The Making of Hematopoiesis: Developmental Ancestry and Environmental Nurture



Abstract
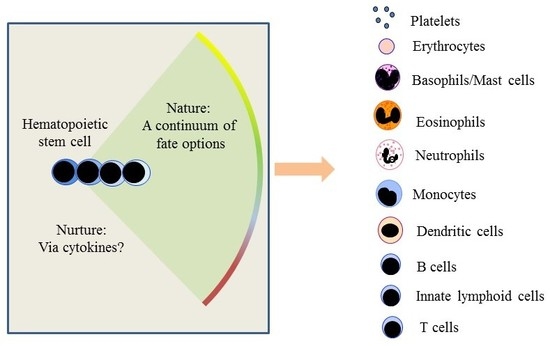
:
{kind=link}
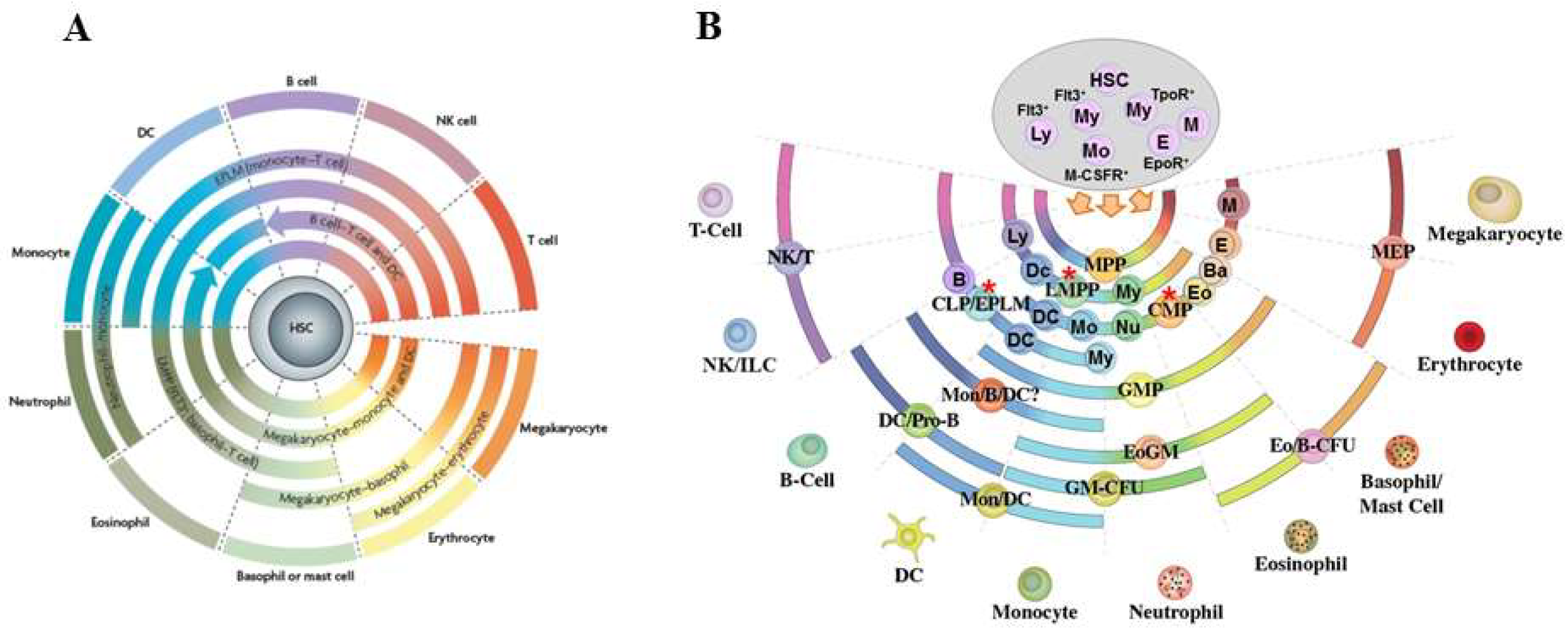{kind=link}
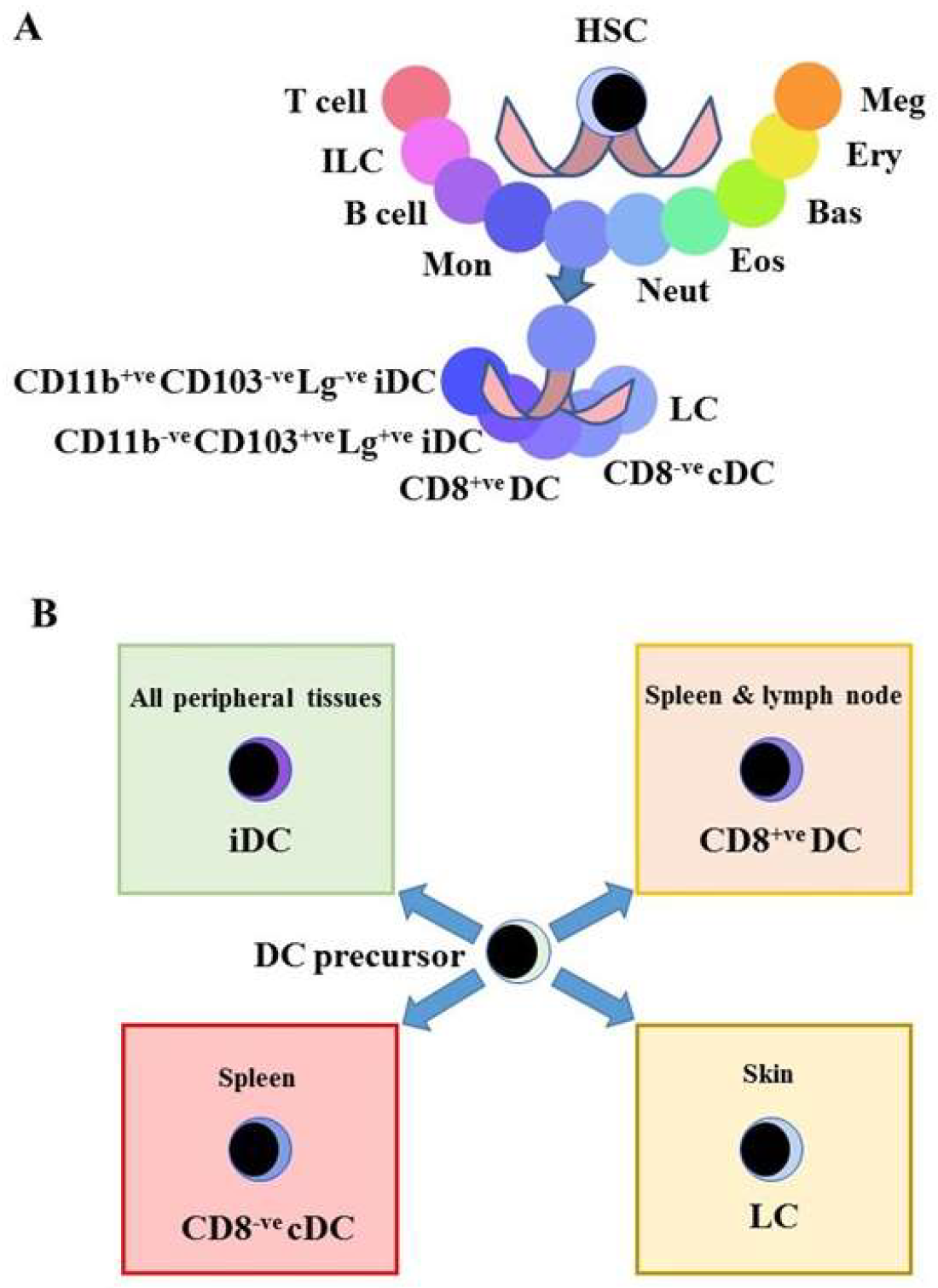{kind=link}
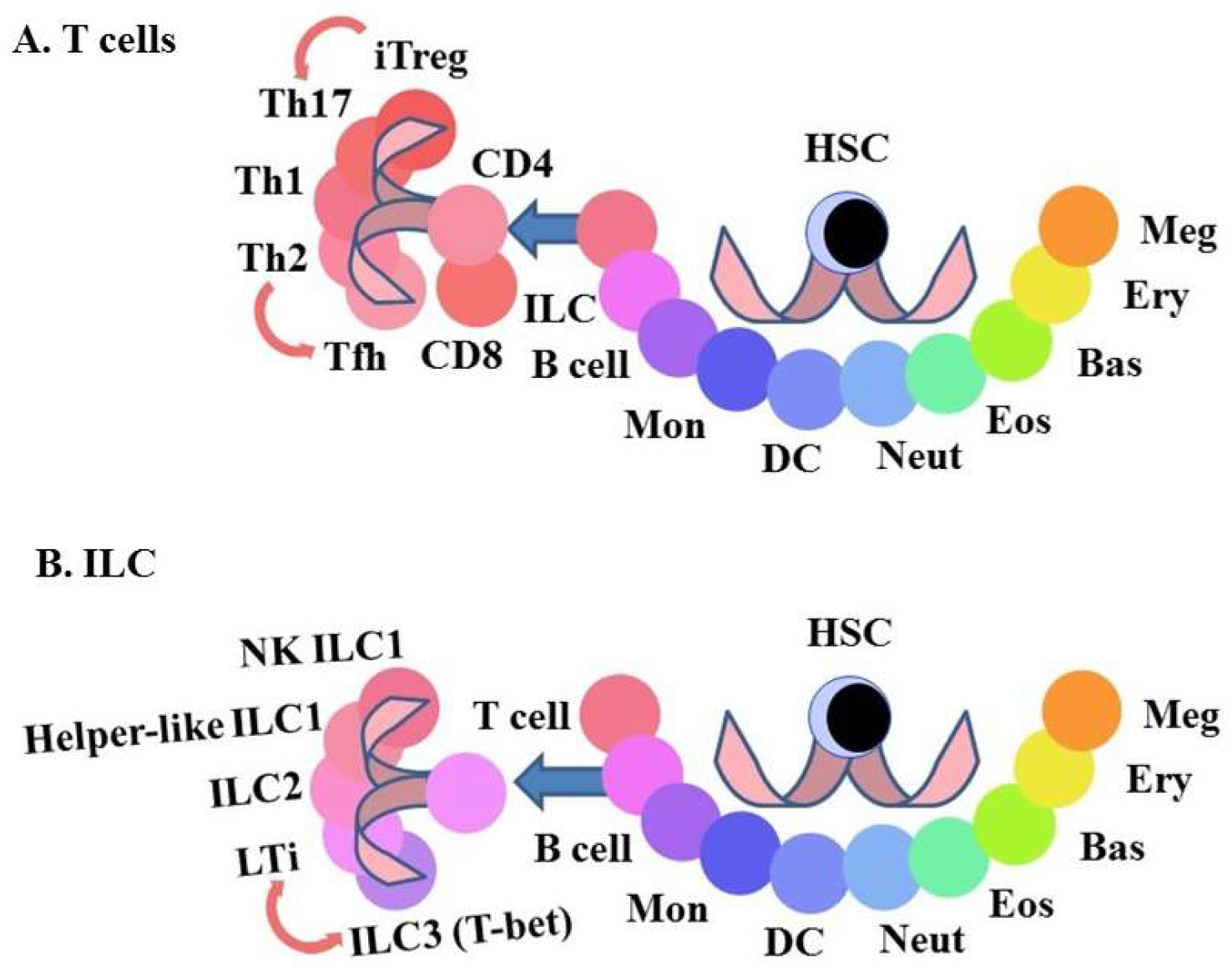{kind=link}
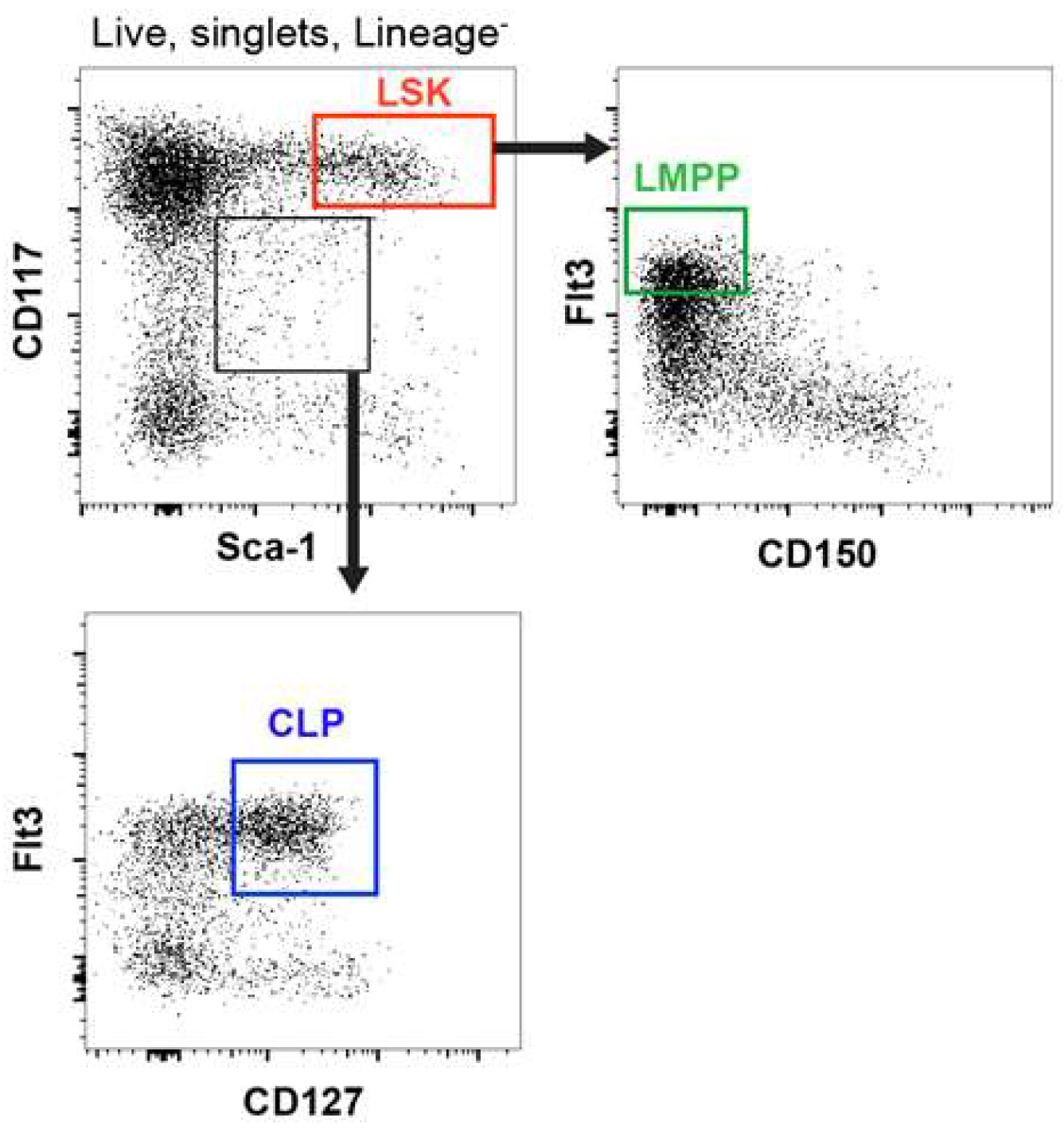{kind=link}
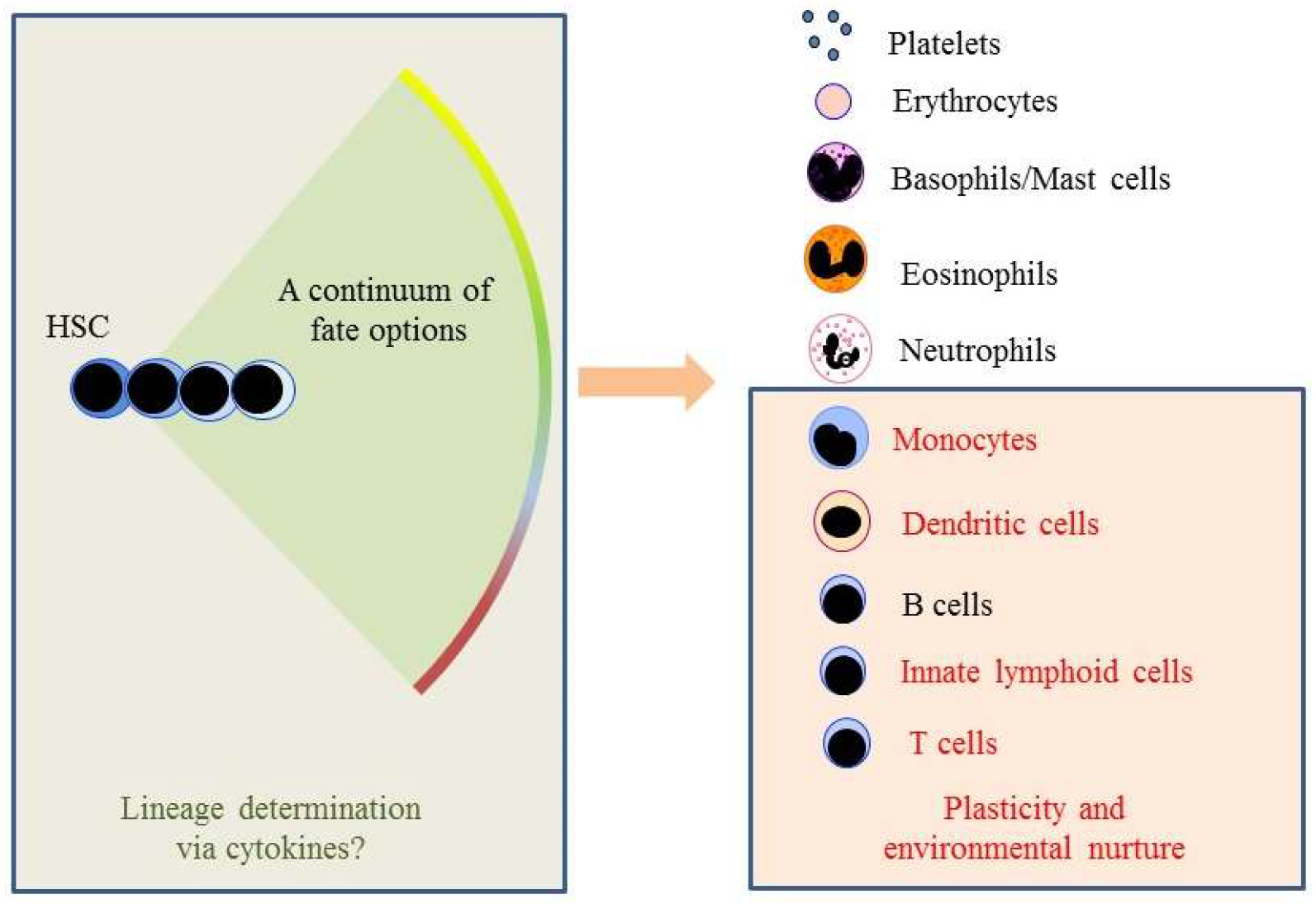{kind=link}
1. Introduction
2. The Mature End-Cell Populations Are Not Homogeneous
3. Some End-Cells Are Interconvertible
4. How Might We Classify the Types of Cells?
5. What Are the Differences between Types of Cells?
6. Does the Environment That Cells Reside in Instruct Cell Fate?
7. The Events That Shape Cell Identity
8. Implications for Leukemia
9. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Sulston, J.E.; Schierenberg, E.; White, J.G.; Thomson, J.N. The embryonic cell lineage of the nematode caenorhabditis elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef]
- Weissman, I.L.; Anderson, D.J.; Gage, F. Stem and progenitor cells: Origins, phenotypes, lineage commitments and transdifferentiations. Annu. Rev. Cell. Dev. Biol. 2001, 17, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.A. Motile and social behaviour of neural crest cells. In Cell Behavior; Bellairs, R., Curtis, A., Dunn, G., Eds.; Cambridge University Press: London, UK, 1981; pp. 429–470. [Google Scholar]
- Metcalf, D.; Burgess, A.W. Clonal analysis of progenitor cell commitment of granulocyte or macrophage production. J. Cell. Physiol. 1982, 111, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Weiss, M.J.; Poncz, M. Megakaryocyte biology and related disorders. J. Clin. Invest. 2005, 115, 3332–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceredig, R.; Rolink, A.G.; Brown, G. Models of haematopoiesis: Seeing the wood for the trees. Nat. Rev. Immunol. 2009, 9, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Grinenko, T.; Eugster, A.; Thielecke, L.; Ramasz, B.; Kruger, A.; Dietz, S.; Glauche, I.; Gerbaulet, A.; von Bonin, M.; Basak, O.; et al. Hematopoietic stem cells can differentiate into restricted myeloid progenitors before cell division in mice. Nat. Commun. 2018, 9, 1898. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Hughes, P.J.; Michell, R.H. Cell differentiation and proliferation—Simultaneous but independent? Exp. Cell. Res. 2003, 291, 282–288. [Google Scholar] [CrossRef]
- Drayson, M.T.; Michell, R.H.; Durham, J.; Brown, G. Cell proliferation and cd11b expression are controlled independently during hl60 cell differentiation initiated by 1,25α-dihydroxyvitamin D3 or all-trans-retinoic acid. Exp. Cell. Res. 2001, 266, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kueh, H.Y.; Champhekhar, A.; Nutt, S.L.; Elowitz, M.B.; Rothenberg, E.V. Positive feedback between pu.1 and the cell cycle controls myeloid differentiation. Science 2013, 341, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Hough, S.R.; Laslett, A.L.; Grimmond, S.B.; Kolle, G.; Pera, M.F. A continuum of cell states spans pluripotency and lineage commitment in human embryonic stem cells. PLoS ONE 2009, 4, e7708. [Google Scholar] [CrossRef] [PubMed]
- Morgani, S.; Nichols, J.; Hadjantonakis, A.K. The many faces of pluripotency: In vitro adaptations of a continuum of in vivo states. BMC Dev. Biol. 2017, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Elghetany, M.T. Surface antigen changes during normal neutrophilic development: A critical review. Blood Cells Mol. Dis. 2002, 28, 260–274. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.E.; Catherman, S.C.; Palis, J. Delineating stages of erythropoiesis using imaging flow cytometry. Methods 2017, 112, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Ciarletta, P.; Ben Amar, M.; Labouesse, M. Continuum model of epithelial morphogenesis during caenorhabditis elegans embryonic elongation. Philos. T. Roy. Soc. A. 2009, 367, 3379–3400. [Google Scholar] [CrossRef] [PubMed]
- Alberti-Servera, L.; von Muenchow, L.; Tsapogas, P.; Capoferri, G.; Eschbach, K.; Beisel, C.; Ceredig, R.; Ivanek, R.; Rolink, A. Single-cell RNA sequencing reveals developmental heterogeneity among early lymphoid progenitors. EMBO J. 2017, 36, 3619–3633. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, P.S.; Schwarzfischer, M.; Loeffler, D.; Kokkaliaris, K.D.; Hilsenbeck, O.; Moritz, N.; Endele, M.; Filipczyk, A.; Gambardella, A.; Ahmed, N.; et al. Early myeloid lineage choice is not initiated by random pu.1 to gata1 protein ratios. Nature 2016, 535, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.H.; Perie, L.; Swart, E.; Gerlach, C.; van Rooij, N.; de Boer, R.J.; Schumacher, T.N. Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature 2013, 496, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, aab2116. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.; Arkin, Y.; Giladi, A.; Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Winter, D.; Lara-Astiaso, D.; Gury, M.; Weiner, A.; et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 2015, 163, 1663–1677. [Google Scholar] [CrossRef] [PubMed]
- Balciunaite, G.; Ceredig, R.; Massa, S.; Rolink, A.G. A b220+ cd117+ cd19- hematopoietic progenitor with potent lymphoid and myeloid developmental potential. Eur. J. Immunol. 2005, 35, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Hughes, P.J.; Michell, R.H.; Ceredig, R. The versatility of haematopoietic stem cells: Implications for leukaemia. Crit. Rev. Clin. Lab. Sci. 2010, 47, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Ema, H.; Morita, Y.; Suda, T. Heterogeneity and hierarchy of hematopoietic stem cells. Exp. Hematol. 2014, 42, 74–82.e2. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Iida, R.; Zhang, Q.; Welner, R.S.; Medina, K.L.; Alberola-Lla, J.; Kincade, P.W. Cd86 is expressed on murine hematopoietic stem cells and denotes lymphopoietic potential. Blood 2012, 119, 4889–4897. [Google Scholar] [CrossRef] [PubMed]
- Mooney, C.J.; Cunningham, A.; Tsapogas, P.; Toellner, K.M.; Brown, G. Selective expression of flt3 within the mouse hematopoietic stem cell compartment. Int. J. Mol. Sci. 2017, 18, 1037. [Google Scholar] [CrossRef] [PubMed]
- Gekas, C.; Graf, T. Cd41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 2013, 121, 4463–4472. [Google Scholar] [CrossRef] [PubMed]
- Oguro, H.; Ding, L.; Morrison, S.J. Slam family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 2013, 13, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan-Pla, A.; Macaulay, I.C.; Jensen, C.T.; Woll, P.S.; Luis, T.C.; Mead, A.; Moore, S.; Carella, C.; Matsuoka, S.; Bouriez Jones, T.; et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 2013, 502, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Mancini, E.; Moore, S.; Mead, A.J.; Atkinson, D.; Rasmussen, K.D.; O’Carroll, D.; Jacobsen, S.E.; Nerlov, C. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J. Exp. Med. 2014, 211, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossadegh-Keller, N.; Sarrazin, S.; Kandalla, P.K.; Espinosa, L.; Stanley, E.R.; Nutt, S.L.; Moore, J.; Sieweke, M.H. M-csf instructs myeloid lineage fate in single haematopoietic stem cells. Nature 2013, 497, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Ichii, M.; Shimazu, T.; Welner, R.S.; Garrett, K.P.; Zhang, Q.; Esplin, B.L.; Kincade, P.W. Functional diversity of stem and progenitor cells with b-lymphopoietic potential. Immunol. Rev. 2010, 237, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D.; Rossi, D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 5465–5470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challen, G.A.; Boles, N.C.; Chambers, S.M.; Goodell, M.A. Distinct hematopoietic stem cell subtypes are differentially regulated by tgf-beta1. Cell Stem Cell 2010, 6, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Tsapogas, P.; Swee, L.K.; Nusser, A.; Nuber, N.; Kreuzaler, M.; Capoferri, G.; Rolink, H.; Ceredig, R.; Rolink, A. In vivo evidence for an instructive role of fms-like tyrosine kinase-3 (flt3) ligand in hematopoietic development. Haematologica 2014, 99, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Collin, M.P.; Bogunovic, M.; Abel, M.; Leboeuf, M.; Helft, J.; Ochando, J.; Kissenpfennig, A.; Malissen, B.; Grisotto, M.; et al. Blood-derived dermal langerin+ dendritic cells survey the skin in the steady state. J. Exp. Med. 2007, 204, 3133–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestle, F.O.; Zheng, X.G.; Thompson, C.B.; Turka, L.A.; Nickoloff, B.J. Characterization of dermal dendritic cells obtained from normal human skin reveals phenotypic and functionally distinctive subsets. J. Immunol. 1993, 151, 6535–6545. [Google Scholar] [PubMed]
- De Smedt, T.; Pajak, B.; Muraille, E.; Lespagnard, L.; Heinen, E.; De Baetselier, P.; Urbain, J.; Leo, O.; Moser, M. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J. Exp. Med. 1996, 184, 1413–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.J. Ipc: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Gordon, S. Monocyte heterogeneity and innate immunity. Immunity 2003, 19, 2–4. [Google Scholar] [CrossRef]
- Satpathy, A.T.; Murphy, K.M.; Kc, W. Transcription factor networks in dendritic cell development. Semin. Immunol. 2011, 23, 388–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, S.H. Demystifying the development of dendritic cell subtypes, a little. Immunol. Cell Biol. 2008, 86, 439–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hume, D.A. Differentiation and heterogeneity in the mononuclear phagocyte system. Mucosal Immunol. 2008, 1, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Hume, D.A. The mononuclear phagocyte system. Curr. Opin. Immunol. 2006, 18, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, R.L.; Shakhar, G.; Dudziak, D.; Wardemann, H.; Eisenreich, T.; Dustin, M.L.; Nussenzweig, M.C. Visualizing dendritic cell networks in vivo. Nat. Immunol. 2004, 5, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Bursch, L.S.; Wang, L.; Igyarto, B.; Kissenpfennig, A.; Malissen, B.; Kaplan, D.H.; Hogquist, K.A. Identification of a novel population of langerin+ dendritic cells. J. Exp. Med. 2007, 204, 3147–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravasi, T.; Wells, C.; Forest, A.; Underhill, D.M.; Wainwright, B.J.; Aderem, A.; Grimmond, S.; Hume, D.A. Generation of diversity in the innate immune system: Macrophage heterogeneity arises from gene-autonomous transcriptional probability of individual inducible genes. J. Immunol. 2002, 168, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Fogg, D.K.; Sibon, C.; Miled, C.; Jung, S.; Aucouturier, P.; Littman, D.R.; Cumano, A.; Geissmann, F. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 2006, 311, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Yamane, H.; Paul, W.E. Cytokines of the γC family control CD4+ t cell differentiation and function. Nat. Immunol. 2012, 13, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Murphy, K.M.; Sher, A. Functional diversity of helper t lymphocytes. Nature 1996, 383, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Wurster, A.L.; Rodgers, V.L.; Satoskar, A.R.; Whitters, M.J.; Young, D.A.; Collins, M.; Grusby, M.J. Interleukin 21 is a t helper (th) cell 2 cytokine that specifically inhibits the differentiation of naive th cells into interferon gamma-producing th1 cells. J. Exp. Med. 2002, 196, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.I.; Chung, Y.; Hwang, D.; Yang, X.O.; Kang, H.S.; Ma, L.; Wang, Y.H.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Generation of t follicular helper cells is mediated by interleukin-21 but independent of t helper 1, 2, or 17 cell lineages. Immunity 2008, 29, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Glatman Zaretsky, A.; Taylor, J.J.; King, I.L.; Marshall, F.A.; Mohrs, M.; Pearce, E.J. T follicular helper cells differentiate from th2 cells in response to helminth antigens. J. Exp. Med. 2009, 206, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Gatzka, M.; Peters, T.; Borkner, L.; Hainzl, A.; Wang, H.; Sindrilaru, A.; Scharffetter-Kochanek, K. Reduced cd18 levels drive regulatory t cell conversion into th17 cells in the cd18hypo pl/j mouse model of psoriasis. J. Immunol. 2013, 190, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, M.; Komatsu, N.; Kawamoto, S.; Suzuki, K.; Kanagawa, O.; Honjo, T.; Hori, S.; Fagarasan, S. Preferential generation of follicular b helper t cells from foxp3+ T cells in gut peyer’s patches. Science 2009, 323, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Souabni, A.; Flavell, R.A.; Wan, Y.Y. An intrinsic mechanism predisposes foxp3-expressing regulatory t cells to th2 conversion in vivo. J. Immunol. 2010, 185, 5983–5992. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Kim, I.K.; Park, Y.J.; Kim, Y.S.; Kim, Y.J.; Chang, W.S.; Lee, Y.S.; Kweon, M.N.; Chung, Y.; Kang, C.Y. Conversion of th2 memory cells into foxp3+ regulatory t cells suppressing th2-mediated allergic asthma. Proc. Natl. Acad. Sci. USA 2010, 107, 8742–8747. [Google Scholar] [CrossRef] [PubMed]
- Eizenberg-Magar, I.; Rimer, J.; Zaretsky, I.; Lara-Astiaso, D.; Reich-Zeliger, S.; Friedman, N. Diverse continuum of CD4+ T-cell states is determined by hierarchical additive integration of cytokine signals. Proc. Natl. Acad. Sci. USA 2017, 114, E6447–E6456. [Google Scholar] [CrossRef] [PubMed]
- Gronke, K.; Kofoed-Nielsen, M.; Diefenbach, A. Innate lymphoid cells, precursors and plasticity. Immunol. Lett. 2016, 179, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Rankin, L.C.; Groom, J.R.; Chopin, M.; Herold, M.J.; Walker, J.A.; Mielke, L.A.; McKenzie, A.N.; Carotta, S.; Nutt, S.L.; Belz, G.T. The transcription factor t-bet is essential for the development of nkp46+ innate lymphocytes via the notch pathway. Nat. Immunol. 2013, 14, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Rankin, L.C.; Girard-Madoux, M.J.; Seillet, C.; Mielke, L.A.; Kerdiles, Y.; Fenis, A.; Wieduwild, E.; Putoczki, T.; Mondot, S.; Lantz, O.; et al. Complementarity and redundancy of il-22-producing innate lymphoid cells. Nat. Immunol. 2016, 17, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Sawa, S.; Cherrier, M.; Lochner, M.; Satoh-Takayama, N.; Fehling, H.J.; Langa, F.; Di Santo, J.P.; Eberl, G. Lineage relationship analysis of rorgammat+ innate lymphoid cells. Science 2010, 330, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.S.; Kearley, J.; Copenhaver, A.M.; Sanden, C.; Mori, M.; Yu, L.; Pritchard, G.H.; Berlin, A.A.; Hunter, C.A.; Bowler, R.; et al. Inflammatory triggers associated with exacerbations of copd orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nature immunology 2016, 17, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, F.; Niiro, H.; Iino, T.; Yoshida, S.; Saito, N.; Onohara, S.; Miyamoto, T.; Minagawa, H.; Fujii, S.; Shultz, L.D.; et al. The developmental program of human dendritic cells is operated independently of conventional myeloid and lymphoid pathways. Blood 2007, 110, 3591–3660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Victora, G.D.; Schwickert, T.A.; Guermonprez, P.; Meredith, M.M.; Yao, K.; Chu, F.F.; Randolph, G.J.; Rudensky, A.Y.; Nussenzweig, M. In vivo analysis of dendritic cell development and homeostasis. Science 2009, 324, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.H.; Sathe, P.; Park, H.Y.; Metcalf, D.; Proietto, A.I.; Dakic, A.; Carotta, S.; O’Keeffe, M.; Bahlo, M.; Papenfuss, A.; et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat. Immunol. 2007, 8, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Onai, N.; Kurabayashi, K.; Hosoi-Amaike, M.; Toyama-Sorimachi, N.; Matsushima, K.; Inaba, K.; Ohteki, T. A clonogenic progenitor with prominent plasmacytoid dendritic cell developmental potential. Immunity 2013, 38, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.F.; Alberti-Servera, L.; Eremin, A.; Grajales-Reyes, G.E.; Ivanek, R.; Tussiwand, R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat. Immunol. 2018, 19, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Schlitzer, A.; Loschko, J.; Mair, K.; Vogelmann, R.; Henkel, L.; Einwachter, H.; Schiemann, M.; Niess, J.H.; Reindl, W.; Krug, A. Identification of CCR9- murine plasmacytoid DC precursors with plasticity to differentiate into conventional DCs. Blood 2011, 117, 6562–6570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathe, P.; Vremec, D.; Wu, L.; Corcoran, L.; Shortman, K. Convergent differentiation: Myeloid and lymphoid pathways to murine plasmacytoid dendritic cells. Blood 2013, 121, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, H.; Reizis, B.; Iwasaki, H.; Mizuno, S.; Hu, D.; Traver, D.; Leder, P.; Sakaguchi, N.; Akashi, K. Plasmacytoid dendritic cells activate lymphoid-specific genetic programs irrespective of their cellular origin. Immunity 2004, 21, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Adolfsson, J.; Mansson, R.; Buza-Vidas, N.; Hultquist, A.; Liuba, K.; Jensen, C.T.; Bryder, D.; Yang, L.; Borge, O.J.; Thoren, L.A.; et al. Identification of flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell 2005, 121, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Karsunky, H.; Inlay, M.A.; Serwold, T.; Bhattacharya, D.; Weissman, I.L. Flk2+ common lymphoid progenitors possess equivalent differentiation potential for the b and t lineages. Blood 2008, 111, 5562–5570. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Weissman, I.L.; Akashi, K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 1997, 91, 661–672. [Google Scholar] [CrossRef]
- Elsasser, W.M. Outline of a theory of cellular heterogeneity. Proc. Natl. Acad. Sci. USA 1984, 81, 5126–5129. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Hemberg, M.; Barahona, M.; Ingber, D.E.; Huang, S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 2008, 453, 544–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suslov, O.N.; Kukekov, V.G.; Ignatova, T.N.; Steindler, D.A. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc. Natl. Acad. Sci. USA 2002, 99, 14506–14511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porritt, H.E.; Rumfelt, L.L.; Tabrizifard, S.; Schmitt, T.M.; Zuniga-Pflucker, J.C.; Petrie, H.T. Heterogeneity among dn1 prothymocytes reveals multiple progenitors with different capacities to generate t cell and non-t cell lineages. Immunity 2004, 20, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Enver, T.; Heyworth, C.M.; Dexter, T.M. Do stem cells play dice? Blood 1998, 92, 348–351. [Google Scholar] [PubMed]
- Enver, T.; Jacobsen, S.E. Developmental biology: Instructions writ in blood. Nature 2009, 461, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D. Lineage commitment and maturation in hematopoietic cells: The case for extrinsic regulation. Blood 1998, 92, 345–347. [Google Scholar] [PubMed]
- Endele, M.; Etzrodt, M.; Schroeder, T. Instruction of hematopoietic lineage choice by cytokine signaling. Exp. Cell Res. 2014, 329, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D. Hematopoietic cytokines. Blood 2008, 111, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrazin, S.; Sieweke, M. Integration of cytokine and transcription factor signals in hematopoietic stem cell commitment. Semin. Immunol. 2011, 23, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Akashi, K.; Kondo, M.; von Freeden-Jeffry, U.; Murray, R.; Weissman, I.L. Bcl-2 rescues t lymphopoiesis in interleukin-7 receptor-deficient mice. Cell 1997, 89, 1033–1041. [Google Scholar] [CrossRef]
- Kondo, M.; Akashi, K.; Domen, J.; Sugamura, K.; Weissman, I.L. Bcl-2 rescues t lymphopoiesis but not b or nk cell development, in common gamma chain-deficient mice. Immunity 1997, 7, 155–162. [Google Scholar] [CrossRef]
- Maraskovsky, E.; O’Reilly, L.A.; Teepe, M.; Corcoran, L.M.; Peschon, J.J.; Strasser, A. Bcl-2 can rescue t lymphocyte development in interleukin-7 receptor-deficient mice but not in mutant rag-1-/- mice. Cell 1997, 89, 1011–1019. [Google Scholar] [CrossRef]
- Malin, S.; McManus, S.; Cobaleda, C.; Novatchkova, M.; Delogu, A.; Bouillet, P.; Strasser, A.; Busslinger, M. Role of stat5 in controlling cell survival and immunoglobulin gene recombination during pro-b cell development. Nat. Immunol. 2010, 11, 171–179. [Google Scholar] [CrossRef] [PubMed]
- von Muenchow, L.; Alberti-Servera, L.; Klein, F.; Capoferri, G.; Finke, D.; Ceredig, R.; Rolink, A.; Tsapogas, P. Permissive roles of cytokines interleukin-7 and flt3 ligand in mouse B-cell lineage commitment. Proc. Natl. Acad. Sci. USA 2016, 113, E8122–E8130. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, R.; Ziegler, S.; Ghilardi, N.; Ledermann, B.; de Sauvage, F.J.; Skoda, R.C. Permissive role of thrombopoietin and granulocyte colony-stimulating factor receptors in hematopoietic cell fate decisions in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 698–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semerad, C.L.; Poursine-Laurent, J.; Liu, F.; Link, D.C. A role for g-csf receptor signaling in the regulation of hematopoietic cell function but not lineage commitment or differentiation. Immunity 1999, 11, 153–161. [Google Scholar] [CrossRef]
- Borzillo, G.V.; Ashmun, R.A.; Sherr, C.J. Macrophage lineage switching of murine early pre-b lymphoid cells expressing transduced fms genes. Mol. Cell. Biol. 1990, 10, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, G.; Grasset, M.F.; Arnaud, S.; Blanchet, J.P.; Mouchiroud, G. Receptor for macrophage colony-stimulating factor transduces a signal decreasing erythroid potential in the multipotent hematopoietic eml cell line. Exp. Hematol. 2000, 28, 1164–1173. [Google Scholar] [CrossRef]
- Iwasaki-Arai, J.; Iwasaki, H.; Miyamoto, T.; Watanabe, S.; Akashi, K. Enforced granulocyte/macrophage colony-stimulating factor signals do not support lymphopoiesis but instruct lymphoid to myelomonocytic lineage conversion. J. Exp. Med. 2003, 197, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- King, A.G.; Kondo, M.; Scherer, D.C.; Weissman, I.L. Lineage infidelity in myeloid cells with tcr gene rearrangement: A latent developmental potential of prot cells revealed by ectopic cytokine receptor signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 4508–4513. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Scherer, D.C.; Miyamoto, T.; King, A.G.; Akashi, K.; Sugamura, K.; Weissman, I.L. Cell-fate conversion of lymphoid-committed progenitors by instructive actions of cytokines. Nature 2000, 407, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Onai, N.; Obata-Onai, A.; Tussiwand, R.; Lanzavecchia, A.; Manz, M.G. Activation of the flt3 signal transduction cascade rescues and enhances type i interferon-producing and dendritic cell development. J. Exp. Med. 2006, 203, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Rieger, M.A.; Hoppe, P.S.; Smejkal, B.M.; Eitelhuber, A.C.; Schroeder, T. Hematopoietic cytokines can instruct lineage choice. Science 2009, 325, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Tsapogas, P.; Ceredig, R. The changing face of hematopoiesis: A spectrum of options is available to stem cells. Immunol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Grinenko, T.; Ramasz, B.; Franke, K.; Lesche, M.; Dahl, A.; Gassmann, M.; Chavakis, T.; Henry, I.; Wielockx, B. Hematopoietic stem cells but not multipotent progenitors drive erythropoiesis during chronic erythroid stress in epo transgenic mice. Stem Cell Rep. 2018, 10, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M.; Dzierzak, E. The many faces of hematopoietic stem cell heterogeneity. Development 2016, 143, 4571–4581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddington, C.H. The Strategy of the Genes; A Discussion of Some Aspects of Theoretical Biology; Allen & Unwin: London, UK, 1957. [Google Scholar]
- Ferrell, J.E., Jr. Bistability, bifurcations and waddington’s epigenetic landscape. Curr. Biol. 2012, 22, R458–R466. [Google Scholar] [CrossRef] [PubMed]
- Sieweke, M.H. Waddington’s valleys and captain cook’s islands. Cell Stem Cell 2015, 16, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Sanchez-Garcia, I. Is lineage decision-making restricted during tumoral reprograming of haematopoietic stem cells? Oncotarget 2015, 6, 43326–43341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.; Sánchez-García, I. Diversity, Versatility and Leukaemia; Nova Biomedical: New York, NY, USA, 2016. [Google Scholar]
- Gonzalez-Herrero, I.; Rodriguez-Hernandez, G.; Luengas-Martinez, A.; Isidro-Hernandez, M.; Jimenez, R.; Garcia-Cenador, M.B.; Garcia-Criado, F.J.; Sanchez-Garcia, I.; Vicente-Duenas, C. The making of leukemia. Int. J. Mol. Sci. 2018, 19, 1494. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, G.; Ceredig, R.; Tsapogas, P. The Making of Hematopoiesis: Developmental Ancestry and Environmental Nurture. Int. J. Mol. Sci. 2018, 19, 2122. https://doi.org/10.3390/ijms19072122
Brown G, Ceredig R, Tsapogas P. The Making of Hematopoiesis: Developmental Ancestry and Environmental Nurture. International Journal of Molecular Sciences. 2018; 19(7):2122. https://doi.org/10.3390/ijms19072122
Chicago/Turabian StyleBrown, Geoffrey, Rhodri Ceredig, and Panagiotis Tsapogas. 2018. "The Making of Hematopoiesis: Developmental Ancestry and Environmental Nurture" International Journal of Molecular Sciences 19, no. 7: 2122. https://doi.org/10.3390/ijms19072122
APA StyleBrown, G., Ceredig, R., & Tsapogas, P. (2018). The Making of Hematopoiesis: Developmental Ancestry and Environmental Nurture. International Journal of Molecular Sciences, 19(7), 2122. https://doi.org/10.3390/ijms19072122