The Involvement of PPARs in the Peculiar Energetic Metabolism of Tumor Cells

 ,
,  ,
,  , ,
, ,  and
and
Abstract
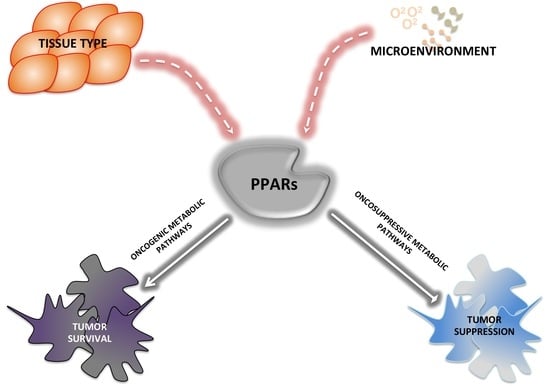
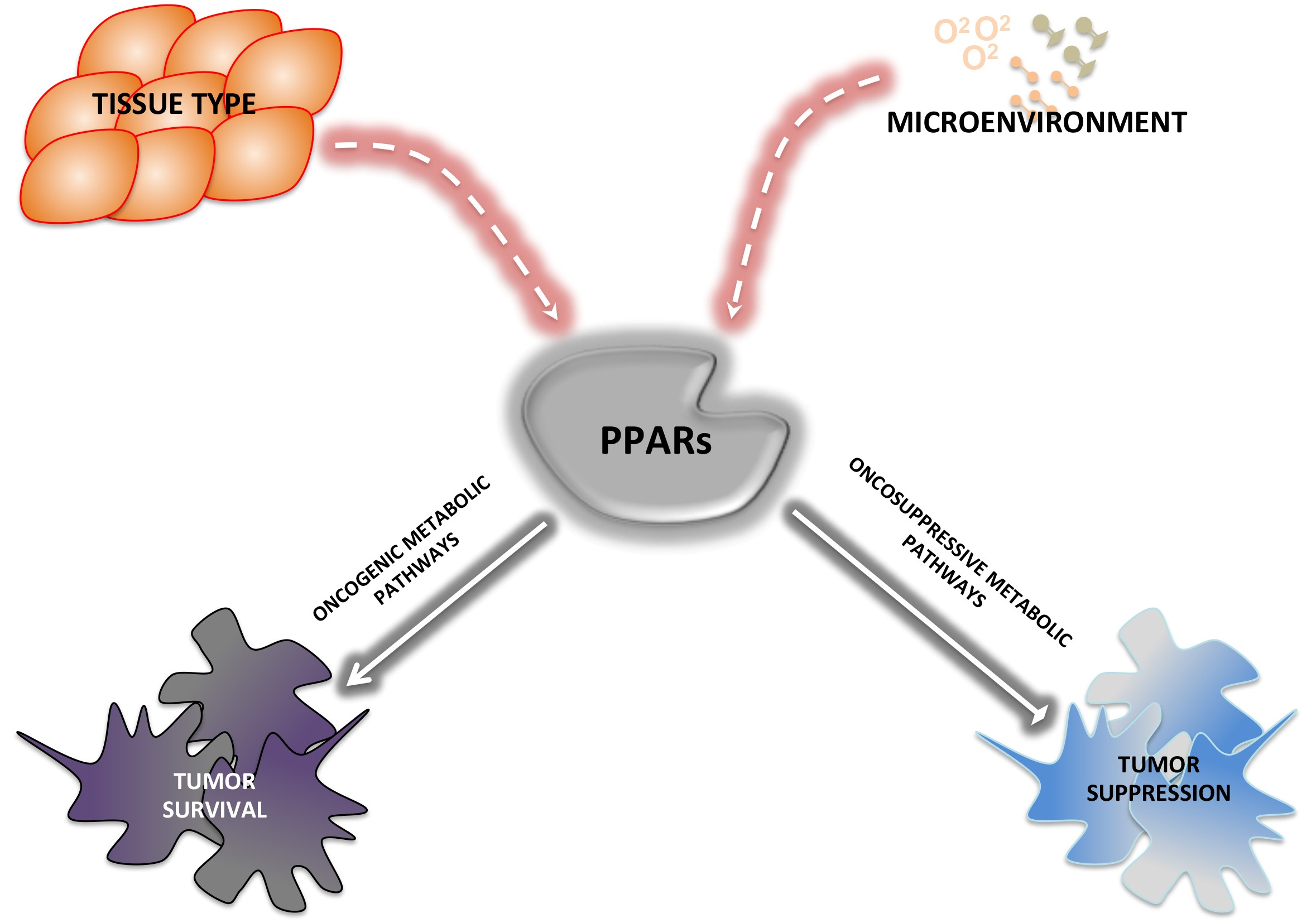
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Glucose Metabolism and OXPHOS in Cancer Cells
1.2. Lipid, Cholesterol, and Glutamine Metabolism in Cancer Cells
2. PPARs
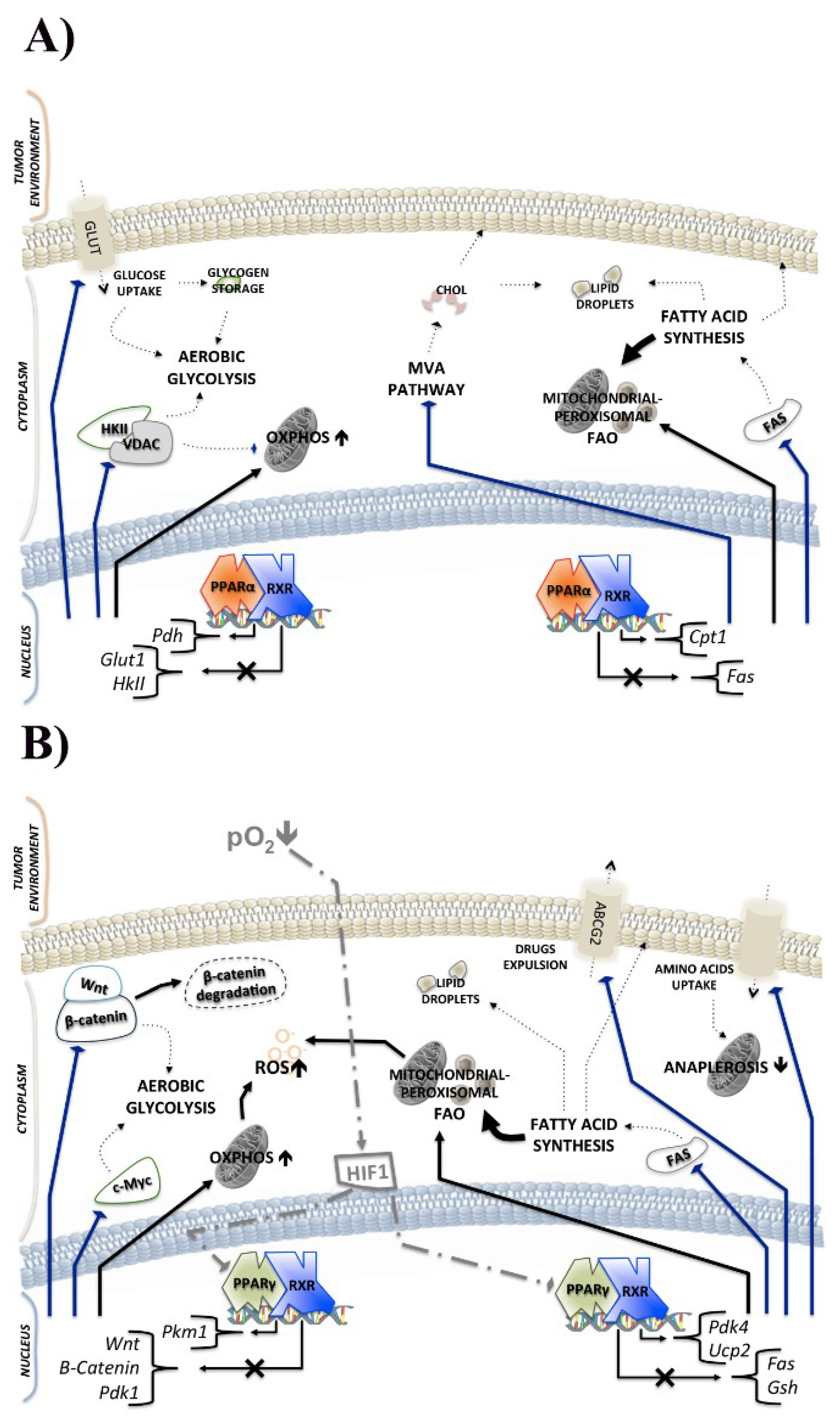
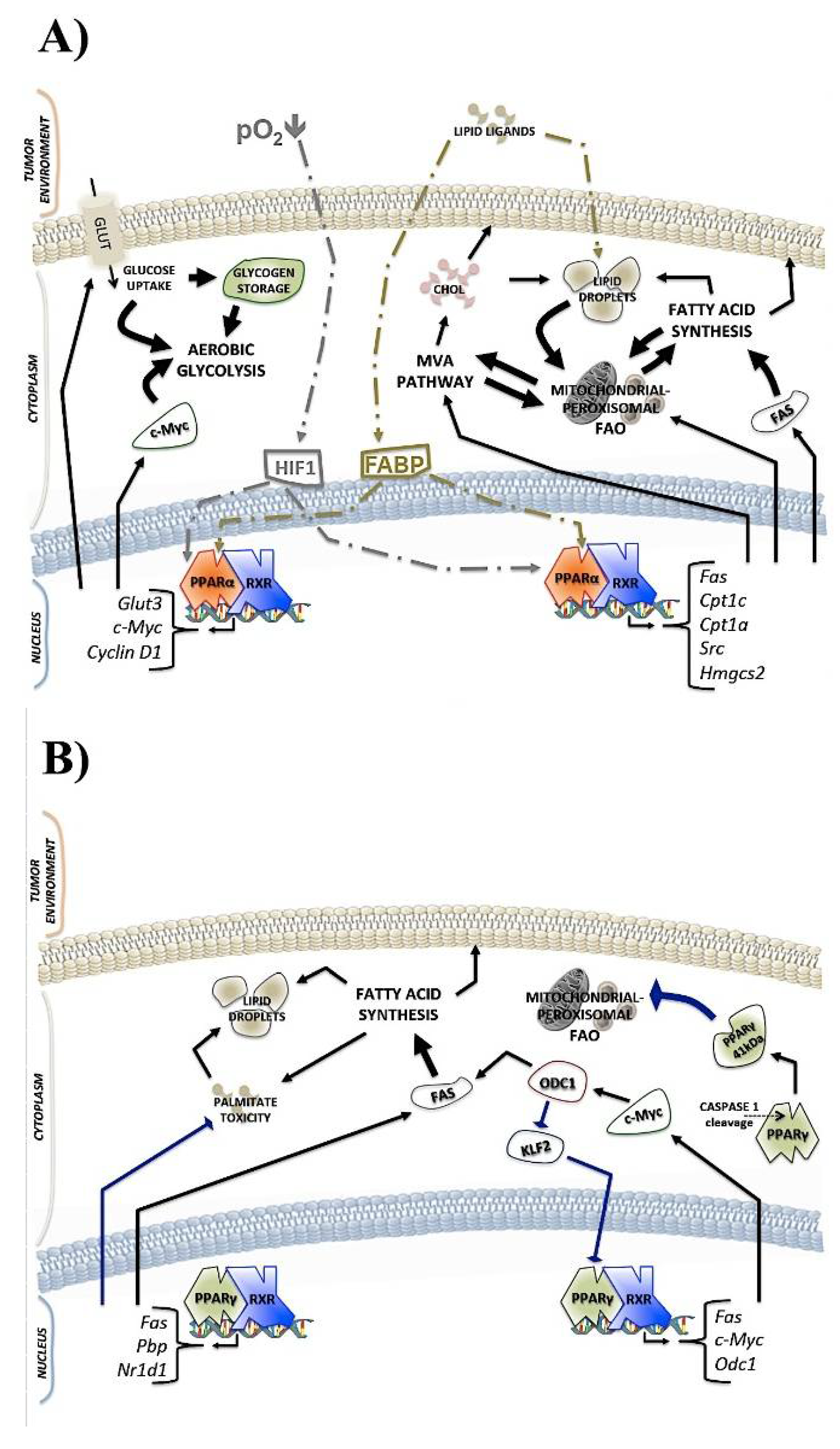
2.1. PPARα and Cancer Metabolism
2.2. PPARγ and Cancer Metabolism
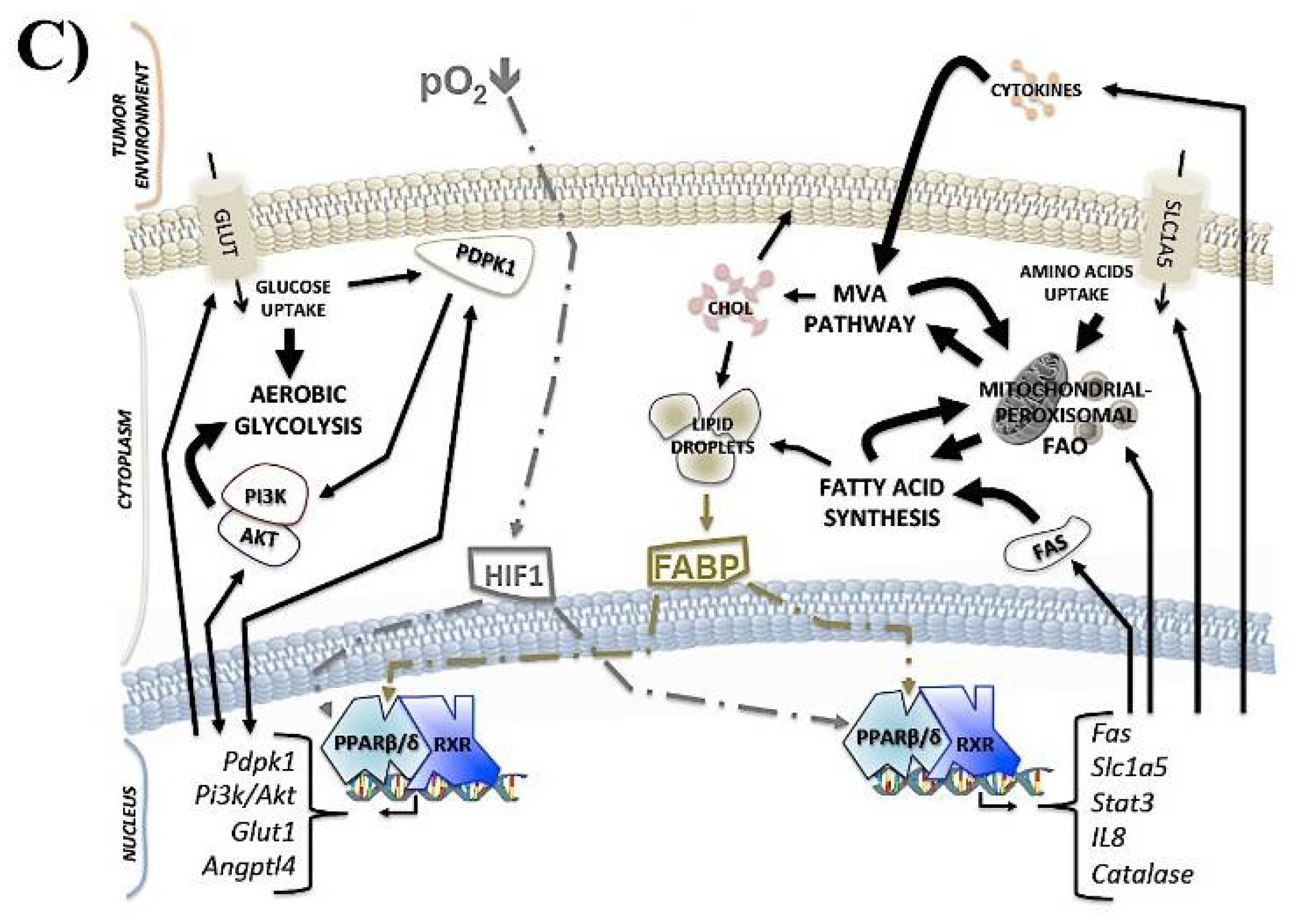
2.3. PPARβ/δ and Cancer Metabolism
3. Conclusions
Funding
Conflicts of Interest
References
- Nelson, D.; Lehninger, A.; Cox, M. Lehninger Principles of Biochemistry; W.H. Freeman: New York, NY, USA, 2008. [Google Scholar]
- Poirier, Y.; Antonenkov, V.D.; Glumoff, T.; Hiltunen, J.K. Peroxisomal β-oxidation—A metabolic pathway with multiple functions. Biochim. Biophys. Acta 2006, 1763, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, S.G.; Zechner, R. Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev. 2013, 27, 459–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cermelli, S.; Guo, Y.; Gross, S.P.; Welte, M.A. The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr Biol. 2006, 16, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Farese, R.V., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [PubMed]
- Khor, V.K.; Shen, W.J.; Kraemer, F.B. Lipid droplet metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 632–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Trédan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Persano, L.; Rampazzo, E.; Della Puppa, A.; Pistollato, F.; Basso, G. The three-layer concentric model of glioblastoma: Cancer stem cells, microenvironmental regulation, and therapeutic implications. Sci. World J. 2011, 11, 1829–1841. [Google Scholar] [CrossRef] [PubMed]
- Fidoamore, A.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; Di Giacomo, E.; Astarita, C.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. Glioblastoma Stem Cells Microenvironment: The Paracrine Roles of the Niche in Drug and Radioresistance. Stem Cells Int. 2016, 2016, 6809105. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Beauvoit, B.; Voisin, P.J.; Canioni, P.; Guérin, B.; Rigoulet, M. Energetic and morphological plasticity of C6 glioma cells grown on 3-D support; effect of transient glutamine deprivation. J. Bioenergy Biomembr. 1998, 30, 565–578. [Google Scholar] [CrossRef]
- Guppy, M.; Leedman, P.; Zu, X.; Russell, V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem. J. 2002, 364, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasdois, P.; Deveaud, C.; Voisin, P.; Bouchaud, V.; Rigoulet, M.; Beauvoit, B. Contribution of the phosphorylable complex I in the growth phase-dependent respiration of C6 glioma cells in vitro. J. Bioenergy Biomembr. 2003, 35, 439–450. [Google Scholar] [CrossRef]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, H.G. Observations on the carbohydrate metabolism of tumours. Biochem. J. 1929, 23, 536–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolková, K.; Plecitá-Hlavatá, L.; Bellance, N.; Benard, G.; Rossignol, R.; Ježek, P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol. 2011, 43, 950–968. [Google Scholar] [CrossRef] [PubMed]
- Epstein, T.; Gatenby, R.A.; Brown, J.S. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE 2017, 12, e0185085. [Google Scholar] [CrossRef] [PubMed]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Majewski, N.; Nogueira, V.; Bhaskar, P.; Coy, P.E.; Skeen, J.E.; Gottlob, K.; Chandel, N.S.; Thompson, C.B.; Robey, R.B.; Hay, N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 2004, 16, 819–830. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Makino, A.; Hullin-Matsuda, F.; Kobayashi, T.; Furihata, M.; Chung, S.; Ashida, S.; Miki, T.; Fujioka, T.; Shuin, T.; et al. Novel lipogenic enzyme ELOVL7 is involved in prostate cancer growth through saturated long-chain fatty acid metabolism. Cancer Res. 2009, 69, 8133–8140. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Karamouzis, M.V.; Papavassiliou, A.G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 2007, 6, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Gray-Bablin, J.; Rao, S.; Keyomarsi, K. Lovastatin induction of cyclin-dependent kinase inhibitors in human breast cells occurs in a cell cycle-independent fashion. Cancer Res. 1997, 57, 604–609. [Google Scholar] [PubMed]
- Newman, A.; Clutterbuck, R.D.; Powles, R.L.; Catovsky, D.; Millar, J.L. A comparison of the effect of the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors simvastatin, lovastatin and pravastatin on leukaemic and normal bone marrow progenitors. Leuk Lymphoma. 1997, 24, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Kodach, L.L.; Jacobs, R.J.; Voorneveld, P.W.; Wildenberg, M.E.; Verspaget, H.W.; van Wezel, T.; Morreau, H.; Hommes, D.W.; Peppelenbosch, M.P.; van den Brink, G.R.; et al. Statins augment the chemosensitivity of colorectal cancer cells inducing epigenetic reprogramming and reducing colorectal cancer cell ‘stemness’ via the bone morphogenetic protein pathway. Gut 2011, 60, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Hager, M.H.; Solomon, K.R.; Freeman, M.R. The role of cholesterol in prostate cancer. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramczyk, H.; Surmacki, J.; Kopeć, M.; Olejnik, A.K.; Lubecka-Pietruszewska, K.; Fabianowska-Majewska, K. The role of lipid droplets and adipocytes in cancer. Raman imaging of cell cultures: MCF10A, MCF7, and MDA-MB-231 compared to adipocytes in cancerous human breast tissue. Analyst 2015, 140, 2224–2235. [Google Scholar] [CrossRef] [PubMed]
- Tirinato, L.; Liberale, C.; Di Franco, S.; Candeloro, P.; Benfante, A.; La Rocca, R.; Potze, L.; Marotta, R.; Ruffilli, R.; Rajamanickam, V.P.; et al. Lipid droplets: A new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells 2015, 33, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedetti, E.; Galzio, R.; Laurenti, G.; D’Angelo, B.; Melchiorre, E.; Cifone, M.G.; Fanelli, F.; Muzi, P.; Coletti, G.; Alecci, M.; et al. Lipid metabolism impairment in human gliomas: Expression of peroxisomal proteins in human gliomas at different grades of malignancy. Int. J. Immunopathol. Pharmacol. 2010, 23, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zuckier, L.S.; Ghesani, N.V. Dominant uptake of fatty acid over glucose by prostate cells: A potential new diagnostic and therapeutic approach. Anticancer Res. 2010, 30, 369–374. [Google Scholar] [PubMed]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Khasawneh, J.; Schulz, M.D.; Walch, A.; Rozman, J.; Hrabe de Angelis, M.; Klingenspor, M.; Buck, A.; Schwaiger, M.; Saur, D.; Schmid, R.M.; et al. Inflammation and mitochondrial fatty acid β-oxidation link obesity to early tumor promotion. Proc. Natl. Acad. Sci. USA 2009, 106, 3354–3359. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta 2011, 1807, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schödel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010, 24, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Michalik, L.; Noy, N.; Yasmin, R.; Pacot, C.; Heim, M.; Flühmann, B.; Desvergne, B.; Wahli, W. Critical roles of PPARβ/δ in keratinocyte response to inflammation. Genes Dev. 2001, 15, 3263–3277. [Google Scholar] [CrossRef] [PubMed]
- Mandard, S.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor α target genes. Cell. Mol. Life Sci. 2004, 61, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Lehrke, M.; Lazar, M.A. The many faces of PPARγ. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Gelman, L.; Tudor, C.; Engelborghs, Y.; Wahli, W.; Desvergne, B. Fluorescence imaging reveals the nuclear behavior of peroxisome proliferator-activated receptor/retinoid X receptor heterodimers in the absence and presence of ligand. J. Biol. Chem. 2005, 280, 17880–17890. [Google Scholar] [CrossRef] [PubMed]
- Juge-Aubry, C.; Pernin, A.; Favez, T.; Burger, A.G.; Wahli, W.; Meier, C.A.; Desvergne, B. DNA binding properties of peroxisome proliferator-activated receptor subtypes on various natural peroxisome proliferator response elements. Importance of the 5’-flanking region. J. Biol. Chem. 1997, 272, 25252–25259. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M. Nuclear receptor minireview series. J. Biol. Chem. 2001, 276, 36863–36864. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Shaw, N.S.; Vinckenbosch, N.; Liu, P.; Yasmin, R.; Desvergne, B.; Wahli, W.; Noy, N. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol. Cell. Biol. 2002, 22, 5114–5127. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef] [PubMed]
- Krey, G.; Braissant, O.; L’Horset, F.; Kalkhoven, E.; Perroud, M.; Parker, M.G.; Wahli, W. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol. Endocrinol. 1997, 11, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Ibanez, M.R.; Gatica, A.E.; Yang, S.; Wei, S.; Mei, S.; Falck, J.R.; Capdevila, J.H. Peroxisomal proliferator-activated receptor-α-dependent inhibition of endothelial cell proliferation and tumorigenesis. J. Biol. Chem. 2007, 282, 17685–17695. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Moreau, F.; Chadee, K. PPARγ is an E3 ligase that induces the degradation of NFκB/p65. Nat. Commun. 2012, 3, 1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Gao, J.; Xu, H.; Xu, Y.; Zhang, Z.; Xu, Q.; Zhang, C. PPARγ E3 ubiquitin ligase regulates MUC1-C oncoprotein stability. Oncogene 2014, 33, 5619–5625. [Google Scholar] [CrossRef] [PubMed]
- Skrypnyk, N.; Chen, X.; Hu, W.; Su, Y.; Mont, S.; Yang, S.; Gangadhariah, M.; Wei, S.; Falck, J.R.; Jat, J.L.; et al. PPARα activation can help prevent and treat non-small cell lung cancer. Cancer Res. 2014, 74, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Liu, Q.; Xu, Y.; Gong, X.; Zhang, R.; Zhou, C.; Su, Z.; Jin, J.; Shi, H.; Shi, J.; et al. PPARα induces cell apoptosis by destructing Bcl2. Oncotarget 2015, 6, 44635–44642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Jin, J.; Zhang, W.; Zhang, Z.; Gao, J.; Liu, Q.; Zhou, C.; Xu, Q.; Shi, H.; Hou, Y.; et al. EGFR/MDM2 signaling promotes NF-κB activation via PPARγ degradation. Carcinogenesis 2016, 37, 215–222. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Jin, J.; Liu, Q.; Xu, Q.; Shi, J.; Hou, Y. PPARα Promotes Cancer Cell Glut1 Transcription Repression. J. Cell. Biochem. 2017, 118, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Kaipainen, A.; Kieran, M.W.; Huang, S.; Butterfield, C.; Bielenberg, D.; Mostoslavsky, G.; Mulligan, R.; Folkman, J.; Panigrahy, D. PPARα deficiency in inflammatory cells suppresses tumor growth. PLoS ONE 2007, 2, e260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaner, D.E.; Lee, E.; Shi, Y.; Wen, F.; Li, Y.; Tung, S.; McCaw, L.; Wong, K.; Gary-Gouy, H.; Dalloul, A.; et al. PPAR-alpha is a therapeutic target for chronic lymphocytic leukemia. Leukemia 2013, 27, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Messmer, D.; Lorrain, K.; Stebbins, K.; Bravo, Y.; Stock, N.; Cabrera, G.; Correa, L.; Chen, A.; Jacintho, J.; Chiorazzi, N.; et al. A Selective Novel Peroxisome Proliferator-Activated Receptor (PPAR)-α Antagonist Induces Apoptosis and Inhibits Proliferation of CLL Cells In Vitro and In Vivo. Mol. Med. 2015, 21, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Desvergne, B.; Wahli, W. Peroxisome-proliferator-activated receptors and cancers: Complex stories. Nat. Rev. Cancer 2004, 4, 61–70. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Yuan, S.; Shi, J.; Hou, Y. PPARδ signaling regulates colorectal cancer. Curr. Pharm. Des. 2015, 21, 2956–2959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, Y.; Xu, Q.; Shi, H.; Shi, J.; Hou, Y. PPARδ promotes tumor progression via activation of Glut1 and SLC1-A5 transcription. Carcinogenesis 2017, 38, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Frattini, V.; Pagnotta, S.M.; Tala; Fan, J.J.; Russo, M.V.; Lee, S.B.; Garofano, L.; Zhang, J.; Shi, P.; Lewis, G.; et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature 2018, 553, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Plonka, P.M.; Urbanska, K.; Reiss, K. Peroxisome proliferator-activated receptor α activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin. Cancer Res. 2006, 12, 3028–3036. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.C.; Zang, C.B.; Liu, H.Y.; Possinger, K.; Fan, S.G.; Elstner, E. A novel PPAR alpha/gamma dual agonist inhibits cell growth and induces apoptosis in human glioblastoma T98G cells. Acta Pharmacol. Sin. 2004, 25, 1312–1319. [Google Scholar] [PubMed]
- Suchanek, K.M.; May, F.J.; Robinson, J.A.; Lee, W.J.; Holman, N.A.; Monteith, G.R.; Roberts-Thomson, S.J. Peroxisome proliferator-activated receptor α in the human breast cancer cell lines MCF-7 and MDA-MB-231. Mol. Carcinog. 2002, 34, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Abu Aboud, O.; Wettersten, H.I.; Weiss, R.H. Inhibition of PPARα induces cell cycle arrest and apoptosis, and synergizes with glycolysis inhibition in kidney cancer cells. PLoS ONE 2013, 8, e71115. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Cattley, R.C.; Gonzalez, F.J. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis 1997, 18, 2029–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidoamore, A.; Cristiano, L.; Laezza, C.; Galzio, R.; Benedetti, E.; Cinque, B.; Antonosante, A.; d’Angelo, M.; Castelli, V.; Cifone, M.G.; et al. Energy metabolism in glioblastoma stem cells: PPARα a metabolic adaptor to intratumoral microenvironment. Oncotarget 2017, 8, 108430–108450. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Persano, L.; Rampazzo, E.; Basso, G.; Viola, G. Glioblastoma cancer stem cells: Role of the microenvironment and therapeutic targeting. Biochem. Pharmacol. 2013, 85, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, G.; Benedetti, E.; D’Angelo, B.; Cristiano, L.; Cinque, B.; Raysi, S.; Alecci, M.; Cerù, M.P.; Cifone, M.G.; Galzio, R.; et al. Hypoxia induces peroxisome proliferator-activated receptor α (PPARα) and lipid metabolism peroxisomal enzymes in human glioblastoma cells. J. Cell. Biochem. 2011, 112, 3891–3901. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Bellot, G.; Gounon, P.; Lacas-Gervais, S.; Pouysségur, J.; Mazure, N.M. Glycogen Synthesis is induced in Hypoxia by the Hypoxia-Inducible Factor and Promotes Cancer Cell Survival. Front. Oncol. 2012, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morihiro, Y.; Yasumoto, Y.; Vaidyan, L.K.; Sadahiro, H.; Uchida, T.; Inamura, A.; Sharifi, K.; Ideguchi, M.; Nomura, S.; Tokuda, N.; Kashiwabara, S.; et al. Fatty acid binding protein 7 as a marker of glioma stem cells. Pathol. Int. 2013, 63, 546–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ta, M.T.; Kapterian, T.S.; Fei, W.; Du, X.; Brown, A.J.; Dawes, I.W.; Yang, H. Accumulation of squalene is associated with the clustering of lipid droplets. FEBS J. 2012, 279, 4231–4244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perroud, B.; Ishimaru, T.; Borowsky, A.D.; Weiss, R.H. Grade-dependent proteomics characterization of kidney cancer. Mol. Cell. Proteom. 2009, 8, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Morimura, K.; Yang, Q.; Tanabe, T.; Takagi, M.; Gonzalez, F.J. Peroxisome proliferator-activated receptor α regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Mol. Cell. Biol. 2007, 27, 4238–4247. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lisanti, M.P.; Liao, D.J. Reviewing once more the c-myc and Ras collaboration: Converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle 2011, 10, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Moralli, S.; Tarrado-Castellarnau, M.; Miranda, A.; Cascante, M. Targeting cell cycle regulation in cancer therapy. Pharmacol. Ther. 2013, 138, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Kamarajugadda, S.; Becker, J.R.; Hanse, E.A.; Mashek, D.G.; Mashek, M.T.; Hendrickson, A.M.; Mullany, L.K.; Albrecht, J.H. Cyclin D1 represses peroxisome proliferator-activated receptor alpha and inhibits fatty acid oxidation. Oncotarget 2016, 7, 47674–47686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Rao, M.; Bouras, T.; Wang, C.; Wu, K.; Zhang, X.; Li, Z.; Yao, T.P.; Pestell, R.G. Cyclin D1 inhibits peroxisome proliferator-activated receptor γ-mediated adipogenesis through histone deacetylase recruitment. J. Biol. Chem. 2005, 280, 16934–16941. [Google Scholar] [CrossRef] [PubMed]
- Hanse, E.A.; Mashek, D.G.; Becker, J.R.; Solmonson, A.D.; Mullany, L.K.; Mashek, M.T.; Towle, H.C.; Chau, A.T.; Albrecht, J.H. Cyclin D1 inhibits hepatic lipogenesis via repression of carbohydrate response element binding protein and hepatocyte nuclear factor 4α. Cell Cycle 2012, 11, 2681–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Wang, C.; Rao, M.; Wu, X.; Bouras, T.; Zhang, X.; Li, Z.; Jiao, X.; Yang, J.; Li, A.; et al. Cyclin D1 represses p300 transactivation through a cyclin-dependent kinase-independent mechanism. J. Biol. Chem. 2005, 280, 29728–29742. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.E. Cyclin D1 goes metabolic: Dual functions of cyclin D1 in regulating lipogenesis. Cell Cycle 2012, 11, 3533–3534. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, M.V.; Pan, Z.; Zhu, Y.; Tordjman, K.; Schneider, J.G.; Coleman, T.; Turk, J.; Semenkovich, C.F. “New” hepatic fat activates PPARα to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 2005, 1, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarthy, M.V.; Zhu, Y.; López, M.; Yin, L.; Wozniak, D.F.; Coleman, T.; Hu, Z.; Wolfgang, M.; Vidal-Puig, A.; Lane, M.D.; et al. Brain fatty acid synthase activates PPARα to maintain energy homeostasis. J. Clin. Investig. 2007, 117, 2539–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiguchi, A.; Asano, T.; Asano, T.; Ito, K.; Sumitomo, M.; Hayakawa, M. Fatty acid synthase over expression is an indicator of tumor aggressiveness and poor prognosis in renal cell carcinoma. J. Urol. 2008, 180, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Abu Aboud, O.; Donohoe, D.; Bultman, S.; Fitch, M.; Riiff, T.; Hellerstein, M.; Weiss, R.H. PPARα inhibition modulates multiple reprogrammed metabolic pathways in kidney cancer and attenuates tumor growth. Am. J. Physiol. Cell Physiol. 2015, 308, C890–C898. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, T.; Shimomura, Y.; Bajotto, G.; Kotake, K.; Arikawa, T.; Ito, N.; Yasuda, A.; Nagata, H.; Nonami, T.; Masuko, K. Peroxisome proliferator-activated receptor α (PPARα) mRNA expression in human hepatocellular carcinoma tissue and non-cancerous liver tissue. World J. Surg. Oncol. 2011, 9, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, Y.; Huang, Y.; Zeng, H.; Hu, B.; Guan, L.; Zhang, H.; Yu, A.M.; Johnson, C.H.; Gonzalez, F.J.; et al. PPARα regulates tumor cell proliferation and senescence via a novel target gene carnitine palmitoyltransferase 1C. Carcinogenesis 2017, 38, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, M.J.; Kurama, T.; Dai, Y.; Suwa, A.; Asaumi, M.; Matsumoto, S.; Cha, S.H.; Shimokawa, T.; Lane, M.D. The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 7282–7287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, P.T.; Mak, T.W. Molecular pathways: Tumor cells Co-opt the brain-specific metabolism gene CPT1C to promote survival. Clin. Cancer Res. 2012, 18, 5850–5855. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Chan, C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci. Rep. 2016, 6, 18669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascaró, C.; Acosta, E.; Ortiz, J.A.; Marrero, P.F.; Hegardt, F.G.; Haro, D. Control of human muscle-type carnitine palmitoyltransferase I gene transcription by peroxisome proliferator-activated receptor. J. Biol. Chem. 1998, 273, 8560–8563. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.M.; Djouadi, F.; Kelly, D.P. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor α. J. Biol. Chem. 1998, 273, 23786–23792. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Lisanti, M.P.; Sotgia, F. Ketone bodies and two-compartment tumor metabolism: Stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 2012, 11, 3956–3963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraon, P.; Cretu, D.; Musrap, N.; Karagiannis, G.S.; Batruch, I.; Drabovich, A.P.; van der Kwast, T.; Mizokami, A.; Morrissey, C.; Jarvi, K.; et al. Quantitative proteomics reveals that enzymes of the ketogenic pathway are associated with prostate cancer progression. Mol. Cell. Proteom. 2013, 12, 1589–1601. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Chou, C.T.; Chang, C.C.; Li, Y.J.; Chen, S.T.; Lin, I.C.; Kok, S.H.; Cheng, S.J.; Lee, J.J.; Wu, T.S.; et al. HMGCS2 enhances invasion and metastasis via direct interaction with PPARα to activate Src signaling in colorectal cancer and oral cancer. Oncotarget 2017, 8, 22460–22476. [Google Scholar] [PubMed]
- Spaner, D.E. Oral high-dose glucocorticoids and ofatumumab in fludarabine-resistant chronic lymphocytic leukemia. Leukemia 2012, 26, 1144–1145. [Google Scholar] [CrossRef] [PubMed]
- Tung, S.; Shi, Y.; Wong, K.; Zhu, F.; Gorczynski, R.; Laister, R.C.; Minden, M.; Blechert, A.K.; Genzel, Y.; Reichl, U.; et al. PPARα and fatty acid oxidation mediate glucocorticoid resistance in chronic lymphocytic leukemia. Blood 2013, 122, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Downward, J. Flicking the Warburg switch-tyrosine phosphorylation of pyruvate dehydrogenase kinase regulates mitochondrial activity in cancer cells. Mol. Cell 2011, 44, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Pond, C.M. Adipose tissue and the immune system. Prostaglandins Leukot. Essent. Fatty Acids 2005, 73, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Tzekov, A.; Park, C.; Choi, K.; Bickel, P.E. OP9 mouse stromal cells rapidly differentiate into adipocytes: Characterization of a useful new model of adipogenesis. J. Lipid Res. 2006, 47, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Goswami, S.; Sharma-Walia, N. Implications of a peroxisome proliferator-activated receptor alpha (PPARα) ligand clofibrate in breast cancer. Oncotarget 2016, 7, 15577–15599. [Google Scholar] [CrossRef] [PubMed]
- Lupu, R.; Menendez, J.A. Targeting fatty acid synthase in breast and endometrial cancer: An alternative to selective estrogen receptor modulators? Endocrinology 2006, 147, 4056–4066. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor α target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef] [PubMed]
- Lacasa, D.; Le Liepvre, X.; Ferre, P.; Dugail, I. Progesterone stimulates adipocyte determination and differentiation 1/sterol regulatory element-binding protein 1c gene expression. potential mechanism for the lipogenic effect of progesterone in adipose tissue. J. Biol. Chem. 2001, 276, 11512–11516. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Pierzchalska, M.; Reiss, K. Peroxisome proliferator activated receptor α ligands as anticancer drugs targeting mitochondrial metabolism. Curr. Pharm. Biotechnol. 2013, 14, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Jan, C.I.; Tsai, M.H.; Chiu, C.F.; Huang, Y.P.; Liu, C.J.; Chang, N.W. Fenofibrate Suppresses Oral Tumorigenesis via Reprogramming Metabolic Processes: Potential Drug Repurposing for Oral Cancer. Int. J. Biol. Sci. 2016, 12, 786–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.P.; Chang, N.W. Proteomic analysis of oral cancer reveals new potential therapeutic targets involved in the Warburg effect. Clin. Exp. Pharmacol. Physiol. 2017, 44, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Patel, B.B.; Blagoi, E.L.; Patterson, M.D.; Seeholzer, S.H.; Zhang, T.; Damle, S.; Gao, Z.; Boman, B.; Yeung, A.T. Analyzing alkaline proteins in human colon crypt proteome. J. Proteome Res. 2004, 3, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Kotta-Loizou, I.; Giaginis, C.; Theocharis, S. The role of peroxisome proliferator-activated receptor-γ in breast cancer. Anticancer Agents Med. Chem. 2012, 12, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Wei, Y.; Zhang, Y.; Zhang, M.; Lu, Y.; Shen, P. Peroxisome proliferator-activated receptor-γ activation inhibits hepatocellular carcinoma cell invasion by upregulating plasminogen activator inhibitor-1. Cancer Sci. 2013, 104, 672–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, N.; Kollipara, R.K.; Singh, D.K.; Sudderth, J.; Hu, Z.; Nguyen, H.; Wang, S.; Humphries, C.G.; Carstens, R.; Huffman, K.E.; et al. Inhibition of cancer cell proliferation by PPARγ is mediated by a metabolic switch that increases reactive oxygen species levels. Cell Metab. 2014, 20, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Girnun, G.D.; Smith, W.M.; Drori, S.; Sarraf, P.; Mueller, E.; Eng, C.; Nambiar, P.; Rosenberg, D.W.; Bronson, R.T.; Edelmann, W.; et al. APC-dependent suppression of colon carcinogenesis by PPARγ. Proc. Natl. Acad. Sci. USA 2002, 99, 13771–13776. [Google Scholar] [CrossRef] [PubMed]
- Reka, A.K.; Kurapati, H.; Narala, V.R.; Bommer, G.; Chen, J.; Standiford, T.J.; Keshamouni, V.G. Peroxisome proliferator-activated receptor-γ activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol. Cancer Ther. 2010, 9, 3221–3232. [Google Scholar] [CrossRef] [PubMed]
- Saez, E.; Tontonoz, P.; Nelson, M.C.; Alvarez, J.G.; Ming, U.T.; Baird, S.M.; Thomazy, V.A.; Evans, R.M. Activators of the nuclear receptor PPARγ enhance colon polyp formation. Nat. Med. 1998, 4, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Jansson, E.A.; Are, A.; Greicius, G.; Kuo, I.C.; Kelly, D.; Arulampalam, V.; Pettersson, S. The Wnt/β-catenin signaling pathway targets PPARγ activity in colon cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Fan, K.H.; Lamprecht, S.A.; Edelmann, W.; Kopelovich, L.; Kucherlapati, R.; Lipkin, M. Peroxisome proliferator-activated receptor γ agonist troglitazone induces colon tumors in normal C57BL/6J mice and enhances colonic carcinogenesis in Apc1638 N/+ Mlh1+/− double mutant mice. Int. J. Cancer 2005, 116, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Demetri, G.D.; Mueller, E.; Sarraf, P.; Spiegelman, B.M.; Winer, E.P. Use of the peroxisome proliferator-activated receptor (PPAR) γ ligand troglitazone as treatment for refractory breast cancer: A phase II study. Breast Cancer Res. Treat. 2003, 79, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Saez, E.; Rosenfeld, J.; Livolsi, A.; Olson, P.; Lombardo, E.; Nelson, M.; Banayo, E.; Cardiff, R.D.; Izpisua-Belmonte, J.C.; Evans, R.M. PPARγ signaling exacerbates mammary gland tumor development. Genes Dev. 2004, 18, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Egerod, F.L.; Nielsen, H.S.; Iversen, L.; Thorup, I.; Storgaard, T.; Oleksiewicz, M.B. Biomarkers for early effects of carcinogenic dual-acting PPAR agonists in rat urinary bladder urothelium in vivo. Biomarkers 2005, 10, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Claes, V.; Duthoit, G.; Hébert, J.L. Circadian rhythms, Wnt/beta-catenin pathway and PPAR alpha/gamma profiles in diseases with primary or secondary cardiac dysfunction. Front. Physiol. 2014, 5, 429. [Google Scholar] [CrossRef] [PubMed]
- Abbot, E.L.; McCormack, J.G.; Reynet, C.; Hassall, D.G.; Buchan, K.W.; Yeaman, S.J. Diverging regulation of pyruvate dehydrogenase kinase isoform gene expression in cultured human muscle cells. FEBS J. 2005, 272, 3004–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, Y.; Xie, Y.; Lee, W.; Bookout, A.L.; Girard, L.; Raso, G.; Behrens, C.; Wistuba, I.I.; Gadzar, A.F.; Minna, J.D.; et al. Research resource: Diagnostic and therapeutic potential of nuclear receptor expression in lung cancer. Mol. Endocrinol. 2012, 26, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Heigener, D.F.; Mok, T.; Soria, J.C.; Rabe, K.F. Management of non-small-cell lung cancer: Recent developments. Lancet 2013, 382, 709–719. [Google Scholar] [CrossRef]
- Vadlapatla, R.K.; Vadlapudi, A.D.; Pal, D.; Mitra, A.K. Mechanisms of drug resistance in cancer chemotherapy: Coordinated role and regulation of efflux transporters and metabolizing enzymes. Curr. Pharm. Des. 2013, 19, 7126–7140. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, G.; Yang, Z.; Wang, L.; Zhang, L.; Wang, T.; Zhang, Y.; Zhang, S.; Han, Y.; Jia, L. Uncoupling protein 2 downregulation by hypoxia through repression of peroxisome proliferator-activated receptor γ promotes chemoresistance of non-small cell lung cancer. Oncotarget 2017, 8, 8083–8094. [Google Scholar] [CrossRef] [PubMed]
- Bouillaud, F. UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim. Biophys. Acta 2009, 1787, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, E.J.; Giaccia, A. Dual roles of NRF2 in tumor prevention and progression: Possible implications in cancer treatment. Free Radic. Biol. Med. 2015, 79, 292–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornic, M.; Delva, L.; Guidez, F.; Balitrand, N.; Degos, L.; Chomienne, C. Induction of retinoic acid-binding protein in normal and malignant human myeloid cells by retinoic acid in acute promyelocytic leukemia patients. Cancer Res. 1992, 52, 3329–3334. [Google Scholar] [PubMed]
- Hu, Z.B.; Ma, W.; Uphoff, C.C.; Lanotte, M.; Drexler, H.G. Modulation of gene expression in the acute promyelocytic leukemia cell line NB4. Leukemia 1993, 7, 1817–1823. [Google Scholar] [PubMed]
- Yasugi, E.; Horiuchi, A.; Uemura, I.; Okuma, E.; Nakatsu, M.; Saeki, K.; Kamisaka, Y.; Kagechika, H.; Yasuda, K.; You, A. Peroxisome proliferator-activated receptor γ ligands stimulate myeloid differentiation and lipogenensis in human leukemia NB4 cells. Dev. Growth Differ. 2006, 48, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Taniguchi, K.; Kumazaki, M.; Yamada, N.; Ito, Y.; Otsuki, Y.; Uno, B.; Hayakawa, F.; Minami, Y.; Naoe, T.; et al. Anti-cancer fatty-acid derivative induces autophagic cell death through modulation of PKM isoform expression profile mediated by bcr-abl in chronic myeloid leukemia. Cancer Lett. 2015, 360, 28–38. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chu, I.S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, F.Y.; Gesell, M.S.; Alousi, M.; Luk, G.D. Analysis of ODC and c-myc gene expression in hepatocellular carcinoma by in situ hybridization and immunohistochemistry. J. Histochem. Cytochem. 1993, 41, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.S.; Feinberg, M.W.; Watanabe, M.; Gray, S.; Haspel, R.L.; Denkinger, D.J.; Kawahara, R.; Hauner, H.; Jain, M.K. The Krüppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-γ expression and adipogenesis. J. Biol. Chem. 2003, 278, 2581–2584. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Oh, S.T.; Won, M.A.; Choi, K.M.; Ko, M.J.; Seo, D.; Jeon, T.W.; Baik, I.H.; Ye, S.K.; Park, K.U.; et al. Targeting ODC1 inhibits tumor growth through reduction of lipid metabolism in human hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2016, 478, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Jain, R.; Carkner, R.D.; Eifert, C.; Brosnan, M.J.; Conklin, D.S. An RNA interference screen identifies metabolic regulators NR1D1 and PBP as novel survival factors for breast cancer cells with the ERBB2 signature. Cancer Res. 2010, 70, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qi, C.; Jain, S.; Rao, M.S.; Reddy, J.K. Isolation and characterization of PBP, a protein that interacts with peroxisome proliferator-activated receptor. J. Biol. Chem. 1997, 272, 25500–25506. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Borie, N.; Ginestier, C.; Groulet, A.; Charafe-Jauffret, E.; Adélaïde, J.; Geneix, J.; Bachelart, L.; Finetti, P.; Koki, A.; et al. Identification and validation of an ERBB2 gene expression signature in breast cancers. Oncogene 2004, 23, 2564–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Srinivasaiah, R.; Carkner, R.D.; Brosnan, M.J.; Conklin, D.S. Peroxisome proliferator-activated receptor-γ protects ERBB2-positive breast cancer cells from palmitate toxicity. Breast Cancer Res. 2009, 11, R16. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, Y.; Wong, J.; Conklin, D.S. PPARγ maintains ERBB2-positive breast cancer stem cells. Oncogene 2013, 32, 5512–5521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- March, H.N.; Rust, A.G.; Wright, N.A.; ten Hoeve, J.; de Ridder, J.; Eldridge, M.; van der Weyden, L.; Berns, A.; Gadiot, J.; Uren, A.; et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat. Genet. 2011, 43, 1202–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, I.; Mui, E.; Galbraith, L.; Patel, R.; Tan, E.H.; Salji, M.; Rust, A.G.; Repiscak, P.; Hedley, A.; Markert, E.; et al. Sleeping Beauty screen reveals Pparg activation in metastatic prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 8290–8295. [Google Scholar] [CrossRef] [PubMed]
- Kompare, M.; Rizzo, W.B. Mitochondrial fatty-acid oxidation disorders. Semin. Pediatr. Neurol. 2008, 15, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Shi, Q.; Zhang, W.; Shu, Y.; Yang, N.; Chen, B.; Wang, Q.; Zhao, X.; Chen, J.; Cheng, N.; et al. Caspase-1 cleaves PPARγ for potentiating the pro-tumor action of TAMs. Nat. Commun. 2017, 8, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J.M.; Lee, S.S.; Li, W.; Ward, J.M.; Gavrilova, O.; Everett, C.; Reitman, M.L.; Hudson, L.D.; Gonzalez, F.J. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β (δ). Mol. Cell. Biol. 2000, 20, 5119–5128. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.C.; Tan, M.J.; Philippe, V.; Tan, S.H.; Tan, C.K.; Ku, C.W.; Goh, Y.Y.; Wahli, W.; Michalik, L.; Tan, N.S. Regulation of epithelial-mesenchymal IL-1 signaling by PPARβ/δ is essential for skin homeostasis and wound healing. J. Cell Biol. 2009, 184, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 2012, 12, 181–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephen, R.L.; Gustafsson, M.C.; Jarvis, M.; Tatoud, R.; Marshall, B.R.; Knight, D.; Ehrenborg, E.; Harris, A.L.; Wolf, C.R.; Palmer, C.N. Activation of peroxisome proliferator-activated receptor δ stimulates the proliferation of human breast and prostate cancer cell lines. Cancer Res. 2004, 64, 3162–3170. [Google Scholar] [CrossRef] [PubMed]
- Aung, C.S.; Faddy, H.M.; Lister, E.J.; Monteith, G.R.; Roberts-Thomson, S.J. Isoform specific changes in PPARα and β in colon and breast cancer with differentiation. Biochem. Biophys. Res. Commun. 2006, 340, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Buttitta, L.A.; Edgar, B.A. Mechanisms controlling cell cycle exit upon terminal differentiation. Curr. Opin. Cell Biol. 2007, 19, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.L.; Morales, J.L.; Zhu, B.; Kang, B.H.; Gonzalez, F.J.; Peters, J.M. Activation of peroxisome proliferator-activated receptor-β/δ (PPAR-β/δ) inhibits human breast cancer cell line tumorigenicity. Mol. Cancer Ther. 2014, 13, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Genini, D.; Garcia-Escudero, R.; Carbone, G.M.; Catapano, C.V. Transcriptional and Non-Transcriptional Functions of PPARβ/δ in Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e46009. [Google Scholar] [CrossRef] [PubMed]
- Pedchenko, T.V.; Gonzalez, A.L.; Wang, D.; DuBois, R.N.; Massion, P.P. Peroxisome proliferator-activated receptor β/δ expression and activation in lung cancer. Am. J. Respir. Cell Mol. Biol. 2008, 39, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Di-Poï, N.; Michalik, L.; Tan, N.S.; Desvergne, B.; Wahli, W. The anti-apoptotic role of PPARβ contributes to efficient skin wound healing. J. Steroid Biochem. Mol. Biol. 2003, 85, 257–265. [Google Scholar] [CrossRef]
- Pollock, C.B.; Yin, Y.; Yuan, H.; Zeng, X.; King, S.; Li, X.; Kopelovich, L.; Albanese, C.; Glazer, R.I. PPARδ activation acts cooperatively with 3-phosphoinositide-dependent protein kinase-1 to enhance mammary tumorigenesis. PLoS ONE 2011, 6, e16215. [Google Scholar] [CrossRef] [PubMed]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. AMPK and PPARdelta agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, G.; Shi, Y.; Sun, L.; Gorczynski, R.; Li, Y.J.; Xu, Z.; Spaner, D.E. PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis 2016, 5, e232. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.C.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.J.; Sun, L.; Shi, Y.; Wang, G.; Wang, X.; Dunn, S.E.; Iorio, C.; Screaton, R.A.; Spaner, D.E. PPAR-delta promotes survival of chronic lymphocytic leukemia cells in energetically unfavorable conditions. Leukemia 2017, 31, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Shi, Y.; Wang, G.; Wang, X.; Zeng, S.; Dunn, S.E.; Fairn, G.D.; Li, Y.J.; Spaner, D.E. PPAR-delta modulates membrane cholesterol and cytokine signaling in malignant B cells. Leukemia 2018, 32, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.; Koo, J.E.; Yeon, S.H.; Kwak, M.K.; Hwang, D.H.; Lee, J.Y. PPARδ deficiency disrupts hypoxia-mediated tumorigenic potential of colon cancer cells. Mol. Carcinog. 2014, 53, 926–937. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Chan, T.A.; Vogelstein, B.; Kinzler, K.W. PPARδ is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999, 99, 335–345. [Google Scholar] [CrossRef]
- Zuo, X.; Peng, Z.; Moussalli, M.J.; Morris, J.S.; Broaddus, R.R.; Fischer, S.M.; Shureiqi, I. Targeted genetic disruption of peroxisome proliferator-activated receptor-δ and colonic tumorigenesis. J. Natl. Cancer Inst. 2009, 101, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Schumann, T.; Adhikary, T.; Wortmann, A.; Finkernagel, F.; Lieber, S.; Schnitzer, E.; Legrand, N.; Schober, Y.; Nockher, W.A.; Toth, P.M.; et al. Deregulation of PPARβ/δ target genes in tumor-associated macrophages by fatty acid ligands in the ovarian cancer microenvironment. Oncotarget 2015, 6, 13416–13433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Tan, M.J.; Huang, R.L.; Tan, C.K.; Chong, H.C.; Pal, M.; Lam, C.R.; Boukamp, P.; Pan, J.Y.; Tan, S.H.; et al. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2−:H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell 2011, 19, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Goh, Y.Y.; Chin, H.F.; Kersten, S.; Tan, N.S. Angiopoietin-like 4: A decade of research. Biosci. Rep. 2012, 32, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Martín-Martín, N.; Zabala-Letona, A.; Fernández-Ruiz, S.; Arreal, L.; Camacho, L.; Castillo-Martin, M.; Cortazar, A.R.; Torrano, V.; Astobiza, I.; Zúñiga-García, P.; et al. PPARδ Elicits Ligand-Independent Repression of Trefoil Factor Family to Limit Prostate Cancer Growth. Cancer Res. 2018, 78, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, S.; Strokin, M.; Sergeeva, M.; Reiser, G. Peroxisome proliferator-activated receptor (PPAR)β/δ, a possible nexus of PPARα- and PPARγ-dependent molecular pathways in neurodegenerative diseases: Review and novel hypotheses. Neurochem. Int. 2013, 63, 322–330. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonosante, A.; D’Angelo, M.; Castelli, V.; Catanesi, M.; Iannotta, D.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. The Involvement of PPARs in the Peculiar Energetic Metabolism of Tumor Cells. Int. J. Mol. Sci. 2018, 19, 1907. https://doi.org/10.3390/ijms19071907
Antonosante A, D’Angelo M, Castelli V, Catanesi M, Iannotta D, Giordano A, Ippoliti R, Benedetti E, Cimini A. The Involvement of PPARs in the Peculiar Energetic Metabolism of Tumor Cells. International Journal of Molecular Sciences. 2018; 19(7):1907. https://doi.org/10.3390/ijms19071907
Chicago/Turabian StyleAntonosante, Andrea, Michele D’Angelo, Vanessa Castelli, Mariano Catanesi, Dalila Iannotta, Antonio Giordano, Rodolfo Ippoliti, Elisabetta Benedetti, and Annamaria Cimini. 2018. "The Involvement of PPARs in the Peculiar Energetic Metabolism of Tumor Cells" International Journal of Molecular Sciences 19, no. 7: 1907. https://doi.org/10.3390/ijms19071907
APA StyleAntonosante, A., D’Angelo, M., Castelli, V., Catanesi, M., Iannotta, D., Giordano, A., Ippoliti, R., Benedetti, E., & Cimini, A. (2018). The Involvement of PPARs in the Peculiar Energetic Metabolism of Tumor Cells. International Journal of Molecular Sciences, 19(7), 1907. https://doi.org/10.3390/ijms19071907