol. 2008, 110, 57. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, V.A.; Jeruschke, S.; Eitner, F.; Becker, J.U.; Pitschke, G.; Ince, Y.; Miner, J.H.; Leuschner, I.; Engers, R.; Everding, A.S.; et al. Impaired glomerular maturation and lack of vegf165b in denys-drash syndrome. J. Am. Soc. Nephrol. 2007, 18, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Qiu, Y.; Ferguson, J.K.; Stevens, M.; Neal, C.; Russell, A.; Kaura, A.; Arkill, K.P.; Harris, K.; Symonds, C.; et al. Vascular endothelial growth factor-a165b is protective and restores endothelial glycocalyx in diabetic nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1889–1904. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Neal, C.R.; Mavrou, A.; Patel, P.; Ahad, T.; Alsop, C.; Lee, T.; Sison, K.; Qiu, Y.; Harper, S.J.; et al. Vegf165b overexpression restores normal glomerular water permeability in vegf164-overexpressing adult mice. Am. J. Physiol. Ren. Physiol. 2012, 303, F1026–F1036. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Oltean, S. Modulation of vegf-a alternative splicing as a novel treatment in chronic kidney disease. Genes 2018, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Bills, V.L.; Varet, J.; Millar, A.; Harper, S.J.; Soothill, P.W.; Bates, D.O. Failure to up-regulate vegf165b in maternal plasma is a first trimester predictive marker for pre-eclampsia. Clin. Sci. 2009, 116, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Guiducci, S.; Romano, E.; Bellando-Randone, S.; Lepri, G.; Bruni, C.; Conforti, M.L.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Increased plasma levels of the vegf165b splice variant are associated with the severity of nailfold capillary loss in systemic sclerosis. Ann. Rheum. Dis. 2013, 72, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Yoshihisa, A.; Yokokawa, T.; Misaka, T.; Sakamoto, N.; Sugimoto, K.; Yamaki, T.; Kunii, H.; Nakazato, K.; Saitoh, S.I.; et al. Association between levels of anti-angiogenic isoform of vascular endothelial growth factor a and pulmonary hypertension. Int. J. Cardiol. 2016, 222, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; Yasuda, Y.; Nakatochi, M.; Shibata, Y.; Hara, T.; Suzuki, A.; Imaizumi, T.; Suzuki, S.; Ishii, H.; Takeshita, K.; et al. Urinary and circulating levels of the anti-angiogenic isoform of vascular endothelial growth factor-a in patients with chronic kidney disease. Clin. Chim. Acta Int. J. Clin. Chem. 2017, 475, 102–108. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Anand-Apte, B.; Lee, M.; Sasaki, T.; Fukai, N.; Shapiro, R.; Que, I.; Lowik, C.; Timpl, R.; Olsen, B.R. Endostatin inhibits vegf-induced endothelial cell migration and tumor growth independently of zinc binding. EMBO J. 1999, 18, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, A.; Sugimoto, H.; Yang, C.; Lively, J.; Zeisberg, M.; Kalluri, R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc. Natl. Acad. Sci. USA 2003, 100, 4766–4771. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M.; Hirata, Y. Antiangiogenesis signals by endostatin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 1044–1053. [Google Scholar] [CrossRef]
- Folkman, J. Antiangiogenesis in cancer therapy--endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Saishin, Y.; Saishin, Y.; Silva, R.L.; Oshima, Y.; Oshima, S.; Melia, M.; Paszkiet, B.; Zerby, D.; Kadan, M.J.; et al. Intraocular expression of endostatin reduces vegf-induced retinal vascular permeability, neovascularization, and retinal detachment. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 896–898. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Maeshima, Y.; Ichinose, K.; Kitayama, H.; Takazawa, Y.; Hirokoshi, K.; Kinomura, M.; Sugiyama, H.; Makino, H. Endostatin peptide, an inhibitor of angiogenesis, prevents the progression of peritoneal sclerosis in a mouse experimental model. Kidney Int. 2007, 71, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yao, Q.; Huang, M.; Wang, B.; Zhang, J.; Wang, T.; Ming, Y.; Zhou, X.; Jia, Q.; Huan, Y.; et al. A randomized phase iii trial of neoadjuvant recombinant human endostatin, docetaxel and epirubicin as first-line therapy for patients with breast cancer (cbcrt01). Int. J. Cancer 2018, 142, 2130–2138. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Lv, W. Endostar (rh-endostatin) versus placebo in combination with vinorelbine plus cisplatin chemotherapy regimen in treatment of advanced non-small cell lung cancer: A meta-analysis. Thorac. Cancer 2018, 9, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, A.I.; Sormunen, R.; Elamaa, H.; Seppinen, L.; Miller, R.T.; Ninomiya, Y.; Janmey, P.A.; Pihlajaniemi, T. Lack of collagen xviii long isoforms affects kidney podocytes, whereas the short form is needed in the proximal tubular basement membrane. J. Biol. Chem. 2011, 286, 7755–7764. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chen, J.; Ziman, B.; Marshall, S.; Maizel, J.; Goligorsky, M.S. Endostatin and kidney fibrosis in aging: A case for antagonistic pleiotropy? Am. J. Physiol. Heart Circ. Phys. 2014, 306, H1692–H1699. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chen, J.; Zhang, Z.; Johnson, G.V.; Cooper, A.J.; Feola, J.; Bank, A.; Shein, J.; Ruotsalainen, H.J.; Pihlajaniemi, T.A.; et al. Endostatin and transglutaminase 2 are involved in fibrosis of the aging kidney. Kidney Int. 2016, 89, 1281–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimoto, T.; Mlakar, L.; Takihara, T.; Feghali-Bostwick, C. An endostatin-derived peptide orally exerts anti-fibrotic activity in a murine pulmonary fibrosis model. Int. Immunopharmacol. 2015, 28, 1102–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, A.L.; Tamarkin, L.; Paciotti, G.F.; Simpson, B.W.; Linehan, W.M.; Yang, J.C.; Fogler, W.E.; Turner, E.M.; Alexander, H.R., Jr.; Libutti, S.K. Serum endostatin levels are elevated and correlate with serum vascular endothelial growth factor levels in patients with stage iv clear cell renal cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 4628–4634. [Google Scholar]
- Dudek, A.Z.; Mahaseth, H. Circulating angiogenic cytokines in patients with advanced non-small cell lung cancer: Correlation with treatment response and survival. Cancer Investig. 2005, 23, 193–200. [Google Scholar] [CrossRef]
- Schips, L.; Dalpiaz, O.; Lipsky, K.; Langner, C.; Rehak, P.; Puerstner, P.; Pummer, K.; Zigeuner, R. Serum levels of vascular endothelial growth factor (vegf) and endostatin in renal cell carcinoma patients compared to a control group. Eur. Urol. 2007, 51, 168–173; discussion 174. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hamm, L.L.; Kleinpeter, M.A.; Husserl, F.; Khan, I.E.; Chen, C.S.; Liu, Y.; Mills, K.T.; He, C.; Rifai, N.; et al. Elevated plasma levels of endostatin are associated with chronic kidney disease. Am. J. Nephrol. 2012, 35, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Afsar, B.; Siriopol, D.; Unal, H.U.; Karaman, M.; Saglam, M.; Gezer, M.; Tas, A.; Eyileten, T.; Guler, A.K.; et al. Endostatin in chronic kidney disease: Associations with inflammation, vascular abnormalities, cardiovascular events and survival. Eur. J. Intern. Med. 2016, 33, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Ueland, T.; Aukrust, P.; Nymo, S.H.; Kjekshus, J.; McMurray, J.J.; Wikstrand, J.; Wienhues-Thelen, U.H.; Block, D.; Zaugg, C.; Gullestad, L. Predictive value of endostatin in chronic heart failure patients with poor kidney function. Cardiology 2015, 130, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Ruge, T.; Carlsson, A.C.; Larsson, T.E.; Carrero, J.J.; Larsson, A.; Lind, L.; Arnlov, J. Endostatin level is associated with kidney injury in the elderly: Findings from two community-based cohorts. Am. J. Nephrol. 2014, 40, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.C.; Ostgren, C.J.; Lanne, T.; Larsson, A.; Nystrom, F.H.; Arnlov, J. The association between endostatin and kidney disease and mortality in patients with type 2 diabetes. Diabetes Metab. 2016, 42, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Hasegawa, Y.; Yamashita, H.; Shimizu, K.; Ding, Y.; Abe, M.; Ohta, H.; Imagawa, K.; Hojo, K.; Maki, H.; et al. Vasohibin as an endothelium-derived negative feedback regulator of angiogenesis. J. Clin. Investig. 2004, 114, 898–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Kobayashi, M.; Miyashita, H.; Ohta, H.; Sonoda, H.; Sato, Y. Isolation of a small vasohibin-binding protein (svbp) and its role in vasohibin secretion. J. Cell Sci. 2010, 123, 3094–3101. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, J.; Adamopoulos, A.; Bleijerveld, O.B.; Mazouzi, A.; Stickel, E.; Celie, P.; Altelaar, M.; Knipscheer, P.; Perrakis, A.; Blomen, V.A.; et al. Vasohibins encode tubulin detyrosinating activity. Science 2017, 358, 1453–1456. [Google Scholar] [CrossRef] [PubMed]
- Aillaud, C.; Bosc, C.; Peris, L.; Bosson, A.; Heemeryck, P.; Van Dijk, J.; Le Friec, J.; Boulan, B.; Vossier, F.; Sanman, L.E.; et al. Vasohibins/svbp are tubulin carboxypeptidases (tcps) that regulate neuron differentiation. Science 2017, 358, 1448–1453. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, H.; Watanabe, T.; Hayashi, H.; Suzuki, Y.; Nakamura, T.; Ito, S.; Ono, M.; Hoshikawa, Y.; Okada, Y.; Kondo, T.; et al. Angiogenesis inhibitor vasohibin-1 enhances stress resistance of endothelial cells via induction of sod2 and sirt1. PLoS ONE 2012, 7, e46459. [Google Scholar] [CrossRef] [PubMed]
- Takeda, E.; Suzuki, Y.; Sato, Y. Age-associated downregulation of vasohibin-1 in vascular endothelial cells. Aging Cell 2016, 15, 885–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosaka, T.; Kimura, H.; Heishi, T.; Suzuki, Y.; Miyashita, H.; Ohta, H.; Sonoda, H.; Moriya, T.; Suzuki, S.; Kondo, T.; et al. Vasohibin-1 expression in endothelium of tumor blood vessels regulates angiogenesis. Am. J. Pathol. 2009, 175, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, T.; Toiyama, Y.; Tanaka, K.; Saigusa, S.; Kobayashi, M.; Inoue, Y.; Mohri, Y.; Kusunoki, M. Vasohibin-1 increases the malignant potential of colorectal cancer and is a biomarker of poor prognosis. Anticancer Res. 2014, 34, 5321–5329. [Google Scholar] [PubMed]
- Takahashi, Y.; Saga, Y.; Koyanagi, T.; Takei, Y.; Machida, S.; Taneichi, A.; Mizukami, H.; Sato, Y.; Matsubara, S.; Fujiwara, H. The angiogenesis regulator vasohibin-1 inhibits ovarian cancer growth and peritoneal dissemination and prolongs host survival. Int. J. Oncol. 2015, 47, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Mikami, S.; Oya, M.; Kosaka, T.; Mizuno, R.; Miyazaki, Y.; Sato, Y.; Okada, Y. Increased vasohibin-1 expression is associated with metastasis and poor prognosis of renal cell carcinoma patients. Lab. Investig. J. Tech. Methods Pathol. 2017, 97, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Fukumitsu, R.; Minami, M.; Yoshida, K.; Nagata, M.; Yasui, M.; Higuchi, S.; Fujikawa, R.; Ikedo, T.; Yamagata, S.; Sato, Y.; et al. Expression of vasohibin-1 in human carotid atherosclerotic plaque. J. Atheroscler. Thromb. 2015, 22, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Wakusawa, R.; Abe, T.; Sato, H.; Yoshida, M.; Kunikata, H.; Sato, Y.; Nishida, K. Expression of vasohibin, an antiangiogenic factor, in human choroidal neovascular membranes. Am. J. Ophthalmol. 2008, 146, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Nishida, K.; Kadota, Y.; Yamasaki, H.; Nasu, T.; Saitou, D.; Tanabe, K.; Sonoda, H.; Sato, Y.; Maeshima, Y.; et al. Inflammatory cytokine-induced expression of vasohibin-1 by rheumatoid synovial fibroblasts. Acta Med. Okayama 2009, 63, 349–358. [Google Scholar] [PubMed]
- Nasu, T.; Maeshima, Y.; Kinomura, M.; Hirokoshi-Kawahara, K.; Tanabe, K.; Sugiyama, H.; Sonoda, H.; Sato, Y.; Makino, H. Vasohibin-1, a negative feedback regulator of angiogenesis, ameliorates renal alterations in a mouse model of diabetic nephropathy. Diabetes 2009, 58, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Saito, D.; Maeshima, Y.; Nasu, T.; Yamasaki, H.; Tanabe, K.; Sugiyama, H.; Sonoda, H.; Sato, Y.; Makino, H. Amelioration of renal alterations in obese type 2 diabetic mice by vasohibin-1, a negative feedback regulator of angiogenesis. Am. J. Physiol. Ren. Physiol. 2011, 300, F873–F886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinamoto, N.; Maeshima, Y.; Yamasaki, H.; Nasu, T.; Saito, D.; Watatani, H.; Ujike, H.; Tanabe, K.; Masuda, K.; Arata, Y.; et al. Exacerbation of diabetic renal alterations in mice lacking vasohibin-1. PLoS ONE 2014, 9, e107934. [Google Scholar] [CrossRef] [PubMed]
- Watatani, H.; Maeshima, Y.; Hinamoto, N.; Yamasaki, H.; Ujike, H.; Tanabe, K.; Sugiyama, H.; Otsuka, F.; Sato, Y.; Makino, H. Vasohibin-1 deficiency enhances renal fibrosis and inflammation after unilateral ureteral obstruction. Physiol. Rep. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Hinamoto, N.; Maeshima, Y.; Saito, D.; Yamasaki, H.; Tanabe, K.; Nasu, T.; Watatani, H.; Ujike, H.; Kinomura, M.; Sugiyama, H.; et al. Urinary and plasma levels of vasohibin-1 can predict renal functional deterioration in patients with renal disorders. PLoS ONE 2014, 9, e96932. [Google Scholar] [CrossRef] [PubMed]
- Hinamoto, N.; Maeshima, Y.; Saito, D.; Yamasaki, H.; Tanabe, K.; Nasu, T.; Watatani, H.; Ujike, H.; Kinomura, M.; Sugiyama, H.; et al. Renal distribution of vasohibin-1 in patients with chronic kidney disease. Acta Med. Okayama 2014, 68, 219–233. [Google Scholar] [PubMed]
- Shibuya, T.; Watanabe, K.; Yamashita, H.; Shimizu, K.; Miyashita, H.; Abe, M.; Moriya, T.; Ohta, H.; Sonoda, H.; Shimosegawa, T.; et al. Isolation and characterization of vasohibin-2 as a homologue of vegf-inducible endothelium-derived angiogenesis inhibitor vasohibin. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Koyanagi, T.; Suzuki, Y.; Saga, Y.; Kanomata, N.; Moriya, T.; Suzuki, M.; Sato, Y. Vasohibin-2 expressed in human serous ovarian adenocarcinoma accelerates tumor growth by promoting angiogenesis. Mol. Cancer Res. 2012, 10, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Olmer, R.; Haase, A.; Merkert, S.; Cui, W.; Palecek, J.; Ran, C.; Kirschning, A.; Scheper, T.; Glage, S.; Miller, K.; et al. Long term expansion of undifferentiated human ips and es cells in suspension culture using a defined medium. Stem Cell Res. 2010, 5, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Norita, R.; Suzuki, Y.; Furutani, Y.; Takahashi, K.; Yoshimatsu, Y.; Podyma-Inoue, K.A.; Watabe, T.; Sato, Y. Vasohibin-2 is required for epithelial-mesenchymal transition of ovarian cancer cells by modulating transforming growth factor-beta signaling. Cancer Sci. 2017, 108, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Miyashita, H.; Suzuki, Y.; Kobayashi, M.; Watanabe, K.; Sonoda, H.; Ohta, H.; Fujiwara, T.; Shimosegawa, T.; Sato, Y. Distinctive localization and opposed roles of vasohibin-1 and vasohibin-2 in the regulation of angiogenesis. Blood 2009, 113, 4810–4818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, K.; Tanabe, K.; Ujike, H.; Hinamoto, N.; Miyake, H.; Tanimura, S.; Sugiyama, H.; Sato, Y.; Maeshima, Y.; Wada, J. Deletion of pro-angiogenic factor vasohibin-2 ameliorates glomerular alterations in a mouse diabetic nephropathy model. PLoS ONE 2018, 13, e0195779. [Google Scholar] [CrossRef] [PubMed]
- Arata, Y.; Tanabe, K.; Hinamoto, N.; Yamasaki, H.; Sugiyama, H.; Maeshima, Y.; Kanomata, N.; Sato, Y.; Wada, J. Immunohistochemistry of vasohibin-2 in human kidney disease : Implications in impaired glucose tolerance and reduced renal function. Acta Med. Okayama 2017, 71, 369–380. [Google Scholar] [PubMed]


{kind=link}
| Factors | Patients (n) 1 | Descriptions | Predictive Ability 2 | Reference |
|---|---|---|---|---|
| Soluble Flt-1 | CKD (130) | Plasma level was significantly associated with decreased estimated GFR. | - | [21] |
| CVD (586) | Plasma level was negatively correlated with estimated GFR before heparinization. | - | [22] | |
| HF (1403) | Estimated GFR decreased with increasing quartile of plasma level. | - | [23] | |
| CVD (329) | Plasma level after establishment of artery access with heparinized saline flush was positively correlated with estimated GFR. | - | [24] | |
| CKD (291) | Plasma levels were weakly negative and strongly positive correlation with estimated GFR pre- and post-heparin injection, respectively. | - | [25] | |
| Soluble endoglin | DM/HT (223) | There was no association between plasma level and renal dysfunction. | - | [39] |
| CKD (216) | Serum levels showed no significant association with CKD stage and estimated GFR. | - | [40] | |
| PEDF | DM (1071) | Plasma level increased with CKD staging, and predicted decline in GFR category, with >25% deterioration in estimated GFR over 4 years. | Yes | [57] |
| DM (246) | Serum level had no association with decline in renal function, defined as sCr ≥176.8 μmol/L or estimated GFR <60 mL/min/1.73 m2 over 3.1 years. | No | [58] | |
| CVD (289) | Plasma level was significantly higher in CKD, defined as estimated GFR <60 mL/min/1.73 m2. | - | [59] | |
| VEGF-A165b | PH (39) | There was no association between plasma level and estimated GFR. | - | [73] |
| CKD (92) | Urinary level, but not serum level, was significantly correlated with decreased GFR based on inulin clearance. | - | [74] | |
| Endostatin | CKD (201) | Plasma level was negatively correlated with estimated GFR. | - | [91] |
| Elderly (786/815) | Serum level was associated with increased risk of incident CKD, defined as estimated GFR <60 mL/min/1.73 m2, over 5 years in independent two cohorts. | Yes | [94] | |
| DM (607) | Serum level was associated with high risk of ≥20% decline in estimated GFR over 4 years. | Yes | [95] | |
| Vasohibin-1 | CKD (67) | Plasma level predicted composite renal events, defined as >30% decline in estimated GFR, initiation of renal replacement therapy or renal disorder-related death, over 3 years. | Yes | [113] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanabe, K.; Sato, Y.; Wada, J. Endogenous Antiangiogenic Factors in Chronic Kidney Disease: Potential Biomarkers of Progression. Int. J. Mol. Sci. 2018, 19, 1859. https://doi.org/10.3390/ijms19071859
Tanabe K, Sato Y, Wada J. Endogenous Antiangiogenic Factors in Chronic Kidney Disease: Potential Biomarkers of Progression. International Journal of Molecular Sciences. 2018; 19(7):1859. https://doi.org/10.3390/ijms19071859
Chicago/Turabian StyleTanabe, Katsuyuki, Yasufumi Sato, and Jun Wada. 2018. "Endogenous Antiangiogenic Factors in Chronic Kidney Disease: Potential Biomarkers of Progression" International Journal of Molecular Sciences 19, no. 7: 1859. https://doi.org/10.3390/ijms19071859
APA StyleTanabe, K., Sato, Y., & Wada, J. (2018). Endogenous Antiangiogenic Factors in Chronic Kidney Disease: Potential Biomarkers of Progression. International Journal of Molecular Sciences, 19(7), 1859. https://doi.org/10.3390/ijms19071859